
Abstract
Background
The study of late-onset/age-related Alzheimer’s disease (AD)(sporadic AD, 95% of AD cases) has been hampered by a paucity of animal models. Oxidative stress is considered a causative factor in late onset/age-related AD, and aldehyde dehydrogenase 2 (ALDH2) is important for the catabolism of toxic aldehydes associated with oxidative stress. One such toxic aldehyde, the lipid peroxidation product 4-hydroxynonenal (HNE), accumulates in AD brain and is associated with AD pathology. Given this linkage, we hypothesized that in mice lacking ALDH2, there would be increases in HNE and the appearance of AD-like pathological changes.
Results
Changes in relevant AD markers in Aldh2 -/- mice and their wildtype littermates were assessed over a 1 year period. Marked increases in HNE adducts arise in hippocampi from Aldh2 -/- mice, as well as age-related increases in amyloid-beta, p-tau, and activated caspases. Also observed were age-related decreases in pGSK3β, PSD95, synaptophysin, CREB and pCREB. Age-related memory deficits in the novel object recognition and Y maze tasks begin at 3.5-4 months and are maximal at 6.5-7 months. There was decreased performance in the Morris Water Maze task in 6 month old Aldh2 -/- mice. These mice exhibited endothelial dysfunction, increased amyloid-beta in cerebral microvessels, decreases in carbachol-induced pCREB and pERK formation in hippocampal slices, and brain atrophy. These AD-associated pathological changes are rarely observed as a constellation in current AD animal models.
Conclusions
We believe that this new model of age-related cognitive impairment will provide new insight into the pathogenesis and molecular/cellular mechanisms driving neurodegenerative diseases of aging such as AD, and will prove useful for assessing the efficacy of therapeutic agents for improving memory and for slowing, preventing, or reversing AD progression.
Similar content being viewed by others

Background
Alzheimer’s disease (AD) is characterized by intraneuronal and extracellular accumulation of amyloid-β peptide (Aβ), intracellular neurofibrillary tangles (NFTs), the major component of which is hyperphosphorylated tau protein (p-tau), disruption of both excitatory amino acid and cholinergic neurotransmission, and loss of vulnerable neurons, notably forebrain cholinergic neurons that project to the cerebral cortex and hippocampus. The search for critically needed AD therapeutics has been dominated by use of a variety of transgenic mouse models of AD, which exhibit neuropathological changes dependent on the overexpression of mutant human genes linked to early-onset, familial AD. However, genetic aberrations account for only a small proportion of AD cases (<5%). The models exhibit Aβ pathology, but rarely together with neuronal loss and aberrant tau phosphorylation. In contrast, the study of late-onset/age-related AD (sporadic AD, >95% of AD cases) has been hampered by a paucity of animal models that mirror age-related progression of AD pathologies. Animal models that develop pathology because the animals age, rather than because the pathology is genetically programmed, would be a valuable addition to currently available transgenic models and would be helpful in assessing intervention strategies that slow or reverse the underlying disease process in addition to relieving symptoms.
The early appearance of oxidative stress markers in AD patients and in animal models of AD that precedes the onset of cognitive decline and appearance of Aβ plaques and NFTs [1-5], suggests that oxidative damage may be a primary driving force in AD pathogenesis, especially synaptic dysfunction. This forms the basis of the oxidative stress hypothesis of AD [6-8]. 4-hydroxynonenal (HNE) is an important lipid peroxidation product formed during periods of oxidative stress. Both free HNE and HNE protein adducts accumulate in the brains of AD patients [9-16] and in certain transgenic mouse models of AD [5,17]. Many of these proteins have been identified, and oxidative modifications by HNE adduct formation often results in altered activity [reviewed in 18]. These include proteins involved in the regulation of energy metabolism, antioxidant defense, neuronal communication, stress responses, cytoskeletal integrity and cell signaling. This provides the basis for mechanisms of oxidative stress-induced damage that complement or contribute to Aβ-mediated toxicity. HNE also alters the function and disposition of Aβ, through changes in Aβ formation, aggregation, catabolism and clearance [19-27].
The enzyme aldehyde dehydrogenase 2 (ALDH2) is important for the detoxification of endogenous aldehydes such as HNE [28,29], and inhibition of ALDH2 increases vulnerability to HNE-induced damage [30]. ALDH2 is expressed in the frontal and temporal cortex, hippocampus, mid-brain, basal ganglia and cerebellum, primarily in glial cells and neuropil [28]. Importantly, ALDH2 has been localized to reactive glia within senile plaques in the cerebral cortex and hippocampus, and its expression/activity is increased in the temporal cortex and putamen in AD brains [28,31].
Cross-sectional studies have examined the association of AD risk for individuals possessing the Glu504Lys loss of function mutation of ALDH2 (present in 30-50% of the East Asian population). Although meta-analysis of these studies indicated no increased risk of AD associated with the variant ALDH2, subgroup analysis did indicate that the association was significant for males [32]. In one of the few animal studies reported, Ohsawa et al. [33] found that introduction of this mutation into mice resulted in increased HNE formation associated with age-dependent neurodegeneration and memory loss. Taken together these data suggest that not only is the ALDH2 pathway critical for the detoxification of HNE in the brain, but also that levels of toxic aldehydes derived from oxidative stress are sufficient to cause neuronal loss and cognitive impairment.
Given the potential importance of HNE in AD pathogenesis and Aβ disposition, we hypothesized that genetic manipulations that increase HNE levels would result in biochemical, histopathological, and cognitive changes that mirror those found in AD. This was assessed using Aldh2 -/- mice as the model.
Results
Behavioural analyses
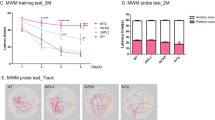
Using the open field novel object recognition (NOR) test and spontaneous alternations in the Y-maze, Aldh2 -/- mice showed a progressive decrease in performance in both memory tasks compared to no change in performance in their wildtype littermates (Figures 1 and 2). Memory deficits began at 3.5-4 months of age and reached a plateau in 6.5 to 7 month old animals. All data were initially tested using a three-way analysis of variance in order to determine if there were sex differences in any of the memory tasks, and this not being the case, data from male and female mice were combined. We also assessed the reproducibility of performance measures between three consecutive generations of 5.5-6 month old animals (Figure 3) and noted very little variation between cohorts. In the Morris Water Maze (MWM) task, there was a significant difference in escape latency for Aldh2 -/- mice compared to wildtype mice in all trial blocks in which the platform was hidden (Figure 4A, trial blocks 4-8). One might not have expected a difference in performance in the first block of hidden platform training, since this was the first instance of the animals having to use spatial cues to find the hidden platform. However, in the first trial of the six trials in this block, there was no difference in escape latency between wildtype and Aldh2 -/- mice (37.3 ± 21.1 s and 41.1 ± 22.1 s, respectively, p > 0.05, Students t-test for unpaired data), indicating that the basis for the better overall performance of the wildtype mice in the first block was due shorter escape latencies in the latter trials of the block. In the probe trial, Aldh2 -/- mice spent less time in the target quadrant and had fewer platform crosses compared to wildtype mice (Figure 4B, C). Indeed, Aldh2 -/- mice spent essentially the same amount of time in each quadrant, whereas wildtype mice spent about three times more time in the target quadrant than the other quadrants. Differences in locomotor activity and coordination, assessed using the balance beam task (Additional file 1: Figure S1), as well as behavioural phenotype assessment using the SHIRPA standardized battery (Tables 1 and 2) [34] were not observed in either 2-3 month old or 5-6 month old wildtype and Aldh2 -/- mice, suggesting that diminished cognitive performance was the result of impaired memory and not due to confounding impairments in motor function. This was reinforced by the finding of no difference in latency times in the MWM task during the 3 day cued platform training, in which mice swim to a visible platform (and also suggests no differences in eyesight, swim speed, basic strategies and motivation between wildtype and Aldh2 -/- mice). Also, we did not observe nonspecific behavioural changes such as floating or thygmotaxis in the Aldh2 -/- mice.

Age-dependent, progressive decline in the Novel Object Recognition (NOR) task performance in Aldh2 -/- mice. Male and female mice were subjected to the NOR task once per month beginning at three months of age, and the frequency of visits to the objects and the time spent exploring each object was recorded. A and B. Frequency of visits to the novel and familiar objects for wildtype mice (A) and Aldh2 -/- mice (B). (C). Ratio of time spent with the novel object in relation to the familiar object. (D). Discrimination index. Data are presented as the mean ± SD (wildtype n = 18, Aldh2 -/- n = 17) and were analyzed by two-way ANOVA with a Bonferroni post-hoc test. In A and B, ***, significant difference to the novel object (p < 0.001). In C and D, *, significant difference from wildtype as indicated (*p < 0.05, **, P < 0.01, ***p < 0.001); ψψψ, significant difference from 3.5-4 month old Aldh2 -/- mice (p < 0.001).

Age-dependent, progressive decline in Y-maze task performance in Aldh2 -/- mice. The spontaneous alternation rate in the Y-maze task was assessed in male and female mice once per month beginning at three months of age. Data are presented as the mean ± SD (wildtype n = 18, Aldh2 -/- n = 17) and were analyzed by two-way ANOVA with a Bonferroni post-hoc test. *, significant difference from wildtype as indicated (*p < 0.05, **, p < 0.01, ***p < 0.001); ψψψ, significant difference from 3.5-4 month old Aldh2 -/- mice (p < 0.001).

Consistency of NOR and Y-maze tasks in successive cohorts of wildtype and Aldh2 -/- mice. Three generations of 5.5-6 month old wildtype and Aldh2 -/- mice were tested in the NOR (A) and Y-maze (B) tasks. Data are presented as the mean ± SD (wildtype n = 8-18, Aldh2 -/- n = 11-17) and were analyzed by two-way ANOVA. There were no differences between cohorts of either wildtype or Aldh2 -/- mice for either behavioural test.

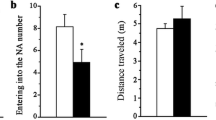
Six month old Aldh2 -/- mice exhibit decreased performance in the Morris Water Maze task. (A). Escape latency (time to reach the hidden platform) was determined in a 3 day cued trial block (4 trials per day) followed by a 5 day hidden trial block (6 trials per day). Day 9 was a probe trial, in which the time spent in the target quadrant (NE)(B) and number of platform crosses (C) were determined (total time of the trial was 60 s). In (A), data are expressed as the mean ± SD of the average scores in each trial block (n = 18). In B and C, data are expressed as the mean ± SD (n = 18). In B, the dotted line represents the expected time spent in each quadrant by chance alone. Data were analyzed by 2-way ANOVA with a Bonferroni post-hoc test in A and B, and by a Student’s t-test for unpaired data in C. In A and C, *, significant difference from wildtype as indicated (*p < 0.05, ** p < 0.01, ***p < 0.001). In B, ***, significant difference from wildtype in the other quadrants; (p < 0.001); ψψψ, significant difference from Aldh2 -/- in the NE quadrant (p < 0.001).
Immunoblot analysis of AD markers
HNE adduct formation in Aldh2 -/- mice was characterized by increases in both the density and number of immunoreactive bands (Figure 5). Because of this, we used dot-blot analysis for HNE adduct assessment in subsequent experiments. We followed the age-related changes of a number of relevant markers over a 12 month period at 3 month intervals in hippocampal homogenates from Aldh2 -/- mice (Figure 6; representative immunoblots and HNE dot-blots from 9 month old animals in Figure 7). Marked increases in HNE adduct formation occur as early as 3 months (Figure 6A). Age-related increases in monomeric Aβ and phospho-tau protein (p-tau) were significant at 6 months of age, increasing over the next 6 months (Figure 6B and C). Tau phosphorylation was assessed using the AT-8 antibody which recognizes phosphorylated tau at Ser202, one of the epitopes considered critical for AD progression. Amyloid precursor protein (APP) and total tau expression were unchanged (Figure 7). In addition, a number of oligomeric Aβ (oAβ) species were present in hippocampi from Aldh2 -/- mice (Figure 8). We observed age-related increases in cleaved (activated) caspases 3 and 6 as early as 3 months of age (Figure 6D and E), and age-related decreases in the postsynaptic marker PSD95, the presynaptic marker, synaptophysin, and both total and phosphorylated (Ser133) cAMP-response element binding protein (CREB) (Figure 6F-I). Glycogen synthase kinase 3β (GSK3β) is the most active tau kinase, and Aldh2 -/- mice exhibit age-related decreases in phosphorylation at the major inhibitory site (Ser9) of GSK3β (Figure 9). Increased expression of nicastrin, the γ-secretase substrate receptor, and decreased expression of neprilysin, a major Aβ degrading enzyme, were observed in the hippocampus of older Aldh2 -/- mice (Figure 10).

Increased HNE adduct formation in hippocampi from Aldh2 -/- mice. Immunoblot analysis of hippocampal homogenates (30 μg protein) from 9 month old wildtype or Aldh2 -/- mice. Proteins were resolved on a 10% SDS-PAGE gel under non-reducing conditions. Immunoreactive bands were quantitated by densitometry. Data are presented as the mean ± SD (n = 3-4) and were analyzed by a Student’s t-test for unpaired data. *** significant difference from wildtype (p < 0.001).

Summary data of immunoblot and dot-blot (HNE) analysis of age-related changes in various AD- and synapse-related markers (panels A-I, as indicated) in hippocampal homogenates (30 μg protein) from wildtype or Aldh2 -/- mice. Immunoreactive bands or spots were quantitated by densitometry. Data are presented as the mean ± SD (n = 4 mice) and were analyzed by a two-way ANOVA with a Bonferroni post-hoc test. *, significant differences from wildtype (*p < 0.05, ***p < 0.001); ψ, significant age-related differences as indicated (ψ p < 0.05, ψψ p < 0.001, ψψψ p < 0.001).

Immunoblot and dot-blot (HNE) analysis of hippocampal homogenates (30 μg protein) from 9 month old wildtype or Aldh2 -/- mice (n = 4 of each). Note the increase in a number of AD-associated markers in Aldh2 -/- mice (top part of figure) and the decrease in synaptic markers (bottom part of figure).

Differential expression of oligomeric forms of Aβ in the hippocampus and cerebral microvasculature of Aldh2 -/- mice. Immunoblot analysis of hippocampal homogenates (upper panel, 30 μg protein) and cerebral microvessel homogenates (lower panel, 30 μg protein) from 12 month old wildtype or Aldh2 -/- mice. Samples were added to Laemmli buffer containing 6 M urea, incubated at room temperature for 30 minutes and then resolved on a 15% SDS-PAGE gel. Note the increased expression of APP in the vasculature of Aldh2 -/- mice in contrast to the equal expression of APP in the hippocampus.

Age-related decreases in phospho-GSK3β (Ser9) in hippocampi from Aldh2 -/- mice. Immunoblot analysis was performed on hippocampal homogenates (30 μg protein) from 3, 6, 9 and 12 month old wildtype or Aldh2 -/- mice and probed for phosphorylated GSK3β and β-actin. A representative immunoblot is shown from 9 month animals. Immunoreactive bands were quantitated by densitometry. Data are presented as the mean ± SD (n = 4 mice) and were analyzed by a two-way ANOVA with a Bonferroni post-hoc test. *, significant differences from wildtype (**p < 0.01, ***p < 0.001).

Increased nicastrin and decreased neprilysin expression in hippocampi from 12 month Aldh2 -/- mice. Immunoblot analysis of hippocampal homogenates (30 μg protein) from 12 month old wildtype and Aldh2 -/- mice probed for nicastrin (A) and neprilysin (B). Immunoreactive bands were quantitated by densitometry. Data are presented as the mean ± SD (n = 4 mice) and were analyzed by Student’s t-test for unpaired data. ***, significant difference from wildtype (p < 0.001).
CREB and extracellular signal-regulated kinase (ERK) activation in Hippocampal slices
In hippocampal slices from 6 month old animals, the increases in pCREB and pERK seen in wildtype mice in response to the cholinergic agonist, carbachol, were absent in Aldh2 -/- mice (Figures 11 and 12), indicating a deficit in cholinergic receptor signaling. In contrast, levels of total CREB and total ERK were unchanged in both wildtype and Aldh2 -/- mice after carbachol treatment (Additional file 2: Figures S2 and Additional file 3: Figure S3).

Lack of carbachol-induced phosphorylation of CREB in hippocampi from Aldh2 -/- mice. Hippocampal slices from 6 month old wildtype and Aldh2 -/- mice were incubated with 50 μM carbachol (+) or vehicle (-) for 30 min and snap frozen. Immunoblot analysis was performed using 30 μg protein of hippocampal homogenate, and immunoreactive bands were quantitated by densitometry. Data are presented as the mean ± SD (n = 6 mice) and were analyzed by Student’s t-test for unpaired data. ***, significant difference from basal (p < 0.001).

Lack of carbachol-induced phosphorylation of ERK1/2 in hippocampi from Aldh2 -/- mice. Hippocampal slices from 6 month old wildtype and Aldh2 -/- mice were incubated with 50 μM carbachol (+) or vehicle (-) for 30 min and snap frozen. Immunoblot analysis was performed using 30 μg protein of hippocampal homogenate, and immunoreactive bands were quantitated by densitometry. Data are presented as the mean ± SD (n = 6 mice) and were analyzed by Student’s t-test for unpaired data. ***, significant difference from basal (p < 0.001).
Assessment of hippocampal atrophy
The calculated area (mean ± SD) of sections of the hippocampus and overlying neocortex in 33 sections from 6 wildtype mice was 7.4 ± 1.0 mm3 and in 27 sections from 7 Aldh2 -/- mice was 6.3 ± 0.6 mm3 (p < 0.05, Student’s t-test for unpaired data).
Vascular pathologies in Aldh2 -/- mice
There was a four-fold increase in HNE adducts in homogenates of mouse cerebral microvessels from 3-12 month old Aldh2 -/- mice (Figure 13A). We observed age-related increases in monomeric Aβ (Figure 13B) as well as a number of oAβ assemblies ranging from 3-4-mers to 20-mers (Figure 8). In addition, endothelial dysfunction and hypercontractility was clearly evident in Aldh2 -/- mice, as indicated by the reduced aortic relaxation responses to acetylcholine (ACh) (Figure 13C), and enhanced contractile responses to the adrenoceptor agonist, phenylephrine (Figure 13D).

Vascular pathologies in Aldh2 -/- mice. Cerebral microvessels from Aldh2 -/- mice exhibit increased HNE adduct formation (A) and age-related increases in Aβ formation (B). Representative dot-blots (HNE, 20 μg protein) and immunoblots (Aβ, 30 μg protein) are from 9 month old animals. Immunoreactive spots or bands were quantitated by densitometry. Data are presented as the mean ± SD (n = 4 mice) and were analyzed by a two-way ANOVA with a Bonferroni post-hoc test. ***, significant difference from wildtype (p < 0.001). ψ ψ ψ, significant age-related differences as indicated (p < 0.001). In aortic ring preparations from 12 month old Aldh2 -/- mice there was a significant decrease in both the potency and maximal relaxation response to the endothelium-dependent vasodilator, acetylcholine (ACh)(C), and a significant increase in the potency of the adrenoceptor agonist, phenylephrine (Phe) (D). Data are presented as the mean ± SD (n = 4 mice) and EC50 values for relaxation or contraction were analyzed by Student’s t-test for unpaired data.
Discussion
The Aldh2 -/- mouse exhibits progressive, age-related cognitive deficits together with a constellation of AD-associated pathological changes rarely observed in current transgenic AD animal models. These include increases in Aβ, p-tau and activated caspases, synaptic loss, defective CREB signalling, and several vascular pathologies. Although synaptic failure is an early event in AD progression, overt neurodegeneration is either not exhibited or is not an early event in most transgenic AD models [35]. For example, in the 5XFAD mouse, increases in intraneuronal Aβ and Aβ plaques occur at 1.5-2 months of age, whereas the loss of the post-synaptic marker, PSD95, is not evident until animals are 9 months old, and is preceded by the cognitive deficits that occur in 4-5 month old animals [36]. In contrast, in Aldh2 -/- mice the loss of synaptic markers and the initial deficits in cognitive performance appear together early on in life (at 3 months of age). One advantage of Aldh2 -/- mice compared to the few other models of sporadic AD that have been described are the availability of appropriate genetic controls (unlike SAMP8 mice [37]). Also, cognitive deficits appear early in life (unlike the degus and rat models [38,39]) but in an age-related manner. A third advantage is that the pathological changes are based upon elevations in a mediator of oxidative stress (HNE) that is considered to be an important contributor to the development of AD pathology. Although there are no rodent models that completely reflect the complexity of human AD, we believe the Aldh2 -/- mouse represents a valuable addition to currently available transgenic models.
Many studies support the formation of HNE as an early event in the pathogenesis of AD [9-16]. Three detoxification pathways for HNE have been described: conjugation with glutathione by glutathione transferases, reduction by aldo-keto reductases, and oxidation by ALDH2. Studies investigating the expression and activity of ALDH2 and the 3 major aldo-keto reductases responsible for HNE metabolism in the brain found that only ALDH2 expression and activity is increased in AD brains [28,31]. This up-regulation of ALDH2 suggests a protective response to the increases in lipid peroxidation and reactive aldehydes associated with AD pathogenesis. In contrast, glutathione transferase activity is decreased in several brain regions in AD, including the hippocampus [40], and brain levels of several antioxidant enzymes (superoxide dismutase, catalase, glutathione reductase) are also decreased in AD brains [18].
As an oxidative stress-based model of age-dependent cognitive impairment and neurodegeneration, the Aldh2 -/- mice have distinct advantages over mice expressing the Glu504Lys mutation of ALDH2 (ALDH2*2) [33]. Unlike Aldh2 -/- mice, no pathological or cognitive changes were seen in 6 month old ALDH2*2 animals, and memory impairment did not occur until 12 months (vs 3.5-4 months in Aldh2 -/- mice). In 12 month old ALDH2*2 mice there was less than a 2-fold increase in HNE adduct formation (vs 3-fold increases at 3 months in Aldh2 -/- mice). Also <50% of ALDH2*2 mice exhibited neurodegeneration or hyperphosphorylated tau vs increased p-tau in all 6 month old Aldh2 -/- mice, as well as decreased PSD95, synaptophysin and pCREB in all 3 month old Aldh2 -/- mice. Thus Aldh2 -/- mice demonstrate more consistent expression of pathological changes, and at a much younger age, than do transgenic ALDH2*2 mice, highlighting their greater utility, predictability, and cost-effectiveness.
We assessed cognitive performance using three different memory tests. The NOR task is considered a ‘pure’ working memory test, free of any reference memory component. This behavioural test is particularly relevant since visual association tests have been reported to detect, with high specificity, a substantial proportion of AD patients up to a year before diagnosis [42]. The Y-maze task and the version of Morris Water Maze task used here, are tests for spatial working memory and spatial reference memory, respectively. Aldh2 -/- mice showed a progressive decrease in performance in both the NOR task and the Y-maze tasks, compared to age and sex-matched littermates (Figures 1 and 2). Also, younger (6 month old) animals exhibited decreased performance in the MWM task (Figure 4). Importantly, this progressive decline in memory performance was accompanied by a parallel loss of synaptic markers and of phosphorylated and total CREB (Figure 6F-I).
Age-related increases in monomeric Aβ occur in hippocampi from Aldh2 -/- mice (Figure 6B). In addition, Aβ dimers and oligomeric Aβ (oAβ) of 45 KDa and 75 KDa (corresponding to 10-12-mers and 16-20 mers, respectively) were present in hippocampi from Aldh2 -/- mice (Figure 8). Oligomeric Aβ assemblies are known to induce neurotoxicity in vitro and in vivo [42-44], and both soluble Aβ and oAβ correlate with disease progression in AD patients [45-47]. Because of sequence differences in the N-terminus of human vs. mouse Aβ, the latter is less prone to aggregate, and endogenous mouse Aβ does not readily form amyloid plaques. Since most studies utilize transgenic mouse models, the formation of mouse oAβ has rarely been investigated or reported. In one study utilizing ApoE4 mice, activation of the amyloid cascade after inhibition of neprilysin did result in the accumulation of prefibrillar oAβ, demonstrating that mouse oAβ can indeed be formed endogenously [48].
Significant increases in HNE adducts of neprilysin [49], one of the principal amyloid-degrading enzymes in the brain, and nicastrin [22], the γ-secretase substrate receptor, occur in AD brain. HNE increases the expression and activity of beta-site APP cleaving enzyme (BACE) and γ-secretase resulting in increased Aβ production [24,50,51]. HNE also forms adducts with neprilysin, resulting in decreased enzymatic activity and Aβ turnover [49,52]. We found increases in nicastrin expression as well as decreases in neprilysin expression in the hippocampus of older Aldh2 -/- mice (Figure 10), which likely contribute to the increased levels of Aβ observed in these mice.
Age-dependent increases in p-tau were observed in Aldh2 -/- mice using the AT8 antibody (Figure 6C). This antibody recognizes phosphorylated tau at Ser202, an epitope associated with intracellular and extracellular filamentous tau [53], and one of the epitopes considered critical for AD progression [54]. Increased tau phosphorylation is seldom observed in transgenic models of AD, and therefore its presence in Aldh2 -/- mice suggests increased HNE contributes to tau pathology. One transgenic mouse model that does exhibit hyperphosphorylated tau and NFTs, the 3xTg-AD mouse, also exhibits oxidative stress, increased lipid peroxidation, and HNE formation as early events, prior to plaque and tangle formation. Furthermore, HNE modifies p-tau, which may play a critical role in the formation of NFTs [55]. GSK3β is the most active tau kinase [54], and has been shown to phosphorylate tau at 42 different sites, 29 of which are found in AD brain [56]. Our data (Figure 9) indicate that Aldh2 -/- mice exhibit age-related decreases in phosphorylation at the major inhibitory site (Ser9) of GSK3β in the hippocampus, suggesting GSK3β activity is increased in Aldh2 -/- mice.
Activation of apoptotic pathways through caspase-mediated cell death contributes to progressive neuronal death in AD [57-60], and we observed age-related increases in cleaved (activated) caspases 3 and 6 as early as 3 months of age in Aldh2 -/- mice (Figure 6D, 6E). Caspase 6 activation has been reported in hippocampus and cerebral cortex of cases of mild cognitive impairment, and of familial and sporadic AD [58,61,62]. In sporadic AD, activated caspase 6 is only present in non-nuclear compartments, and it has been suggested that caspase 6 activation is related to neurodegeneration rather than apoptosis (58). Further, a recent study identified a novel mechanism whereby soluble Aβ-induced caspase 3 activation leads to the downregulation of PSD95 and synaptophysin, resulting in synaptic dysfunction [63].
Synaptic failure and loss of synapses, a hallmark of AD pathology, occurs early in disease progression, prior to plaque and NFT formation and neuronal death. Synaptic loss is a key event in early cognitive decline [64] and a highly relevant correlate of cognitive deficits in AD [65]. As an index of neurodegeneration and synaptic loss, we assessed hippocampal levels of the postsynaptic protein, PSD95, a major scaffolding molecule localized at the postsynaptic density of excitatory glutamatergic synapses, and of synaptophysin, a synaptic vesicle protein and presynaptic marker. There were marked, age-related decreases in both PSD95 and synaptophysin beginning at 3 months (Figure 6F, 6G), suggesting that hippocampal synaptic loss in these mice is an early event that occurs concomitant with the development of cognitive deficits. There was also evidence of brain atrophy in these mice; measurements of the area of hippocampal formation and overlying neocortex indicated a 15% reduction in area in Aldh2 -/- mice.
The induction of structural changes at synapses requires activation of gene networks and transcription factors such as CREB, and an association between activity-dependent gene expression and CREB activation is well established [66,67]. Activation of CREB by phosphorylation at Ser133 is critical for memory formation and synaptic strengthening [68] and mediates long term potentiation (LTP) by acting upon downstream genes involved in synaptic formation and maintenance, neuronal plasticity and neurogenesis [69]. Decreased levels of pCREB are seen in the hippocampus of AD brain [70], and molecular network analysis suggests a central role of aberrant CREB-mediated gene regulation in AD [71]. Restoration of CREB signalling restores LTP and regulates gene products, notably BDNF, leading to neuroprotection and restoration of neuronal function. Studies have also reported reciprocal regulation of amyloidogenesis by CREB and direct dysregulation of CREB activation by Aβ [72-75]. We observed marked decreases in basal pCREB and total CREB levels in Aldh2 -/- mice (Figure 6H, 6I). Furthermore, the increases in pCREB and pERK seen in hippocampal slices of the wildtype mice in response to cholinergic receptor activation by carbachol are absent in Aldh2 -/- mice (Figures 11 and 12), indicating a deficit in cholinergic receptor signaling.
In addition to the observed AD-like pathologies in the brains of Aldh2 -/- mice, we found significant vascular alterations in cerebral microvessels (CMVs) of these mice. Marked increases in HNE adducts and age-related increases in monomeric Aβ were found in CMVs from Aldh2 -/- mice compared to wildtype (Figure 13A, 13B), as well as a number of oAβ assemblies ranging from 3-4-mers to 20-mers (Figure 8). Cerebral amyloid angiopathy (CAA) is characterized by the accumulation of Aβ in CMVs. It is a common pathological feature in AD occurring in 60-90% of AD patients [76] and also occurs in some transgenic animal models [77]. The presence of CAA significantly worsens cognitive performance in the early stages of AD [78]. Endothelial dysfunction and arterial hypercontractility have been associated with the AD phenotype [79,80], and the degree of endothelial dysfunction correlates with the AD severity [79]. Endothelial dysfunction and hypercontractility were clearly evident in Aldh2 -/- mice, as indicated by the reduced aortic relaxation responses to acetylcholine that are dependent on endothelial NO release, and by enhanced contractile responses to the adrenoceptor agonist, phenylephrine (Figure 13C, 13D). Thus, this mouse model incorporates both neural and vascular pathological changes associated with AD.
Conclusions
We have characterized a novel oxidative stress-based mouse model of cognitive impairment with AD-like biochemical and structural pathologies based on increased HNE formation due to the genetic deletion of ALDH2. These mice exhibit age-dependent cognitive impairment, synaptic loss, neurodegeneration, altered CREB signalling, increased Aβ, hyperphosphorylated tau, activated caspases, and vascular pathologies. The Aldh2 -/- animal model exhibits elevated Aβ, and more importantly, elevated p-tau and neurodegeneration, significant human pathologies rarely seen together in current transgenic models. We believe that this new model of age-related cognitive impairment is a valuable addition to currently available transgenic models and will provide new insight into the pathogenesis and molecular/cellular mechanisms driving neurodegenerative diseases of aging such as AD. It may be useful for delineating the chronology of appearance of AD biomarkers, identifying targets for early intervention, and assessing of the efficacy of therapeutic agents for improving memory and for slowing, preventing, or reversing the synaptic failure and neurodegeneration associated with AD.
Methods
Generation of Aldh2 -/- mice
All procedures for animal experimentation were undertaken in accordance with the principles and guidelines of the Canadian Council on Animal Care and were approved by the Queen’s University Animal Care Committee. Animals were maintained under a 12 h light/dark cycle, with free access to food and water. The Aldh2 -/- mice have a C57BL/6 background and were generated by gene targeting knockout as previously described [81] and kindly provided by Dr. T. Kawamoto (University of Occupational and Environmental Health, Kitakyushu, Japan). Wildtype male C57BL/6 mice (20 -30 g) were obtained from Jackson Laboratory, (Bar Harbor ME) and backcrossed with Aldh2 -/- mice for more than 10 generations. Wildtype and Aldh2 -/- cohorts were generated by mating heterozygotes, and genotyping of the progeny by PCR analysis of genomic DNA extracted from tail tips or ear punches using the primers as reported [82].
Behavioural analyses
Open-field Novel Object Recognition (NOR) task. The NOR task consisted of three consecutive days of testing per trial; habituation, training with two identical objects, and testing with one familiar and one novel object. On the testing day, animals were allowed to explore the objects until they accumulated 30 seconds of total object exploration time (exploration was recorded when the nose of the mouse was within approximately 1cm of the object). This was done rather than using a set exposure time in the test environment in order to account for any variability in movement and exploration that may occur between mice. Two measures of behaviour were assessed: frequency of visits to the objects, and the time spent exploring each object. From the latter measure the discrimination index (difference in time exploring the novel and familiar object, divided by total exploration time) and the ratio of time spent with the novel object in relation to the familiar object were calculated. Y-maze task. Mice were placed in the center of the maze and allowed to explore the three maze arms freely for 10 min. The maze was surrounded by distinct spatial cues so that the mice could distinguish between arms. The spontaneous alternation rate was calculated as the total triads containing entries into each of the three arms without repeated entry into a previously visited arm, divided by the total number of arm entries. Morris Water Maze task. The maze consisted of a circular pool (1.2m in diameter, [83]) filled with water (approximately 23°C) made opaque by non-toxic white paint. A circular platform (15cm in diameter) was submerged approximately 1cm below the surface in the northeast (NE) quadrant of the maze and, thus, hidden from view. The escape latency (time to reach the hidden platform) was determined in in a 3 day cued trial block (4 trials per day) followed by a 5 day hidden trial block (6 trials per day) [84]. For cued platform training, a black-and-white striped pole (2.2 cm diameter and 15 cm height) was attached to the center of the platform. Each mouse was allowed 60 seconds to locate the platform per trial. Mice that could not find the platform within 60 seconds were gently guided towards it. Day 9 was a 60 second probe trial, in which the time spent in each quadrant and number of platform crosses were recorded. Balance beam task. This task consisted of three consecutive days of testing on a 1 meter long beam with a flat surface (1.8 cm wide) resting about 50cm above the floor on two supports. An empty cage with nesting material served as a finish point at the end of the beam to attract the mouse. Day 1 and day 2 were training days in which the mouse was encouraged to cross the beam 3 times with a 10 minute break between each trial. On day 3, the mice were again given 3 trials, and the two best times required to cross the beam and enter the finish box were averaged [85].
Immunoblot and dot-blot analysis
Mouse hippocampi were isolated and homogenized in lysis buffer (25 mM HEPES pH 7.0, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 0.1% SDS, protease inhibitors and phosphatase inhibitors (Roche Diagnostics, Mannheim, Germany)) and centrifuged at 10000g for 10 minutes. Proteins in the supernatant fraction were separated by SDS-PAGE on 10-15% gels (depending on protein of interest) and transferred electrophoretically to PVDF membranes. For the immunoblot analysis of HNE adducts in Figure 5, proteins were resolved on a 10% SDS-PAGE gel under non-reducing conditions. In all other experiments, HNE adducts were assessed by dot-blot analysis, in which homogenates were spotted onto a PVDF membrane and dried before probing with antibody. For immunoblot analysis of Aβ, samples were added to Laemmli buffer containing 6 M urea and incubated at room temperature for 30 minutes prior to electrophoresis. Immunoblots or HNE dot-blots were probed with primary antibody followed by secondary antibody, and immunoreactive bands or spots visualized by enhanced chemiluminescence. Immunoreactive bands or spots were quantified by optical densitometry using ImageJ software (version 1.43). Blots were stripped and reprobed for β-actin to confirm equal loading. The following antibodies were used: mouse monoclonal to Aβ (W0-2, Millipore, recognizes residues 4-10 of Aβ), mouse monoclonal to APP (2C11, Millipore), rabbit polyclonal to 4-hydroxynonenal (HNE11-S, Alpha Diagnostics International), mouse monoclonal to PHF-tau (AT8, Pierce Thermo Scientific), mouse monoclonal to total tau (ab64193, Abcam), rabbit polyclonal to caspase-3 (ab90437, Abcam), rabbit polyclonal to caspase-6 (ab52295, Abcam), mouse monoclonal to PSD95 (Pierce Thermo Scientific), mouse monoclonal to synaptophysin (ab8049, Abcam), rabbit polyclonal to phospho-CREB (Ser133) (06-519, Millipore), rabbit polyclonal to total CREB (06-863, Millipore), mouse monoclonal to phospho-ERK1/2 (05-481, Millipore), rabbit polyclonal to ERK1/2 (06-182, Millipore), rabbit polyclonal to phospho-GSK3β (Ser9)(ab131097, Abcam), rabbit polyclonal to nicastrin (ab45425, Abcam) mouse monoclonal to neprilysin (ab951, Abcam) and mouse monoclonal to β-actin (Sigma).
CREB and ERK activation in hippocampal slices
Mouse hippocampal mini-slices containing the CA1 region were prepared as we have described for rat [86]. After a 3 h equilibration in Krebs’ buffer, slices were exposed to the cholinergic receptor agonist, carbachol (50 μM) for 30 min, conditions in which we have observed increases in pERK in rat hippocampal mini-slices [86]. After incubation, slices were homogenized in lysis buffer (50mM TRIS, pH 7.4, 50mM sodium pyrophosphate, 1mM EDTA, 1% Triton X-100, 1mM dithiothreitol, 1mM PMSF, 1mM NaF, 1mM NaVO4 and protease and phosphatase inhibitors), and total and phosphorylated ERK and CREB assessed by immunoblot analysis.
Preparation of cerebral microvessels
Mouse cerebral microvessels were prepared using a modified mechanical dispersion and filtration technique as previously described [87]. Briefly, cortices were isolated, cleaned, and carefully homogenized by hand using a Dounce homogenizer in 5-fold excess volume of phosphate-buffered saline with protease and phosphatase inhibitors (Roche Diagnostics, Mannheim, Germany). Dextran was added to a final concentration of 15% and the homogenate was centrifuged (6200g for 30 minutes at 4°C). The pellet was resuspended in the PBS solution and passed through a 40 μm mesh filter to capture microvessels. The microvessels were homogenized in lysis buffer (25 mM HEPES pH 7.0, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 0.1% SDS, protease inhibitors and phosphatase inhibitors (Roche Diagnostics, Mannheim, Germany)) and used for immunoblot and dot-blot analysis.
Assessment of hippocampal atrophy
For assessment of hippocampal atrophy, measurements were made of coronal sections cut at -2.30 to -2.10 Bregma. The neocortical surface and base of the hippocampal formation were outlined and connected by 2 lines bounding the hippocampal formation and meeting the neocortical surface at ninety degrees. The area bounded by these perimeter lines was calculated using the imaging software associated with the microscope.
Isolated blood vessel preparations
Isolated rings of aorta (2-3 mm) were prepared for isometric tension measurements and were equilibrated for 1 h at an optimal resting tension of 5 mN. Cumulative concentration-response curves for phenylephrine (0.1 nM-30 μM) were then obtained. After washout, aortic rings were contracted submaximally with phenylephrine (0.2–5 μM), and after the induced tone had stabilized, cumulative concentration-response curves were obtained for acetylcholine (ACh) (1 nM–10 μM).
Data analysis
Data is expressed as the mean ± SD and was analyzed by one-, two- or three-way analysis of variance with Bonferroni’s post-hoc test, and/or a two-tailed Student’s t test for unpaired data, as indicated. A p-value of 0.05 or less was considered statistically significant. Relaxation responses to ACh were measured as the percentage decrease in phenylephrine-induced tone. EC50 values for relaxation and contraction were determined from the concentration-response curves using a sigmoidal dose–response curve-fitting algorithm. Due to inhomogeneity of variance, statistical analysis for the blood vessel experiments was performed using logarithmically transformed data.
Abbreviations
- Aβ:
-
Amyloid beta peptide
- ACh:
-
Acetylcholine
- AD:
-
Alzheimer’s disease
- ALDH2:
-
Aldehyde dehydrogenase 2
- APP:
-
Amyloid precursor protein
- BACE:
-
Beta-site APP cleaving enzyme
- CAA:
-
Cerebral amyloid angiopathy
- CMV:
-
Cerebral microvessels
- CREB:
-
Cyclic AMP response element binding protein
- ERK:
-
Extracellular signal-regulated kinase
- GSK3β:
-
Glycogen synthase kinase 3β
- HNE:
-
4-hydroxynonenal
- NFT:
-
Neurofibrillary tangle
- NOR:
-
Novel object recognition
References
Lovell MA, Ehmann WD, Butler SM, Markesbery WR. Elevated thiobarbituric acid-reactive substances and antioxidant enzyme activity in the brain in Alzheimer’s disease. Neurology. 1995;45:1594–601.
Hensley K, Maidt ML, Yu Z, Sang H, Markesbery WR, Floyd RA. Electrochemical analysis of protein nitrotyrosine and dityrosine in the Alzheimer brain indicates region-specific accumulation. J Neurosci. 1998;18:8126–2.
Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj EK, et al. Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurology. 2001;60:759–67.
Praticò D, Uryu K, Leight S, Trojanoswki JQ, Lee VM. Increased lipid peroxidation precedes amyloid plaque formation in an animal model of Alzheimer amyloidosis. J Neurosci. 2001;21:4183–7.
Resende R, Moreira PI, Proença T, Deshpande A, Busciglio J, Pereira C, et al. Brain oxidative stress in a triple-transgenic mouse model of Alzheimer disease. Free Radic Biol Med. 2008;44:2051–7.
Markesbery WR. Oxidative stress hypothesis in Alzheimer’s Disease. Free Radic Biol Med. 1997;23:134–47.
Butterfield DA. β-Amyloid-associated free radical oxidative stress and neurotoxicity : Implications for Alzheimer’s disease. Chem Res Toxicol. 1997;10:495–506.
Praticò D. Oxidative stress hypothesis in Alzheimer’s disease: a reappraisal. Trends Pharmacol Sci. 2008;29:609–15.
Lovell MA, Ehmann WD, Mattson MP, Markesbery WR. Elevated 4-hydroxynonenal in ventricular fluid in Alzheimer’s disease. Neurobiol Aging. 1997;18:457–61.
Markesbery WR, Lovell MA. Four-hydroxynonenal, a product of lipid peroxidation, is increased in the brain in Alzheimerʼs disease. Neurobiol Aging. 1998;19:33–6.
McGrath LT, McGleenon BM, Brennan S, McColl D, McIlroy S, Passmore AP. Increased oxidative stress in Alzheimer’s disease as assessed with 4-hydroxynonenal but not malondialdehyde. Q J Med. 2001;94:485–90.
Sayre LM, Zelasko DA, Harris PL, Perry G, Salomon RG, Smith MA. 4-Hydroxynonenal-derived advanced lipid peroxidation end products are increased in Alzheimer’s disease. J Neurochem. 1997;68:2092–7.
Montine KS, Olson SJ, Amarnath V, Whetsell WO, Graham DG, Montine TJ. Immunohistochemical detection of 4-hydroxy-2-nonenal adducts in Alzheimerʼs disease is associated with inheritance of APOE4. Am J Path. 1997;150:437–43.
Fukuda M, Kanou F, Shimada N, Sawabe M, Saito Y, Murayama S, et al. Elevated levels of 4-hydroxynonenal-histidine Michael adduct in the hippocampi of patients with Alzheimer’s disease. Biomed Res. 2009;30:227–33.
Reed TT, Pierce WM, Markesbery WR, Butterfield DA. Proteomic identification of HNE-bound proteins in early Alzheimer disease: Insights into the role of lipid peroxidation in the progression of AD. Brain Res. 2009;1274:66–76.
Butterfield DA, Bader Lange ML, Sultana R. Involvements of the lipid peroxidation product, HNE, in the pathogenesis and progression of Alzheimer’s disease. Biochim Biophys Act. 1801;2010:924–9.
Butterfield DA, Galvan V, Lange MB, Tang H, Sowell RA, Spilman P, et al. In vivo oxidative stress in brain of Alzheimer disease transgenic mice: Requirement for methionine 35 in amyloid beta-peptide of APP. Free Radic Biol Med. 2010;48:136–44.
Sultana R, Perluigi M, Allan BD. Lipid peroxidation triggers neurodegeneration: A redox proteomics view into the Alzheimer Disease brain. Free Radic Biol Med. 2013;62:157–69.
Ando Y, Brannstrom T, Uchida K, Nyhlin N, Nasman B, Suhr O, et al. Histochemical detection of 4-hydroxynonenal protein in Alzheimer amyloid. J Neurol Sci. 1998;156:172–6.
Liu L, Komatsu H, Murray IVJ, Axelsen PH. Promotion of amyloid beta protein misfolding and fibrillogenesis by a lipid oxidation product. J Mol Biol. 2008;377:1236–50.
Siegel SJ, Bieschke J, Powers ET, Kelly JW. The oxidative stress metabolite 4-hydroxynonenal promotes Alzheimer protofibril formation. Biochemistry. 2008;46:1503–10.
Gwon A-R, Park J-S, Arumugam TV, Kwon Y-K, Chan SL, Kim S-H, et al. Oxidative lipid modification of nicastrin enhances amyloidogenic γ-secretase activity in Alzheimer’s disease. Aging Cell. 2012;11:559–68.
Chen L, Na R, Gu M, Richardson A, Ran Q. Lipid peroxidation up-regulates BACE1 expression in vivo: a possible early event of amyloidogenesis in Alzheimer’s disease. J Neurochem. 2008;107:197–207.
Tamagno E, Parola M, Bardini P, Piccini A, Borghi R, Guglielmotto M, et al. Beta-site APP cleaving enzyme up-regulation induced by 4-hydroxynonenal is mediated by stress-activated protein kinases pathways. J Neurochem. 2005;92:628–36.
Wang D-S, Iwata N, Hama E, Saido TC, Dickson DW. Oxidized neprilysin in aging and Alzheimer’s disease brains. Biochem Biophys Res Commun. 2003;310:236–41.
Wang R, Wang S, Malter JS, Wang D. Effects of HNE-modification induced by Aβ on neprilysin expression and activity in SH-SY5Y cells. J Neurochem. 2009;108:1072–82.
Owen JB, Sultana R, Aluise CD, Erickson MA, Price TO, Bu G, et al. Oxidative modification to LDL receptor- related protein 1 in hippocampus from subjects with Alzheimer disease: implications for Abeta accumulation in AD brain. Free Radic Biol Med. 2010;49:1798–803.
Picklo MJ, Olson SJ, Markesbery WR, Montine TJ. Expression and activities of aldo-keto oxidoreductases in Alzheimer disease. J Neuropath Exp Neurol. 2001;60:686–95.
Yoval-Sánchez B, Rodríguez-Zavala JS. Differences in susceptibility to inactivation of human aldehyde dehydrogenases by lipid peroxidation byproducts. Chem Res Toxicol. 2012;25:722–9.
Ohsawa I, Nishimaki K, Yasuda C, Kamino K, Ohta S. Deficiency in a mitochondrial aldehyde dehydrogenase increases vulnerability to oxidative stress in PC12 cells. J Neurochem. 2003;84:1110–7.
Michel TM, Gsell W, Käsbauer L, Tatschner T, Sheldrick AJ, Neuner I, et al. Increased activity of mitochondrial aldehyde dehydrogenase (ALDH) in the putamen of individuals with Alzheimer’s disease: a human postmortem study. J Alzheimers Dis. 2010;19:1295–301.
Hao P, Chen Y, Wang J. Meta-analysis of aldehyde dehydrogenase 2 gene polymorphism and Alzheimer’s disease in East Asians. Can J Neurolog Sci. 2011;38:500–6.
Ohsawa I, Nishimaki K, Murakami Y, Suzuki Y, Ishikawa M, Ohta S. Age-dependent neurodegeneration accompanying memory loss in transgenic mice defective in mitochondrial aldehyde dehydrogenase 2 activity. J Neurosci. 2008;28:6239–49.
Rogers DC, Jones DN, Nelson PR, Jones CM, Quilter CA, Robinson TL, et al. Use of SHIRPA and discriminant analysis to characterise marked differences in the behavioural phenotype of six inbred mouse strains. Behav Brain Res. 1999;105:207–17.
Palmer AM. Neuroprotective therapeutics for Alzheimer’s disease: progress and prospects. Trends Pharmacol Sci. 2011;32:141–7.
Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006;26:10129–40.
Pallas M, Camins A, Smith MA, Perry G, Lee H, Casadesus G. From aging to Alzheimer’s Disease : Unveiling “The Switch” with the senescence-accelerated mouse model. J Alzheimer’s Dis. 2008;15:615–024.
Ardiles AO, Tapia-Rojas CC, Mandal M, Alexandre F, Kirkwood A, Inestrosa NC, et al. Postsynaptic dysfunction is associated with spatial and object recognition memory loss in a natural model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2012;109:13835–40.
Cohen RM, Rezai-Zadeh K, Weitz TM, Rentsendorj A, Gate D, Spivak I, et al. A transgenic Alzheimer rat with plaques, tau pathology, behavioral impairment, oligomeric aβ, and frank neuronal loss. J Neurosci. 2013;33:6245–56.
Lovell MA, Xie C, Markesbery WR. Decreased glutathione transferase activity in brain and ventricular fluid in Alzheimer’s disease. Neurology. 1998;51:1562–6.
Lindeboom J, Schmand B, Tulner L, Walstra G, Jonker C. Visual association test to detect early dementia of the Alzheimer type. J Neurol Neurosurg Psychiatry. 2002;73:126–33.
Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8:101–12.
Walsh DM, Selkoe DJ. A beta oligomers - a decade of discovery. J Neurochem. 2007;101:1172–84.
Wilcox KC, Lacor PN, Pitt J, Klein WL. Abeta oligomer-induced synapse degeneration in Alzheimer’s disease. Cell Mol Neurobiol. 2011;31:939–48.
Kuo YM, Emmerling MR, Vigo-Pelfrey C, Kasunic TC, Kirkpatrick JB, Murdoch GH, et al. Water-soluble Abeta (N-40, N-42) oligomers in normal and Alzheimer disease brains. J Biol Chem. 1996;271:4077–81.
Selkoe DJ. Resolving controversies on the path to Alzheimer’s therapeutics. Nat Med. 2011;17:1060–5.
Tomic JL, Pensalfini A, Head E, Glabe CG. Soluble fibrillar oligomer levels are elevated in Alzheimer’s disease brain and correlate with cognitive dysfunction. Neurobiol Dis. 2009;35:352–8.
Belinson H, Kariv-Inbal Z, Kayed R, Masliah E, Michaelson DM. Following activation of the amyloid cascade, apolipoprotein E4 drives the in vivo oligomerization of amyloid-β resulting in neurodegeneration. J Alzheimers Dis. 2010;22:959–70.
Wang D-S, Iwata N, Hama E, Saido TC, Dickson DW. Oxidized neprilysin in aging and Alzheimer’s disease brains. Biochem Biophys Res Commun. 2003;310:236–41.
Tamagno E, Guglielmotto M, Aragno M, Borghi R, Autelli R, Giliberto L, et al. Oxidative stress activates a positive feedback between the gamma- and beta-secretase cleavages of the beta-amyloid precursor protein. J Neurochem. 2008;104:683–95.
Jo DG, Arumugam TV, Woo HN, Park JS, Tang SC, Mughal M, et al. Evidence that gamma-secretase mediates oxidative stress-induced beta-secretase expression in Alzheimer’s disease. Neurobiol Aging. 2010;31:917–25.
Wang R, Wang S, Malter JS, Wang DS. Effects of HNE-modification induced by Abeta on neprilysin expression and activity in SH-SY5Y cells. J Neurochem. 2009;108:1072–82.
Augustinack JC, Schneider A, Mandelkow EM, Hyman BT. Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer’s disease. Acta Neuropathol. 2002;103:26–35.
Cavallini A, Brewerton S, Bell A, Sargent S, Glover S, Hardy C, et al. An unbiased approach to identifying tau kinases that phosphorylate tau at sites associated with Alzheimer disease. J Biol Chem. 2013;288:23331–47.
Liu Q, Smith MA, Avilá J, DeBernardis J, Kansal M, Takeda A, et al. Alzheimer-specific epitopes of tau represent lipid peroxidation-induced conformations. Free Radic Biol Med. 2005;38:746–54.
Hanger DP, Anderton BH, Noble W. Tau phosphorylation: the therapeutic challenge for neurodegenerative disease. Trends Mol Med. 2009;15:112–9.
Louneva N, Cohen JW, Han LY, Talbot K, Wilson RS, Bennett DA, et al. Caspase-3 is enriched in postsynaptic densities and increased in Alzheimer’s disease. Am J Pathol. 2008;173:1488–95.
Guo H, Albrecht S, Bourdeau M, Petzke T, Bergeron C, LeBlanc AC. Active caspase-6 and caspase-6-cleaved tau in neuropil threads, neuritic plaques, and neurofibrillary tangles of Alzheimer’s disease. Am J Pathol. 2004;165:523–31.
Graham RK, Ehrnhoefer DE, Hayden MR. Caspase-6 and neurodegeneration. Trends Neurosci. 2011;34:646–56.
Snigdha S, Smith ED, Prieto GA, Cotman CW. Caspase-3 activation as a bifurcation point between plasticity and cell death. Neurosci Bull. 2012;28:14–24.
Albrecht S, Bourdeau M, Bennett D, Mufson EJ, Bhattacharjee M, LeBlanc AC. Activation of caspase-6 in aging and mild cognitive impairment. Am J Pathol. 2007;170:1200–9.
Albrecht S, Bogdanovic N, Ghetti B, Winblad B, LeBlanc AC. Caspase-6 activation in familial Alzheimer disease brains carrying amyloid precursor protein, presenilin I or presenilin II mutations. J Neuropathol Exp Neurol. 2009;68:1282–93.
Liu J, Chang L, Roselli F, Almeida OF, Gao X, Wang X, et al. Amyloid-β induces caspase-dependent loss of PSD-95 and synaptophysin through NMDA receptors. J Alzheimers Dis. 2010;22:541–56.
Selkoe D. J. Alzheimer’s disease is a synaptic failure. Science. 2002;298:789–91.
Arendt T. Synaptic degeneration in Alzheimer’s disease. Acta Neuropathol. 2009;118:167–79.
Greenberg ME, Ziff EB, Greene LA. Stimulation of neuronal acetylcholine receptors induces rapid gene transcription. Science. 1986;234:80–3.
Montminy MR, Bilezikjian LM. Binding of a nuclear protein to the cyclic-AMP response element of the somatostatin gene. Nature. 1987;328:175–8.
Silva AJ, Kogan JH, Frankland PW, Kida S. CREB and memory. Annu Rev Neurosci. 1998;21:127–48.
Bito H, Deisseroth K, Tsien RW. CREB phosphorylation and dephosphorylation: a Ca(2+)- and stimulus duration-dependent switch for hippocampal gene expression. Cell. 1996;87:1203–14.
Yamamoto-Sasaki M, Ozawa H, Saito T, Rösler M, Riederer P. Impaired phosphorylation of cyclic AMP response element binding protein in the hippocampus of dementia of the Alzheimer type. Brain Res. 1999;824:300–3.
Satoh J, Tabunoki H, Arima K. Molecular network analysis suggests aberrant CREB-mediated gene regulation in the Alzheimer disease hippocampus. Dis Markers. 2009;27:239–52.
Puzzo D, Vitolo O, Trinchese F, Jacob JP, Palmeri A, Arancio O. Amyloid-beta peptide inhibits activation of the nitric oxide/cGMP/cAMP-responsive element-binding protein pathway during hippocampal synaptic plasticity. J Neurosci. 2005;25:6887–97.
Ma QL, Harris-White ME, Ubeda OJ, Simmons M, Beech W, Lim GP, et al. Evidence of Abeta- and transgene-dependent defects in ERK-CREB signaling in Alzheimer’s models. J Neurochem. 2007;103:1594–607.
Zheng Z, Sabirzhanov B, Keifer J. Oligomeric amyloid-{beta} inhibits the proteolytic conversion of brain-derived neurotrophic factor (BDNF), AMPA receptor trafficking, and classical conditioning. J Biol Chem. 2010;285:34708–17.
Saura CA, Valero J. The role of CREB signaling in Alzheimer’s disease and other cognitive disorders. Rev Neurosci. 2011;22:153–69.
Kalaria RN, Ballard C. Overlap between pathology of Alzheimer disease and vascular dementia. Alzheimer Dis Assoc Disord. 1999;13 Suppl 3:S115–23.
Shineman DW, Basi GS, Bizon JL, Colton CA, Greenberg BD, Hollister BA, et al. Accelerating drug discovery for Alzheimer’s disease: best practices for preclinical animal studies. Alzheimers Res Ther. 2011;3:28.
Esiri MM, Nagy Z, Smith MZ, Barnetson L, Smith AD. Cerebrovascular disease and threshold for dementia in the early stages of Alzheimer’s disease. Lancet. 1999;354:919–20.
Dede DS, Yavuz B, Yavuz BB, Cankurtaran M, Halil M, Ulger Z, et al. Assessment of endothelial function in Alzheimer’s disease: is Alzheimer’s disease a vascular disease? J Am Geriatr Soc. 2007;55:1613–7.
Chow N, Bell RD, Deane R, Streb JW, Chen J, Brooks A, et al. Serum response factor and myocardin mediate arterial hyper- contractility and cerebral blood flow dysregulation in Alzheimer’s phenotype. Proc Natl Acad Sci U S A. 2007;104:823–8.
Kitagawa K, Kawamoto T, Kunugita N, Tsukiyama T, Okamoto K, Yoshida A, et al. Aldehyde dehydrogenase (ALDH) 2 associates with oxidation of methoxyacetaldehyde; in vitro analysis with liver subcellular fraction derived from human and Aldh2 gene targeting mouse. FEBS Lett. 2000;476:306–11.
Isse T, Oyama T, Kitagawa K, Matsuno K, Matsumoto A, Yoshida A, et al. Diminished alcohol preference in transgenic mice lacking aldehyde dehydrogenase activity. Pharmacogenetics. 2002;12:621–6.
Van Dam D, Lenders G, De Deyn PP. Effect of Morris water maze diameter on visual-spatial learning in different mouse strains. Neurobiol Learn Mem. 2006;85:164–72.
Delpolyi AR, Fang S, Palop JJ, Yu G, Wang X, Mucke L. Altered navigational strategy use and visuospatial deficits in hAPP transgenic mice. Neurobiol Aging. 2006;29:253–66.
Luong TN, Carlisle HJ, Southwell A, Patterson PH. Assessment of Motor Balance and Coordination in Mice using the Balance Beam. J Vis Exp. 2011; e2376.
Bennett BM, Reynolds JN, Prusky GT, Douglas RM, Sutherland RJ, Thatcher GRJ. Cognitive deficits in rats after forebrain cholinergic depletion are reversed by a novel NO mimetic nitrate ester. Neuropsychopharmacology. 2007;32:505–13.
Brendel K, Meezan E, Carlson EC. Isolated brain microvessels: a purified, metabolically active preparation from bovine cerebral cortex. Science. 1974;185:953–5.
Acknowledgments
The authors wish to thank Mrs. Diane Anderson, Lihua Xue and Margo Poklewska-Koziell for technical assistance. Aldh2 -/- mice were kindly provided by Dr. T. Kawamoto, University of Occupational and Environmental Health, Kitakyushu, Japan. This work was supported by a grant from the Harry Botterell Foundation. Thanks also to Dr. Khem Jhamandas for his critical review of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors have no competing interests.
Authors’ contributions
YD, AH, RDA and BMB conceived and designed the experiments and contributed to writing the manuscript. YD performed the biochemical analyses and associated data analysis. AH performed the behavioural testing and associated data analysis. RS-S performed the hippocampal atrophy assessments. All authors read and approved the final manuscript.
Additional files
Additional file 1: Figure S1.
Performance in the Balance Beam Test is unaltered in Aldh2 -/- mice. Performance on the balance beam was quantified by measuring the total time taken for the mouse to transverse the beam. Differences in performance were not observed in either 2-3 month old (A) or 5-6 month old (B) animals. Data are presented as the mean ± SD (wildtype n = 15, Aldh2 -/- n = 20) and were analyzed using Student’s t-test for unpaired data (p > 0.05).
Additional file 2: Figure S2.
Lack of carbachol-induced changes in total CREB in hippocampi from wildtype or Aldh2 -/- mice. Hippocampal slices from 6 month old wildtype and Aldh2 -/- mice were incubated with 50 μM carbachol (+) or vehicle (-) for 30 min and snap frozen. Immunoblot analysis was performed using 30 μg protein of hippocampal homogenate, and immunoreactive bands were quantitated by densitometry. Data are presented as the mean ± SD (n = 6 mice) and were analyzed by Student’s t-test for unpaired data (p > 0.05).
Additional file 3: Figure S3.
Lack of carbachol-induced changes in total ERK in hippocampi from wildtype or Aldh2 -/- mice. Hippocampal slices from 6 month old wildtype and Aldh2 -/- mice were incubated with 50 μM carbachol (+) or vehicle (-) for 30 min and snap frozen. Immunoblot analysis was performed using 30 μg protein of hippocampal homogenate, and immunoreactive bands were quantitated by densitometry. Data are presented as the mean ± SD (n = 6 mice) and were analyzed by Student’s t-test for unpaired data (p > 0.05).
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
{kind=link}
Cite this article
D’Souza, Y., Elharram, A., Soon-Shiong, R. et al. Characterization of Aldh2 -/- mice as an age-related model of cognitive impairment and Alzheimer’s disease. Mol Brain 8, 27 (2015). https://doi.org/10.1186/s13041-015-0117-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13041-015-0117-y