
Abstract
Background
Children with Down syndrome (DS) have an enhanced risk of developing acute leukemia, with the most common subtype being acute megakaryoblastic leukemia (AMKL). Myeloid leukemia in Down syndrome (ML-DS) is considered a disease with distinct clinical and biological features. There are few studies focusing on the clonal cytogenetic changes during evolution of ML-DS.
Case presentation
Here, we describe a complex karyotype involving a previously unreported set of chromosomal abnormalities acquired during progression of ML-DS in an infant boy: derivative der(1)t(1;15)(q24;q23), translocation t(4;5)(q26;q33) and derivative der(15)t(7;15)(p21;q23). Different molecular cytogenetic probes and probesets including whole chromosome painting (WCP) and locus specific probes, as well as, multicolor-FISH and multicolor chromosome banding (MCB) were performed in order to characterize the chromosomal abnormalities involved in this complex karyotype. The patient was treated according to the acute myeloid leukemia-Berlin-Frankfurt-Munich-2004 (AML-BFM 2004) treatment protocol for patients with Down syndrome; however, he experienced a poor clinical outcome.
Conclusion
The molecular cytogenetic studies performed, allowed the characterization of novel chromosomal abnormalities in ML-DS and possible candidate genes involved in the leukemogenic process. Our findings suggest that the complex karyotype described here was associated with the poor prognosis.
Similar content being viewed by others


Background
Children with Down syndrome (DS) have a higher risk of developing acute leukemia than children without DS, and the most common subtype is acute megakaryoblastic leukemia (AMKL) [1]. Myeloid leukemia in Down syndrome (ML-DS) is a disease with distinct clinical and biological features, encompassing both myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML) [2, 3]. ML-DS can be preceded by transient myeloproliferative disease (TMD) in newborns. Although TMD spontaneously disappears in most cases, approximately 20% of children diagnosed with TMD develop ML-DS [4]. ML-DS is characterized by a higher occurrence in young age, low number of leukemic blasts in the bone marrow, somatic mutations in GATA-1 (a transcription factor that regulates the differentiation of megakaryocyte and erythrocyte precursors) and a better clinical outcome when treated with reduced-intensity chemotherapy protocols [5].
Most cytogenetic studies on DS-related leukemia have been single case reports or relatively small series. However, several studies have shown that the karyotypic patterns of ML-DS are different from those observed in AML of children without DS, e.g. translocations t(8;21), t(15;17), t(9;11), inversion inv.(16), as well as AMKL associated translocations t(1;22) and t(1;3) [1, 5, 6]. The most frequent chromosomal alterations associated with ML-DS are: duplication dup(1q), deletion del(6q), del(7p), dup(7q), trisomy +8, +11, del(16q) and +21. According to Forestier and colleagues [1], the types and frequencies of chromosomal changes occurring in addition to the constitutional +21 in ML-DS may provide important clues to the pathogenesis of acute leukemia in such patients.
Even though, the importance of cytogenetic alterations in classification and risk stratification of non-DS-AML is well recognized, e.g. in World Health Organization classification, there is limited information on cytogenetic alterations and their prognostic impact in ML-DS [1,2,3,4,5]. In a previous collaborative international AML-BFM Group Study, Blink and colleagues [5] described the first study on the prognostic impact of cytogenetic groups in 358 patients with ML-DS. In this study, the cytogenetic risk groups were: normal karyotype, trisomy 8, loss of chromosomes 5 and 7, trisomy 21, dup(1q), del(16q) and other chromosomal alterations. However, complex karyotypes were not categorized. There are a few studies focusing on the clonal cytogenetic changes during the evolution of ML-DS and the prognostic impact of a complex karyotype in ML-DS [4, 7,8,9,10]. The role of acquired chromosomal abnormalities (ACAs) in the progression of ML-DS has been discussed [10]. Some studies indicated that ACAs appear to be a risk factor for progression of disease [9, 10]. However, other studies did not observe this association [4, 5].
Here, we describe a complex karyotype with yet unreported chromosomal abnormalities in a 1-year-old boy presenting ML-DS. These chromosomal abnormalities were defined by molecular cytogenetic approaches and this complex karyotype was associated with a poor prognosis. Taken together with data from the literature, this may be supportive to include complex karyotypes in ML-DS as an additional adverse risk factor in classification and risk stratification of ML-DS.
Case presentation
A DS infant boy (one year of age) with a history of thrombocytopenia was referred to Martagão Gesteira Institute for clinical investigation, Rio de Janeiro, RJ, Brazil. Peripheral blood values were: hemoglobin 8.3 g/dl (age-adjusted range: 13.5–18.0 g/dl), platelet count 10 × 109/l (150–400 × 109/l) and white blood cell count of 48 × 109/l (age-adjusted range: 4–10 × 109/l). Morphologic evaluation of the bone marrow revealed hypocellularity (with granulocytic population decreased), the presence of dysplasia in the erythroid and megakaryocytic lineages, suggesting a diagnosis of ML-DS (myelodysplastic syndrome). Flow cytometry analysis of bone marrow (BM) cells showed a maturation block in granulocytic and monocytic lineages with high expression of CD7 and CD56 in the monocytic maturation. Cytogenetic analysis of bone marrow cells using G-banding showed: 47,XY,add(7)(p?),add(15)(q?), +21c [14]/47,XY,+21c [14].
Three months later, the patient presented with 51.8% of blast cells and the following immunophenotype: CD45lo/+, CD117hi, HLADR−/+(40%), CD13−/+(40%), CD33+, CD36++, CD34−/+(1%), IREM2−/+(10%), CD71+lo, CD11b−, CD16−, CD64−, CD14−, CD15−, MPO−, CD123−, CD9−, CD41a−, TdT−, CD7−, CD56−, cyCD3−, CD3−, CD19− and cyCD79a−, compatible with ML-DS (AML secondary from MDS). Classical cytogenetic analysis was performed, during AML secondary from MDS, using bone marrow cells and G-banding. This analysis revealed a clonal karyotypic evolution with the complex karyotype:
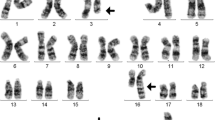
47,XY,t(3;5)(q21;q32),add(7)(p?),add(15)(q?),+21c [14]/ 47,XY,add(7)(p?),add(15)(q?),+21c [5]/47,XY,+21c [3] (Fig. 1a). Different molecular cytogenetic approaches were performed in order to characterize the chromosomal alterations. Initially, we used the whole chromosome painting (WCP) probes for chromosomes 7 and 15 (Fig. 1b). The karyotype was defined as: 47,XY,del(3)(q21),add(5q),der(7)t(7;15)(p15;q21q26),add(15)(q21),+21c [18]/47,XY, der(7)t(7;15)(p15;q21q26),add(15)(q21),+21c [5]/47,XY,+21c [2]. Afterward, more specific probes were used to characterize genes possibly involved in the chromosomal rearrangements: SPEC JAZF in 7p15, SPEC ETV1 in 7p21, SPEC SOX2 in 3q26, and SPEC VHL in 3p25 (Zytovison, Bremerhaven, Germany) and subtelomeric probe for 7pter (Abbott/Vysis, Göttingen, Germany). Thus, we could narrow down the breakpoint between deletion 7p21 and 7p15, revealing the deletion of the ETV1 gene (Fig. 1c). With multicolor-FISH, the complex karyotype was confirmed and refined as 47,XY,der(3)del(3)(p),der(4)t(4;5),der(7)t(7;15),+21c (Fig. 2a). Multicolor banding (MCB) was performed for chromosomes 1, 3, 4, 5, 7 and 15 (Fig. 2b), as described by Liehr and colleagues [11]. The chromosomal rearrangements and the breakpoints involved in derivative chromosomes 1, 3, 4, 5, 7 and 15 were defined (Fig. 2c-e). The final karyotype was characterized as: 46,XY,der(1)t(1;15)(q24;q23),del(3)(q21q25),t(4;5)(q26;q33), del(7)(p21),der(15)t(7;15)(p21;q23),+21c. The karyotypes were described according to the International System for Human Cytogenetic Nomenclature [12].

a G-banding showing the complex karyotype, chromosomal aberrations are pointed by the arrows; (b) Whole chromosome paints (WCP) for chromosomes 7 and 15 confirmed the unbalanced nature of the translocations observed in G-banding; (c) Application of ETV1/CEP7 and JAZF1 break-apart combined with WCP for chromosome 7 probes narrowed the breakpoint down to 7p21 and showed the ETV1 gene deletion

a M-FISH results showing the trisomy 21 and the involvement of chromosomes 3, 4, 5, 7 and 15 in this complex karyotype; (b) Pseudocolor depiction of MCB for the chromosomes involved in the complex rearrangement; (c-e) Schematic drawings of the rearranged chromosomes compared to their formats in G-banding
The patient was treated with AML-BFM 2004 protocol, arm designed for DS. At the end of induction therapy, he had 5% of blasts in bone marrow. After the first two blocks of high doses intensification, he presented with 20% of blasts in peripheral blood, sepsis and died.
Discussion and conclusions
DS leukemogenesis is suggested to be a multistep process in which progenitor cells acquire multiple genetic lesions during progression to acute leukemia. The first event is trisomy 21, but it is not sufficient for malignant cell expansion [13, 14]. At cytogenetic level, the present case illustrates this concept, since during leukemic evolution the patient acquired new cytogenetic abnormalities resulting in a complex karyotype. Present since the initial diagnosis ML-DS (MDS phase), the translocation t(7;15)(p21;q23) was associated with a deletion of the ETV1 gene in 7p21 and a rearrangement of MAP2K5 (mitogen-activated protein kinase 5) located in 15q23. The ETV1 (ETS-translocation variant 1) gene belongs to the ETS (erythroblastosis virus E26 transforming sequences) family of transcription factors. The ETS proteins regulate many target genes that modulate biological processes like cell growth, angiogenesis, migration, proliferation and differentiation [15]. Pathologically, the ETV1 protein is aberrantly expressed through chromosomal translocations in a subset of solid tumors, e.g. prostate, melanoma, gastrointestinal stromal tumors [16]. Interestingly, in this study, the ETV1 gene was deleted, so probably altering the signaling pathways of biological processes which are thought to contribute to the leukemogenesis. Furthermore, MAPK pathways play critical roles in a wide variety of cancer types, from hematologic malignancies to solid tumors [17]. To the best of our knowledge, this translocation has not been described yet in ML-DS.
Besides translocation t(7;15), a derivative der(1)t(1;15)(q24;q23) was also present at the initial stage of ML-DS. Partial trisomy of the long arm of chromosome 1, by means of duplication, has been recurrently reported and appears to represent a non-random chromosomal abnormality in ML-DS [1, 5, 18]. So, we hypothesized that the co-existence of translocation t(7;15)(p21;q23) and derivative der(1)t(1;15)(q24;q23) could be drivers of genetic aberrations, that block normal myeloid differentiation, potentially inducing the leukemic evolution. During this leukemogenesis process, the progenitor cells acquired other chromosomal abnormalities.
When leukemic infiltration was diagnosed, two additional chromosomal abnormalities were observed during disease progression: deletion del(3)(q21q25) and translocation t(4;5)(q26;q33). Translocations or inversions involving 3q21 and 3q26 are associated with high-risk in AML and these patients usually present a poor prognosis [19]. The translocation t(4;5)(q26;q33) has not yet been described in an ML-DS before. Interestingly, the N-deacetylase/N-sulfotransferase (NDST4) gene, is located in 4q26. The encoded enzyme has a dual function, i.e. processing glucosamine and heparan polymers, the latter being key components of cell microenvironment, playing an important role in cell-cell interactions and adhesion. Bone marrow microenvironment has been implicated as a source of chemoresistance and disease relapse [20]. In the second breakpoint 5q33-q34, there are individual genes such as EGR1, CSF1R, and RPS14 that may contribute to malignant transformation [21]. This new complex karyotype originated during the evolution of the disease, associated with poor clinical outcome presented by our patient, reinforces the importance of routinely karyotyping of ML-DS.
Our data have confirmed and extended prior knowledge that ML-DS is cytogenetically characterized mainly by a relatively high frequency of copy number alterations [18]. In the present work, we described a complex karyotype characterized by chromosomal translocations (balanced and unbalanced), suggesting other mechanisms involved in the appearance of these chromosomal abnormalities. Trisomies are quite characteristic for ML-DS, as trisomy 8 and 11, suggesting that DS patients may be more susceptible to non-disjunctional events during cell division [1, 22].
The present case exemplifies the clonal karyotypic evolution being typical for ML-DS and demonstrated the impact of ACAs during the evolution of ML-DS. Thus, defining the cytogenetic and molecular characteristics of disease progression in ML-DS is important to understand the pathogenesis of ML-DS. For this purpose, we emphasize the utility of multicolor FISH, once the involvement of chromosome 1 was only detected using this molecular cytogenetic method. Therefore, further molecular studies involving a larger number of patients are needed to clarify the dilemma of the cytogenetic impact in ML-DS prognosis, making possible the incorporation of the cytogenetic information of leukemia associated with DS in the risk group stratification.
In summary, our study suggests that the leukemogenic process was triggered by the accumulation of ACAs. The molecular cytogenetic methods applied in this highly complex karyotype allowed the characterization of chromosomal regions with high resolution and provided the ability to identify possible candidate genes involved in the leukemogenic process, giving new insights into the biology of ML-DS. As the correlation of cytogenetic changes with the development of the disease and its prognosis in ML-DS is not as clear yet as in children without DS, our study shows that ACAs were clearly associated with the evolution of the disease, resulting in a complex karyotype; thus, the latter may be supportive of including complex karyotypes in ML-DS as an adverse risk factor in classification and risk stratification of ML-DS in near future.
Abbreviations
- +:
-
trisomy
- ACAs:
-
Acquired chromosomal abnormalities
- AMKL:
-
Acute megakaryoblastic leukemia
- AML:
-
Acute myeloid leukemia
- del:
-
deletion
- der:
-
derivative
- DS:
-
Down syndrome
- dup:
-
duplication
- FISH:
-
Fluorescence in situ hybridization
- inv.:
-
inversion
- MDS:
-
Myelodysplastic syndrome
- ML:
-
Myeloid leukemia
- ML-DS:
-
Myeloid leukemia of Down syndrome
- SPEC:
-
Spectrum
- t:
-
translocation
- TMD:
-
Transient myeloproliferative disease
- WCP:
-
Whole chromosome painting
References
Forestier E, Izraeli S, Beverloo B, Haas O, Pession A, Michalová K, Stark B, Harrison CJ, Teigler-Schlegel A, Johansson B. Cytogenetic features of acute lymphoblastic and myeloid leukemias in pediatric patients with Down syndrome: an iBFM-SG study. Blood. 2008;111:1575–83.
Hasle H, Niemeyer CM, Baumann I, Bennett JM, Kerndrup G, Head DRA. Pediatric approach to the WHO classification of myelodysplastic and myeloproliferative diseases. Leukemia. 2003;17:277–82.
Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, Bloomfield CD, Cazzola M, Vardiman JW. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–405.
Klusmann JH, Creutzig U, Zimmermann M, Dworzak M, Jorch N, Langebrake C, Pekrun A, Macakova-Reinhardt K, Reinhardt D. Treatment and prognostic impact of transient leukemia in neonates with Down syndrome. Blood. 2008;111:2991–8.
Blink M, Zimmermann M, von Neuhoff C, Reinhardt D, de Haas V, Hasle H, O'Brien MM, Stark B, Tandonnet J, Pession A, Tousovska K, Cheuk DK, Kudo K, Taga T, Rubnitz JE, Haltrich I, Balwierz W, Pieters R, Johansson B, van den Heuvel-Eibrink MM, Zwaan CM. Normal karyotype is a poor prognostic factor in myeloid leukemia of Down syndrome: a retrospective, international study. Haematologica. 2014;99:299–307.
Bhatnagar N, Nizery L, Tunstall O, Vyas P, Roberts I. Transient abnormal Myelopoiesis and AML in Down syndrome: an update. Curr Hematol Malig Rep. 2016;11:333–41.
Wang N, Leung J, Warrier RP, Shorin M, Kirkpatrick D, Nowak MJ, Strand R. Nonrandon chromosomal aberrations and clonal chromosomal evolution in acute leukemia associated with Down's syndrome. Cancer Genet Cytogenet. 1987;28:155–62.
Duflos-Delaplace D, Laï JL, Nelken B, Genevieve F, Defachelles AS, Zandecki M. Transient leukemoid disorder in a newborn with Down syndrome followed 19 months later by an acute myeloid leukemia: demonstrattion of the same structural change in both instances with clonal evolution. Cancer Genet Cytogenet. 1999;113:166–71.
Massey GV, Zipursky A, Chang MN, Doyle JJ, Nasim S, Taub JW, Ravindranath Y, Dahl G, Weinstein HJA. Prospective study of the natural history of transient leukemia (TL) in neonates with Down syndrome (DS): Children’s oncology group (COG) study POG-9481. Blood. 2006;107:4606–13.
Picos Cárdenas VJ, Meza Espinoza JP, Garibaldi Covarrubias RF, Barajas Torres RL, González García JR. Transient myeloproliferative disorder progression and acquired chromosomal abnormalites in children with Down syndrome. Pediatr Blood Cancer. 2012;59:962–3.
Liehr T, Heller A, Starke H, Rubtsov N, Trifonov V, Mrasek K, et al. Microdissection based high resolution multicolor banding for all 24 human chromosomes. Int J Mol Med. 2002;9:335–9.
Shaffer L, McGowan-Jordan J, Schmid M. ISCN 2013. An International System for Human Cytogenetic Nomenclature. Basel: Karger; 2013.
Zraeli S, Rainis L, Hertzberg L, Smooha G, BirgerY. Trisomy of chromosome 21 in leukemogenesis. Blood Cells Mol Dis. 2007;39:156–9.
Mateo MK, Barbarie D, Byatt S-A, Sutton R, Marshall GM. Down syndrome and leukemia: insigts into leukemogenesis and translational targets. Transl Pediatr. 2015;4:76–92.
Rahim S, Minas T, Hong SH et al. A small molecule inhibitor of ETV1, YK-4-279, prevents prostate cancer growth and metastasis in a mouse Xenograft model. Plos One 2014; 9(12): e114260: 1–20.
Chen Y, Chi P. ETV1 (ets variant 1). Atlas Genet Cytogenet Oncol Haematol. 2013;17:483–6.
Pritchard AL, Hayward NK. Molecular pathways: mitogen-activated protein kinase. Pathway mutations and drug resistance. Clin Cancer Res. 2013;19:2301–9.
Blink M, den Heuvel-Eibrink V, Aalbers A, Balgobind BV, IHIM H, JPP M, VHJ v d V, Beverloo BH, Hass V, Halse H, Reinhardt D, Klusmann J-H, Pieters R, Calado RT, Zwaan CM. High frequency of copy number alterations in myeloid leukaemia of Down syndrome. Br J Haematol. 2012;158:798–814.
Raya JM, Martín-Santos T, Luño E, Sanzo C, Perez-Sirvent ML, Such E, Navarro JT, et al. Acute myeloid leukemia with inv(3)(q21q26.2) or t(3;3)(q21;q26.2): clinical and biological features and comparison with other acute myeloid leukemias with cytogenetic aberrations involving long arm of chromosome 3. Hematology. 2015;20:435–41.
Bakker E, Qattan M, Mutti L, Demonacos C, Krstic-Demonacos M. The role of icroenvironment and immunity in drug response in leukemia. Biochim Biophys Acta. 1863;2016:414–26.
de Souza DC, Otero L, Tavares RC, Dobbin J, Abdelhay E, Fernandez TS. An uncommon case of a child with del(5q) and hypocellular myelodysplastic syndrome. Pediatr blood and. Cancer. 2010;55:767.
Fong C, Brodeur GM. Down's syndroe and leukemia: epidemiology, genetics, cytogenetics and mechanisms of leukemogenesis. Cancer Genet Cytogenet 187. 28:55–76.
Acknowledgments
The authors acknowledge with gratitude Luize Otero, Tarsis P. Vieira and Roberto R. Capela de Matos. This study was supported by CAPES/PROBRAL/DAAD (project no.419/14), FAPERJ (E-26/200.50/2016) and the Brazilian Ministry of Health (National Cancer Institute/INCA, Brazil).
Funding
Funding information is not applicable/No funding was received.
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article. More details are available on request.
Author information
Authors and Affiliations
Contributions
DCS participated in the design of the study and in manuscript writing. AFF, MAKO, TL conducted the molecular cytogenetic analyses; DRNG provided the revision of the clinical history of the child with ESC, who also performed the immunophenotyping studies. TL, EA, MLMS and TSF revised the manuscript critically for important intellectual content. The authors alone are responsible for the content and writing of the paper. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Ethics Committee of the Brazilian National Cancer Institute (CEP #088/07). Written institutional informed consent for the participation of the patient in this study was obtained from the patient’s parents.
Consent for publication
The patient’s parents have given their institutional informed written consent for publication of the present case report.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
de Souza, D.C., de Figueiredo, A.F., Ney Garcia, D.R. et al. A unique set of complex chromosomal abnormalities in an infant with myeloid leukemia associated with Down syndrome. Mol Cytogenet 10, 35 (2017). https://doi.org/10.1186/s13039-017-0335-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13039-017-0335-3