
Abstract
Small supernumerary marker chromosomes (sSMCs) are additional derivative chromosomes present in an otherwise numerically and structurally normal karyotype. They may derive from each of the 24 human chromosomes, and most contain a normal centromeric region with an alphoid sequence from a single chromosome. The majority of human chromosomes have a unique centromeric DNA-sequence enabling their indubitable characterization. However, chromosomes 14 and 22 share a common centromeric sequence D14/22Z1, and sSMCs with this DNA-stretch can derive from either chromosome. Euchromatin-carrying sSMCs(14 or 22) may be further characterized by molecular cytogenetics. However, in most diagnostic laboratories, heterochromatic sSMCs cannot be differentiated between chromosomes 14 or 22 derivation and are often reported as der(14 or 22). Still, heterochromatic sSMC(14 or 22) can be distinguished from each other using the D22Z4 probe (non-commercial) localized to 22p11.2. Herein, 355 sSMC(14 or 22) analyzed in the authors’ laboratory during the last ~ 20 years are summarized to address the questions: (1) What are the true frequencies of chromosome 14- and chromosome 22- derived sSMCs within D14/22Z1-positive cases? (2) Does sub-characterization of sSMC(14) and sSMC(22) make a difference in routine diagnostics? These questions could be answered as follows: (ad 1) within the studied group of sSMCs ~ 40% are derived from chromosome 14 and ~ 60% from chromosome 22; (ad 2) the knowledge on exact sSMC origin can help to save costs in routine diagnostics; i.e. in a clinically abnormal person with sSMC(14) a test for uniparental disomy is indicated, which is not necessary if a chromosome 22 origin for the sSMC was determined.
Similar content being viewed by others


Background
Small supernumerary marker chromosomes (sSMCs) represent a rare aberration as they are simultaneously a numerical and structural rearrangement. sSMCs are additional derivative chromosomes present in an (in most cases) otherwise numerically and structurally normal karyotype. They can have a variety of sizes and structures derived from all 24 human chromosomes. Most carry one or two normal centromeric regions with alphoid sequences [1]. Although recognized since 1961 [2], sSMCs remain an issue in diagnostics. However, in the preceding decade, progress has been made regarding genotype–phenotype correlations for sSMC subgroups [1, 3]; sSMC related clinical syndromes have been identified e.g. cat eye syndrome (OMIM 115470), Emanuel syndrome (OMIM 609029), Pallister-Killian syndrome (OMIM 601803), isochromosome 18p-syndrome (OMIM 614290), supernumerary der(22)t(8;22)-syndrome (OMIM 613700) [4], and others [5]. The clinical outcome of the remaining sSMC cases is largely attributed to euchromatic content of the aberration. In particular, phenotypes have been associated with the presence (or absence) of dosage sensitive genes within pericentromeric euchromatic regions. Nonetheless, specific gene(s) have not yet been identified [6]. Furthermore, the occurrence of de novo sSMC in diagnostics is complicated by the following problems:
-
sister chromosomes of a sSMC may be subject to uniparental disomy (UPD) [7];
-
sSMC may by discontinuous, due to formation by chromothripsis; this makes sSMC cases unique, private events and genotype–phenotype correlations difficult, or nearly impossible [8];
-
sSMCs may be complex, i.e. consist of a centromeric part of chromosome A and a telomeric part of chromosome B [5];
-
sSMC-presence may hint towards a cryptic mosaic in certain body tissues; indeed tissues may harbor cells comprised of complete trisomy of the chromosome from which the sSMC originated (incomplete trisomic rescue) [9].
When an sSMC is detected via banding cytogenetics, nowadays there is often discussion of how to further characterize it. Molecular karyotyping seems at first glance to be optimal, as (if a SNP-based array is used) even isoUPD of an sSMC’s sister chromosomes can be detected, along with copy number gains induced by the sSMC [10]. In addition, discontinuous sSMCs can also be readily detected, along with cryptic mosaic forms of full trisomies. However, there are limitations to this approach as euchromatic sections of an sSMC (and potentially a mosaic trisomy) can only be detected if the percentage of cells containing the aberration is large enough to be detected by the corresponding applied platform. As mosaicism is rather rule than exception in sSMCs [11], which can impact phenotype [12], the authors’ laboratory remains committed to sSMC characterization by molecular cytogenetics. This holds true for the majority of cytogenetic institutions around the world, which given financial constraints often cannot afford modern and expensive newer high throughput approaches [13, 14].
While molecular cytogenetics has clear benefits, such as enabling single cell level studies, and thus has the potential to detect even low level and cryptic mosaics, there are also limitations [13, 14]. For example, sSMCs, clearly or not clearly derived from an acrocentric chromosome (based on silver staining of nucleolus organizer region [15]) cannot be unambiguously resolved for chromosomal origin via fluorescence in situ hybridization (FISH) with centromeric probes. While the chromosome 15 probes D15Z1 in 15p11.2 and D15Z3 in 15q11.1 guarantee clear results if the sSMC is derived from chromosome 15, this resolution is not possible between chromosomes 13 and 21 or 14 and 22, as they have common alphoid sequences, i.e. D13/21Z1 and D14/22Z1. Thus, the chromosome of origin cannot be determined between these chromosomes when the sSMC contains no euchromatin. While this problem is as of yet unresolvable between chromosomes 13 and 21, chromosomes 14 and 22 can be distinguished using the D22Z4 probe localized on 22p11.2, which is not commercially available [16]. Also it has to be added that molecular cytogenetics must be the gold-standard for sSMC-characterization, as molecular karyotyping may miss up to > 80% of sSMC, as recently shown [17].
Herein, 355 sSMC cases positive for D14/22Z1 via FISH analyzed in the authors’ laboratory during the last ~ 20 years (Additional file 2: Tables S1–S3—and sSMC database), were revisited. The main questions are: What are the true frequencies of chromosome 14- and chromosome 22–derived sSMCs among D14/22Z1-positive cases? In addition, we investigated whether sub-characterization of sSMC(14) and sSMC(22) makes a difference in routine diagnostics and counselling.
Results
Overall, 355 sSMC derived from chromosomes 14 or 22 were studied, which comprised 172 clinically normal cases, 110 clinically abnormal, and 73 with unclear clinical correlation. Comparatively, prenatal detection included 45/110 cases (~ 41%) designated clinically abnormal, whereas although postnatal detection included a higher number of cases, there was a slightly lower proportion of clinically affected cases (65/185, ~ 35%). Please note that individuals with ‘infertility’ were considered clinically normal.
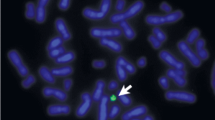
The D22Z4 probe localized to 22p11.2 could only aid in the distinction between an sSMC(14) or sSMC(22) if it contained 22p11.2 material; if this was not the case and/or insufficient material was available for further sSMC-characterization, the sSMC could not be clearly designated as a der(14) or der(22). An example of a case where the sSMC could be clearly characterized as a inv dup(22)(q11.1) after FISH is shown in Fig. 1A. This ambiguity occurred in 19/355 cases (5.4%) (Additional file 2: Table S1a-c). A normal outcome was documented in seven of nine cases, whereas in the ten remaining cases no clinical information was available.

a A typical result after co-hybridization of D14/22Z1 (green), specific for 14p11.1-q11.1 and 22p11.1-q11.1 and D22Z4 (red), staining 22p11.2. On green signal is visible each on both chromosomes 14 and 22, as well as the sSMC (mar(22)); however, red signals are only present on both chromosomes 22 (one time, each), and two red signals on the sSMC. b, c Frequencies of sSMC cases characterized in this study (a) and in the literature (Liehr, 2020a) (b) are depicted. Percentages for cases with normal, abnormal, and unclear clinical outcomes are given for der(14 or 22), der(14) and der(22) carriers. For der(22) cases, the abnormal cases are on the left side and subdivided in cat eye syndrome (CES) and Emanuel syndrome (ES). In all pie charts the number of abnormal cases is higher in the literature (c) than cases from the authors’ single laboratory (b)
sSMCs that lacked detectable signals for the D22Z4 probe, but had D14/22Z1 signals were considered derivatives of chromosome 14. For 101/130 cases clinical correlation was available: 81 cases had a normal outcome, while 20 demonstrated diverse cytogenomic aberrations and clinical outcomes. In 3/20 cases, the sSMC was present in conjunction with UPD(14), and in 8/20 cases, complex sSMCs were present. Interestingly, there were no statistically differences in the percentage of prenatally and postnatally detected abnormal versus normal sSMC carriers.
Heterochromatic sSMCs with detectable D22Z4 and D14/22Z1 FISH-signals were considered derived from chromosome 22. Euchromatic sSMCs anyway were positive for probes derived from 22q11.2 and thus clearly attributed to be derived from #22. While 34/206 cases lacked clinical information, the remaining 172 cases could be placed into several groups. As previously stated, there were two distinct groups composed of 84 clinically normal and 88 clinically abnormal carriers. Abnormal cases accounted for more prenatal (~ 58%) compared to postnatal cases (~ 47%). Within the abnormal cases there were two well defined syndromes: cat eye syndrome ((CES)—56 cases) and Emanuel syndrome ((ES)—15 cases). CES was detected prenatally in ~ 68% and postnatally in ~ 60% of the abnormal cases, and ES was detected prenatally in 22% and postnatally in 14%. In addition, there were 3 cases of prenatally detected sSMC carriers with complex sSMC(22). Interestingly, one clinically normal carrier of an sSMC derived from chromosome 22 also had UPD(22).
Figure 1b depicts the distribution of clinically normal and clinically abnormal cases for all der(14 or 22), in combination with cases with unclear clinical correlation, including those sSMC characterized as der(14) or der(22). Figure 1c summarizes all published cases [3] for comparison, highlighting publication bias. Figure 2 summarizes the 291 cases with known clinical correlation of the sSMC. Figure 3 summarizes cases with chromosome 22 derived sSMCs, with differences in pre- and postnatal detection highlighted. Finally, Fig. 4 compares frequencies of normal and abnormal sSMC carriers with sSMC(14 or 22), sSMC(14) and sSMC(22).

The 291/355 cases with clear clinical result studied herein are subdivided by normal and abnormal phenotypes. In first column all 291 cases are listed (14/22—all), the second and third columns include only sSMCs derived from chromosomes 14 or 22, i.e. der(14) or der(22), respectively; the last column includes unresolvable cases (14/22—unres). Normal cases are highlighted in blue, abnormal in red

These pie charts depict firstly all der(22) cases with clear clinical correlation, which are subsequently compared to the relative frequencies of prenatal and postnatal detection

Normal and abnormal cases before (first double column) and after (second and third double columns) separation into either chromosome 14 or chromosome 22 derived sSMCs, and their correlation with normal and abnormal clinical outcome
Discussion
Here the two questions raised at the beginning of this publication are answered.
What are the true frequencies of chromosome 14- and chromosome 22-derived sSMCs within D14/22Z1-positive cases?
Herein, 355 sSMC cases derived from chromosomes 14 or 22 from a single laboratory are summarized (Fig. 1b). This collective of patients has the advantage that it is not influenced by ‘publication-bias’, as otherwise present in the literature (Fig. 1c). The latter bias is a consequence of the report of only ‘interesting’ cases in scientific papers. Thus, it is more likely that clinically abnormal cases are reported than clinically normal ones; and such cases lost during follow-up they are not reported at all. Consequently, the data presented here should be a true representation of findings expected in a cytogenetic lab during routine diagnostics.
Nearly 40% of sSMC(14 or 22) cases detected in our laboratory were clinically abnormal (Fig. 2). Complete characterization of the sSMC as either sSMC(14) or sSMC(22) further divided the overall proportion of abnormal cases, as only 20% of der(14) compared with 52% of der(22) were clinically abnormal. Distinction between detection period (prenatal or postnatal) for der(14) versus der(22) was associated with only slight differences, which included the prenatal detection of 24% normal vs. 58% abnormal cases (Additional file 1: Fig. S1). Thus, there is a remarkable difference in risk estimation when the lab can make a determination that the sSMC of the fetus is derived from a chromosome 14 or 22 (with the risk for an affected child at 45%), versus those derived from either chromosome 14 (24%) or 22 (58%). It is estimated that 30–50% of fetuses with de novo sSMCs without clear clinical prognoses are terminated, and the assigning of the sSMC to the respective derivative chromosome is critical for pregnancy decision-making [18, 19]. Overall, within the here studied group of sSMC(14 or 22) carriers ~ 40% of sSMCs are derived from chromosome 14 and ~ 60% from chromosome 22 (Fig. 2).
Does sub characterization of sSMC(14) and sSMC(22) make a difference in routine diagnostics?
For routine diagnostics it must be considered that de novo sSMCs may also occur in conjunction with a UPD of sister chromosomes the sSMC derived from [7]. Exclusion of UPD is clinically paramount if the sSMC is derived from chromosome 14, as UPD(14)mat results in Temple syndrome (OMIM 616222), and UPD(14)pat in Kagami-Ogata syndrome (OMIM 608149) [4]. For chromosome 22, given the absence of imprinting defects, it is only the extremely rare homozygote recessive condition that results from (segmental) iso-UPD, which could have a clinical impact. However, only 11 of such cases are known, and none contained an sSMC [20]. Accordingly, a definite characterization of de novo sSMC(14) origin in a prenatal case or in a clinically abnormal person saves costs in routine diagnostics, as the number of patient-probes to be submitted to a UPD-test can markedly be reduced.
Furthermore, if an sSMC has been characterized to derive from chromosome 22 using centromeric probes without knowledge of the clinical presentation, it is more likely that in a postnatal setting a normal sSMC carrier (53%) has been detected, than when such an aberration has been detected in prenatal setting (42%); see Fig. 3. Interestingly, during the postnatal detection of clinically normal sSMC(14 or 22) carriers, when the sSMC is further characterized by the chromosome of origin, there is an approximate 1:1 ratio into mostly heterochromatic derivative chromosomes der(14) or der(22). Thus, during postnatal detection in clinically normal patients studied due to infertility, the clinical impact of detailed sSMC characterization is negligible and can be omitted.
Finally, it must be considered that the characterization of an sSMC being definitely derived from chromosome 14 or 22 is, in a prenatal setting and/ or a definitely clinically abnormal sSMC-carrier only the starting point to further characterize the genetic content of an sSMC. However, it is always possible that a heterochromatic sSMC, where also a possibly meaningful UPD has been excluded, may be only an anecdotal finding, as the real reason for the observed clinical problems of the patient have another genetic reason, like previously reported by us for an sSMC-carrier with fragile-X-syndrome [21].
Conclusion
In conclusion, further characterization of a prenatally detected sSMC(14 or 22) by the heterochromatin-directed D22Z4 probe and/or euchromatin directed probes in the long arm of chromosome 14 or 22 is of the utmost importance; these studies provide essential information on the putative clinical outcome of the unborn child and should be performed. Finally, only cases with the sSMC derived from chromosome 14 require testing to exclude UPD(14).
Material and methods
This retrospective study included 355 sSMC cases. All cases were positive for D14/22Z1 via FISH and were analyzed in the authors’ laboratory during the last ~ 20 years (Additional file 2: Tables S1–S3). All were previously included in the sSMC database [3] and/or other literature referred to in Additional file 2: Tables S1–S3. Nevertheless, all sSMC were further characterized with the D22Z4 probe, specific for 22p11.2, and/or, if euchromatic, by other probes as listed in sSMC database [3].
The Additional file 2: Table S1a-c include sSMCs derived from chromosomes 14 or 22, Additional file 2: Table 2a-c those derived from chromosome 14, and Additional file 2: Table S3a-c those derived from chromosome 22. Additional file 2: Table S1a, S2a and S3a include cases which were clinically normal, Additional file 2: Table S1b, S2b and S3b include clinically abnormal cases (including those with cat eye syndrome and Emanuel syndrome), and Additional file 2: Tables S1c, S2c and S3c include cases which have not been associated with a clear clinical outcome, due to loss during follow-up.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Abbreviations
- FISH:
-
Fluorescence in situ hybridization
- isoUPD:
-
Isodisomy
- OMIM:
-
Online Mendelian Inheritance in Man
- sSMCs:
-
Small supernumerary marker chromosomes
- sSMCs(14 or 22):
-
SSMC derived from chromosome 14 or 22
- UPD:
-
Uniparental disomy
- UPD(14):
-
UPD of chromosomes 14
- UPD(22):
-
UPD of chromosomes 22
References
Liehr T. Small supernumerary marker chromosomes (sSMC). A guide for human geneticists and clinicians; with contributions by UNIQUE (The Rare Chromosome Disorder Support Group). 1st ed. Berlin: Springer; 2012.
Ilberry PLT, Lee CWG, Winn SM. Incomplete trisomy in a mongoloid child exhibiting minimal stigmata. Med J Aust. 1961;48:182–4.
Liehr T. Small supernumerary marker chromosomes. http://cs-tl.de/DB/CA/sSMC/0-Start.html. Accessed 29 Nov 2020.
OMIM Online Mendelian Inheritance in Man®. An online catalog of human genes and genetic disorders. https://omim.org/. Accessed 29 Nov 2020.
Liehr T, Cirkovic S, Lalic T, Guc-Scekic M, de Almeida C, Weimer J, Iourov I, Melaragno MI, Guilherme RS, Stefanou EG, Aktas D, Kreskowski K, Klein E, Ziegler M, Kosyakova N, Volleth M, Hamid AB. Complex small supernumerary marker chromosomes—an update. Mol Cytogenet. 2013;6:46.
Al-Rikabi ABH, Pekova S, Fan X, Jančušková T, Liehr T. Small supernumerary marker chromosome may provide information on dosage-insensitive pericentric regions in human. Curr Genomics. 2018;19:192–9.
Liehr T, Ewers E, Hamid AB, Kosyakova N, Voigt M, Weise A, Manvelyan M. Small supernumerary marker chromosomes and uniparental disomy have a story to tell. J Histochem Cytochem. 2011;59:842–8.
Kurtas NE, Xumerle L, Leonardelli L, Delledonne M, Brusco A, Chrzanowska K, Schinzel A, Larizza D, Guerneri S, Natacci F, Bonaglia MC, Reho P, Manolakos E, Mattina T, Soli F, Provenzano A, Al-Rikabi AH, Errichiello E, Nazaryan-Petersen L, Giglio S, Tommerup N, Liehr T, Zuffardi O. Small supernumerary marker chromosomes: a legacy of trisomy rescue? Hum Mutat. 2019;40:193–200.
Santos M, Mrasek K, Rigola MA, Starke H, Liehr T, Fuster C. Identification of a “cryptic mosaicism” involving at least four different small supernumerary marker chromosomes derived from chromosome 9 in a woman without reproductive success. Fertil Steril. 2007;88(969):e11–7.
Zhou L, Zheng Z, Wu L, Xu C, Wu H, Xu X, Tang S. Molecular delineation of small supernumerary marker chromosomes using a single nucleotide polymorphism array. Mol Cytogenet. 2020;13:19.
Liehr T, Klein E, Mrasek K, Kosyakova N, Guilherme RS, Aust N, Venner C, Weise A, Hamid AB. Clinical impact of somatic mosaicism in cases with small supernumerary marker chromosomes. Cytogenet Genome Res. 2013;139:158–63.
Liehr T, Al-Rikabi A. Mosaicism: reason for normal phenotypes in carriers of small supernumerary marker chromosomes with known adverse outcome. A systematic review. Front Genet. 2019;10:1131.
Liehr T. What about the real costs of next generation sequencing (NGS) in human genetic diagnostics? http://atlasofscience.org/what-about-the-real-costs-of-next-generation-sequencing-ngs-in-human-genetic-diagnostics/. 2017.
Liehr T, Mrasek K, Klein E, Weise A. Modern high throughput approaches are not meant to replace ‘old fashioned’ but robust techniques. J Genet Genomes. 2017;1:e101.
Bloom SE, Goodpasture C. An improved technique for selective silver staining of nucleolar organizer regions in human chromosomes. Hum Genet. 1976;34:199–206.
Liehr T. Benign & pathological chromosomal imbalances. Microscopic and submicroscopic copy number variations (CNVs) in genetics and counseling. 1st ed. London: Academic Press; 2014.
Liehr T, Al-Rikabi ABH. Impaired spermatogenesis due to small supernumerary marker chromosomes: the reason for infertility is only reliably ascertainable by cytogenetics. Sex Dev. 2018;12:281–7.
Cavani S, Malcarne M, Arlanian A, Stagni L, Piombo G, Baldo C, Scaraglio T, Boggio G, Mogni M, Uras A, Alabiso A, Zucca M, Zerrega G, Dagna Briscarelli F, Pierluigi M. Prenatal and postnatal identification of 93 supernumerary small chromosomes. Ann Genetiq. 2003;4:623 (abstract no 7.19).
Shaffer BL, Caughey AB, Cotter PD, Norton ME. Variation in the decision to terminate pregnancy in the setting of an abnormal karyotype with uncertain significance. In: Abstract book of the 54th annual meeting of the American Society of Human Genetics; 2004:494 (abstract no 2756).
Liehr T. Cases with uniparental disomy. http://cs-tl.de/DB/CA/UPD/0-Start.html [accessed November 29, 2020].
Nelle H, Schreyer I, Ewers E, Mrasek K, Kosyakova N, Merkas M, Hamid AB, Fahsold R, Ujfalusi A, Anderson J, Rubtsov N, Küchler A, von Eggeling F, Hentschel J, Weise A, Liehr T. Presence of harmless small supernumerary marker chromosomes hampers molecular genetic diagnosis: a case report. Mol Med Rep. 2010;3(4):571–4.
Acknowledgments
sSMC cases were provided by numerous colleagues from Germany and around the world.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
TL developed the idea for the paper and drafted the paper together with HEW; MZ, SK, NP and AA-R did practical work in sSMC characterization; TL and HEW did the overall data interpretation. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All sSMC cases were studied during routine diagnostics, however, all studies in sSMC patients were also reviewed and approved also by University Clinic Jena, Ethical Commission, Jena, Germany (code: 4738-03/16).
Consent for publication
Were applicable, written informed consent to participate was provided by the participants' legal guardian/next of kin.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Fig. S1.
The 97 prenatal cases with clear clinical results studied herein subdivided by normal and abnormal phenotypes. The first column all 97 cases (14/22 – all) are depicted, in the second and third columns include only sSMCs derived from chromosomes 14 or 22, i.e. der(14) and der(22), respectively; the last column includes the unresolvable cases (14/22 – unres). Normal cases are highlighted in blue, abnormal in red.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liehr, T., Williams, H.E., Ziegler, M. et al. Small supernumerary marker chromosomes derived from chromosome 14 and/or 22. Mol Cytogenet 14, 13 (2021). https://doi.org/10.1186/s13039-021-00533-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13039-021-00533-6