
Abstract
Background
Cytogenetic abnormalities are important prognostic markers in plasma cell myeloma (PCM) and detection is routinely performed by interphase fluorescence in-situ hybridization (FISH) with a panel of probes after enrichment of the plasma cells in the bone marrow specimen. Cell sorting by immunomagnetic beads and concurrent labeling of the cytoplasmic immunoglobulin are the usual enrichment methods. We present an alternative method of plasma cell enrichment termed Target FISH, which is an automated system that combines the images of May-Grünwald- Giemsa (MGG) staining and FISH study on the same plasma cell for analysis.
Results
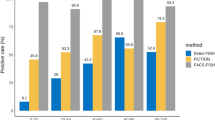
Our experience of Target FISH on 40 PCM patients was described. Briefly, plasma cells were MGG stained, image captured, de-stained, FISH probe hybridized and finally relocated for simultaneous analysis of morphology and FISH signal pattern. The FISH probe panel was TP53/CEP17, t(4;14) IGH/FGFR3, t(14;16) IGH/MAF and CKS1B(1q21)/CDKN2C(P18). Gain of 1q21 was the most common abnormality detected in 18 patients (45 %), to be followed by t(4;14) IGH/FGFR3 detected in 11 patients (27.5 %). Of note, 10 patients showed coexistence of both t(4;14) and 1q21 gain. Two patients showed del(17p)/TP53, one in association with t(4;14) and 1q gain while the other was stand alone. None of this patient cohort showed t(14;16) IGH/MAF. Using the critical binomial function, the normal cutoff FISH positive value for del(17p)/TP53 was 3.4 %, t(4;14) IGH/FGFR3 was 6.8 %, t(14;16) IGH/MAF was 5.6 % and +1q21 was 5.7 %.
Conclusions
The equipment cost notwithstanding, when compared with cell sorting, the total reagent cost was around 10 % lower in Target FISH. The total processing time was longer for Target FISH but manual fluorescence microscopy was no longer necessary. The main advantage of Target FISH was the complete certainty that the cytogenetic abnormality was detected in the cells of interest, and hence a more stringent analytical cutoff value might be considered. Optimization of the cell collection and slide preparation process upfront was required to accrue adequate target cells on each slide for analysis. Our experience suggested that Target FISH was applicable as a routine method of plasma cell enrichment in clinical diagnostic laboratories.
Similar content being viewed by others


Background
The detection of cytogenetic abnormalities in PCM is clinically important as a prognostic marker to risk stratify patients [1]. While conventional metaphase chromosome study detects cytogenetic abnormalities in only one third of PCM patients, interphase FISH improves the detection frequency to around 90 % of patients [2]. The reasons for the discrepancy are attributable to low plasma cell percentage in the bone marrow, low proliferative index of the plasma cells and hence outgrown by granulocytic precursors, and that chromosome translocations such as t(4;14)(p16;q32) may be morphologically cryptic or co-exist with other complex changes and escape detection by the less experienced cytogeneticist.
It is not recommended to perform interphase FISH directly on the PCM bone marrow due to often low plasma cell percentage and admixture by other hemopoietic cells. The plasma cells should be selected either by immunomagnetic beads or flow cytometry based plasma cell sorting or concurrent labeling of the cytoplasmic immunoglobulin (cIg) light chain to allow unambiguous detection of cytogenetic abnormalities in the neoplastic plasma cell population[3, 4]. The enrichment techniques of cell sorting [5] or cIg-FISH [6] however are labor, time and cost intensive. Hence the incorporation of these methods into the routine workflow of a diagnostic cytogenetics laboratory may be challenging. An alternative method of plasma cell enrichment termed Target FISH based on sequential MGG stain to identify plasma cell populations to be followed by FISH analysis was previously reported [7]. Herein, we present an automated Target FISH system for use in routine molecular diagnostics.
Results and discussion
We reported the experience on the initial 40 PCM patients for whom Target FISH was performed (Table 1). These patients were accrued in a 12-month period from April 2015 to March 2016. The patients comprised 20 males and females each at a median age of 61 years (range 47–89 years). The median percentage of plasma cells in the bone marrow was 29 % (range 12–100 %). Monoclonal gammopathy of undetermined significance (MGUS) were excluded. In the same time period, two samples in which FISH was failed (due to heavy background fluorescence and aged sample respectively) and one inadequate sample were also excluded. Conventional cytogenetic study results were available in only 10 patients (no. 2, 7, 9, 13, 17, 21, 22, 25, 33 and 35) for correlation (Table 1).
Gain of 1q21 was the most common abnormality detected in 18 patients (45 %), which was consistent with the high prevalence of this cytogenetic abnormality reported in the literature [8–10]. The t(4;14) IGH/FGFR3 was detected in 11 patients (27.5 %). Of note, 10 patients showed coexistence of both t(4;14) and 1q21 gain. Again this observation was consistent with the much overlap between adverse IgH translocations and 1q21 gain as reported by the UK MRC Myeloma IX Trial [11]. Two patients showed del(17p)/TP53, one in association with t(4;14) and 1q gain while the other was stand alone. None of this patient cohort showed t(14;16) IGH/MAF. However 4 patients tested positive for t(11;14) IGH/CCND1, which was performed selectively when the myeloma cells showed small lymphoplasmacytic morphology and expression of CD20 [12]. Incidental finding of secondary abnormalities i.e. monosomy or deletion of chromosomes 4, 14 or 16 or trisomy or amplification of chromosomes 4, 14 or 16 detected by t(4;14) IGH/FGFR3 and/or t(14;16) IGH/MAF FISH probes were encountered in 17 patients.
The normal cutoff value to define FISH positivity for each probe was determined in the laboratory by studying a cohort of 6 normal control samples together with the negative results in the patient samples for the FISH probes del(17p)/TP53 (n = 38), t(4;14) IGH/FGFR3 (n = 29), t(14;16) IGH/MAF (n = 40) and +1q21 (n = 22). The 6 normal controls were patients with reactive plasmacytosis proven by absence of paraprotein and lack of light chain restriction on the plasma cells. This normal control cohort consisted of 2 male patients and 4 female patients with an age range of 41–81 years. The diagnoses were metastatic colon cancer, adult Still’s disease, anaplastic large cell lymphoma, incidental finding of increased globulins on checkup, primary osseous diffuse large B-cell lymphoma and primary hyperparathyroidism. The range of bone marrow plasma cells was 6–8 %. The definition of false positive signal pattern was less than 2 red signals for TP53/CEP17, any yellow fusion signal for t(4;14) IGH/FGFR3 or t(14;16) IGH/MAF, and more than 2 red signals for chromosome 1q CKS1B/CDKN2C. The critical binomial function of the Microsoft Excel spreadsheet was used to determine the 95 % confidence limit of normal cutoff value [13]. The normal cutoff value for TP53/CEP17 was 3.4 %, t(4;14) IGH/FGFR3 was 6.8 %, t(14;16) IGH/MAF was 5.6 % and chromosome 1q CKS1B/CDKN2C was 5.7 %. However, for clinical reporting, conservative cutoff levels of 10 % for fusions and 20 % for numerical abnormalities were generally recommended [4]. In the literature, other cutoff values were quoted, for example 30 % for chromosome 1q gain [14] and 60 % for del(17p) [15]. Notwithstanding the analytical validation, the clinical significance of the cutoff value should also be considered. For example, a study on bortezomib-based chemotherapy in PCM patients according to the copy number of 1q21 showed that 20 % of involved plasma cells or more had no significant difference on survival, indirectly confirming the validity of 20 % rather than a higher cutoff in defining 1q gain [16]. Similarly another FISH study showed that 50 % for 17p deletion and 20 % for 1q gain were the optimal cutoff values associated with the greatest survival difference for predicting poor clinical outcome [17].
A caveat of Target FISH was that although thousands or ten thousands of cells were captured by the system on each slide, the percentage categorized as plasma cells by the machine before manual visualization and re-classification was uneven and often discordant with the percentage determined by microscopic examination of the bone marrow aspirate smears. Notably, a lower BioView percentage was observed in 32 patients (80 %) in this cohort (Table 1). This resulted in calling the FISH result based on less than the target of at least 100 analyzable cells for each probe. This scenario was seen consistently across all probes in only 5 patients. Further optimization of the density gradient centrifugation and cytospin preparation were required to prevent this apparent “cell loss” in the Target FISH experiments. Following on this, Target FISH might not be applicable to the study of MGUS in which the plasma cell percentage was low, in contrast to cell sorting in which a larger volume of bone marrow blood could be used if available.
Before the implementation of Target FISH, plasma cell enrichment was performed by cell sorting with CD138 immunomagnetic beads (Miltenyi Biotec GmbH, Germany) on 253 patients and this cohort did not include the 40 patients for whom Target FISH was performed. The cohort consisted of 161 male and 92 female patients at a median age of 61 years (range: 38–88 years). The median bone marrow plasma cell percentage was 42 % and cell sorting was only performed in the 140 patients (55 %) with less than 50 % plasma cells in the bone marrow. The purity after cell sorting was checked by cytospin preparation. The volume of bone marrow sample was 1–3 mLs and comparable with Target FISH. Fluorescence microscopy was performed by two observers who analyzed at least 100 cells each. Positive cutoff levels of 10 % for fusions and 20 % for numerical abnormalities were adopted for reporting. For comparison with Target FISH, the frequency of cytogenetic abnormalities was 41.9 % for +1q21, 16.6 % for t(4;14) IGH/FGFR3, 0.9 % for t(14;16) IGH/MAF and 7.5 % for del(17p)/TP53.
The workflow and cost of Target FISH was compared with cell sorting by immunomagnetic beads (Table 2). Unfortunately our laboratory had no experience with cIg-FISH which precluded any comparison. The Target FISH was a 3-day procedure but for urgent cases the first 2 days could be combined into one day to stop at the probe hybridization step overnight to obtain the FISH results the next day. The total reagent cost of Target FISH was around 10 % less expensive than cell sorting because there was no need to employ the immunomagnetic beads. The equipment cost however had to be taken into consideration. The total processing time of Target FISH however was longer than cell sorting. This notwithstanding, the 3 h (4 slides at 45 min each) of BioView slide scanning on day 3 were performed automatically by the machine after the cell matching was verified, instead of the tedious 2-h manual examination under fluorescence microscopy in the dark room. Moreover, the reporting procedure was facilitated since all the captured images could be reviewed on the computer monitor. Under manual examination only the representative images were captured and one would resort to repeat fluorescence microscopy for counterchecking results. The manpower requirement was equivalent for both Target FISH and cell sorting.
Conclusion
Correlation of plasma cell morphology with FISH by manual means [18] was not a new concept. Neither was automated image analysis an entirely new concept for the purpose of plasma cell identification in myeloma FISH [19]. Consistent with a previous report in the literature [7], the Target FISH system presented in our study was capable of combining the two processes in a fully automated fashion for relocation of plasma cells to allow the analysis of the MGG image and FISH signals of the same plasma cell. In this way, it was 100 % ensured that the cytogenetic abnormality - if any was detected - was found in the cells of interest. Target FISH saved the cost on reagent, obviated the need for tedious manual fluorescence microscopy and enriched the sample to completely focus on the analysis of plasma cells. We reckoned that Target FISH was applicable as an attractive alternative to cell sorting and cIg-FISH as a method of plasma cell enrichment for detection of PCM cytogenetic abnormalities in clinical molecular diagnostic laboratories.
Methods
Bone marrow preparation for morphology
Mononuclear cells in the bone marrow were separated by density gradient centrifugation to prepare cytospin slides for staining and morphological examination by BioView system (Abbott Molecular, Des Plaines, IL). Around 2 mL of bone marrow blood in EDTA was processed. Briefly, bone marrow was diluted 1:4 with wash buffer and added slowly to separation reagent prepared from 1 volume of Histopaque 1.077 (Sigma) mixed with 3 volumes of Histopaque 1.119. The mixture was centrifuged at 500 g for 30 min and decelerated slowly for 30 min to zero. Care was taken not to disturb the different layer when removing the tube from the centrifuge. The mononuclear cell layer at the interface between the plasma layer above and the Ficoll-hypaque layer below was carefully pipetted off to another tube and washed twice. The cell pellet was re-suspended in morphology preserver consisting of Fetal Bovine Serum in Ham’s F-10 Nutrient Mix (Life Technologies).
Preparation of cytospin slides and MGG Staining
The cytospin centrifuge produced single-layer cell preparations that also flattened the cells for optimal cytoplasmic and nuclear presentation required for morphology analysis. 10–20 mLs of cell pellet were added to 2–3 mLs of Morphology Preserver to create the cell suspension. Up to 200 μLs of this suspension was loaded to a cuvette and centrifuged at 1500 rpm for 5 min. The slide was then carefully extracted from the cuvette and allowed to dry after marking the area around the cells. The cell density was checked under light microscopy by a 20x dry objective to ensure single layer of cells that showed minimal cellular overlap and no clustering. High concentration caused the cells to overlap hence difficult to scan and classify, and low concentration reduced the analyzable cell number that adversely affected the test result. The concentration of the suspension should be adjusted by dilution or addition of more cell pellet as deemed necessary. At least 8 cytospin slides were made, 4 for the 4-probe myeloma FISH panel used in our laboratory and 4 for backup. Slides could be stored at −20 °C for up to 2 months until staining.
The cytospin slides were stained successively in May-Grünwald stain for 2 min and in 5 % Giemsa stain for 8 min. After drying, the slides were checked under light microscopy to confirm the appropriate staining quality for BioView scanning. Freshly prepared MGG stains were prepared each time to ensure optimal staining quality for plasma cell image recognition by BioView.
Automated scanning of the MGG-stained slides by BioView
The slide was loaded onto the stage of BioView for scanning and time required was around 15–20 min per slide depending on the cell quantity. After scanning, the plasma cell category as automatically selected by the machine was reviewed and bona fide plasma cells were reclassified into ‘my_class’ (Fig. 1). The aim was to obtain 200 plasma cells per slide for FISH to allow for hybridization failure since the reporting target was at least 100 plasma cells with clear FISH signals. If insufficient plasma cells were obtained, other categories were reviewed to move plasma cells into ‘my_class’ category.

a From 5333 cells that were captured on this particular MGG stained slide, the image capture and analysis system automatically assigned 1301 cells to the plasma cell category. b The plasma cell category was manually reviewed to reclassify bona fide or clearly abnormal plasma cells into My_class category, which contained 200 cells in this case
MGG de-staining and interphase FISH procedure
De-staining was performed by immersing slide in ice-cold (−20 °C) Carnoy’s solution (methanol: acetic acid 3:1) for 2 min at room temperature, and then moved to be immersed in another jar for 1 h. The slide was rinsed in 1x phosphate buffered saline for 5 min to be ready for FISH.
The pre-hybridization, probe hybridization and post-hybridization procedures were performed in accordance with standard leukemia lymphoma FISH protocols [20, 21]. Four FISH probes constituted the myeloma panel in our laboratory based on recommendations of the International Myeloma Working Group [1, 3, 22], namely Vysis TP53/CEP17 FISH probe kit (5 N56-20), Vysis IGH/FGFR3 DF FISH probe kit (1 N69-20), Vysis IGH/MAF DF FISH probe kit (5 N32-20) from Abbott Molecular, and CKS1B/CDKN2C(P18) Amplification/Deletion probe kit (LPH 039) from Cytocell (Cambridge, UK). Other FISH probes such as cyclin D1 gene amplification [23] were not routinely applied. However the IGH/CCND1 probe would be used when t(11;14) myeloma was suspected. Probe hybridization was performed on either Vysis HYBrite or ThermoBrite (Abbott Molecular).
Scanning FISH slides by BioView
The slides were mounted on the stage and immersion oil was applied manually. Through the computer terminal, the corresponding case and slide number were documented, and the appropriate scanning task and probe name were selected. The images of the MGG-stained plasma cells were retrieved from the ‘re-visit my_class’ of the scan program and a dual-view screen was opened, in which a live image of the slide under fluorescence microscopy was displayed on the left side and the bright field image of the plasma cells on the right side, thus allowing direct matching of the FISH signal and morphology. The BioView captured images from 13 focal planes spaced 0.65 μm apart for each cell to minimize chance juxtaposition of FISH signals as a cause of false positive fusion signal pattern. Once a relocated plasma cell with clear FISH signals was identified from the live image, the bright field image previous captured was checked for accuracy of localization based on size, shape and surrounding cells. If a match was verified, both images were centered and the process was finished for that particular plasma cell. The process was then repeated for as many cells on whatever number of slides as required by the laboratory. We aimed to select at least 100 cells per slide with clear FISH signals, and 4 slides were used per patient for a myeloma panel of 4 probes. When all the slides for the case were properly configured, the slides were scanned automatically at around 45 min per slide.
When scanning was completed, the results were manually visualized from the BioView software. A statistically summary of the FISH signal patterns of the selected plasma cells of each particular probe was displayed. Moreover, a composite image of FISH on the left side and MGG-stained picture of the same plasma cell on the right side could be prepared for inclusion in the pathology report (Fig. 2).

Images of MGG stained (right) and FISH probe hybridized (left) plasma cells. a t(4;14) IGH/FGFR3 probe, showing the 2F1O1G signal pattern on a positive plasma cell. b t(14;16) IGH/MAF probe, showing the 2O2G signal pattern on a negative plasma cell. c CKS1B/CDKN2C(P18) probe, showing 3O2G signal pattern on the plasma cell positive for +1q21. d TP53/CEP17 probe, showing 1O2G signal pattern on the plasma cell positive for del(17p)/TP53
Abbreviations
FISH, fluorescence in-situ hybridization; MACS, magnetic-activated cell sorting; MGG, May-Grünwald-Giemsa; PCM, plasma cell myeloma; SD, standard deviation
References
Chng WJ, Dispenzieri A, Chim CS, Fonseca R, Goldschmidt H, Lentzsch S, et al. IMWG consensus on risk stratification in multiple myeloma. Leukemia. 2014;28:269–77.
Drach J, Schuster J, Nowotny H, Angerler J, Rosenthal F, Fiegl M, et al. Multiple myeloma: high incidence of chromosomal aneuploidy as detected by interphase fluorescence in situ hybridization. Cancer Res. 1995;55:3854–9.
Munshi NC, Anderson KC, Bergsagel PL, Shaughnessy J, Palumbo A, Durie B, et al. Consensus recommendations for risk stratification in multiple myeloma: report of the International Myeloma Workshop Consensus Panel 2. Blood. 2011;117:4696–700.
Ross FM, Vet-Loiseau H, Ameye G, Gutierrez NC, Liebisch P, O'Connor S, et al. Report from the European Myeloma Network on interphase FISH in multiple myeloma and related disorders. Haematologica. 2012;97:1272–7.
Shin SY, Jang S, Park CJ, Chi HS, Lee JH, Lee JH, et al. Application of an immune-magnetic cell sorting method for CD138-positive plasma cells in FISH analysis of multiple myeloma. Int J Lab Hematol. 2012;34:541–6.
Gole L, Lin A, Chua C, Chng WJ. Modified cIg-FISH protocol for multiple myeloma in routine cytogenetic laboratory practice. Cancer Genet. 2014;207:31–4.
Slovak ML, Bedell V, Pagel K, Chang KL, Smith D, Somlo G. Targeting plasma cells improves detection of cytogenetic aberrations in multiple myeloma: phenotype/genotype fluorescence in situ hybridization. Cancer Genet Cytogenet. 2005;158:99–109.
Hu Y, Chen L, Sun CY, She XM, Ai LS, Qin Y. Clinical significance of chromosomal abnormalities detected by interphase fluorescence in situ hybridization in newly diagnosed multiple myeloma patients. Chin Med J (Engl). 2011;124:2981–5.
Lai YY, Huang XJ, Cai Z, Cao XS, Chen FP, Chen XQ, et al. Prognostic power of abnormal cytogenetics for multiple myeloma: a multicenter study in China. Chin Med J (Engl). 2012;125:2663–70.
Lim AS, Lim TH, See KH, Ng YJ, Tan YM, Choo NS, et al. Cytogenetic and molecular aberrations of multiple myeloma patients: a single-center study in Singapore. Chin Med J (Engl). 2013;126:1872–7.
Boyd KD, Ross FM, Chiecchio L, Dagrada GP, Konn ZJ, Tapper WJ, et al. A novel prognostic model in myeloma based on co-segregating adverse FISH lesions and the ISS: analysis of patients treated in the MRC Myeloma IX trial. Leukemia. 2012;26:349–55.
Fonseca R, Blood EA, Oken MM, Kyle RA, Dewald GW, Bailey RJ, et al. Myeloma and the t(11;14)(q13;q32); evidence for a biologically defined unique subset of patients. Blood. 2002;99:3735–41.
Ciolino AL, Tang ME, Bryant R. Statistical treatment of fluorescence in situ hybridization validation data to generate normal reference ranges using Excel functions. J Mol Diagn. 2009;11:330–3.
Avet-Loiseau H, Attal M, Campion L, Caillot D, Hulin C, Marit G, et al. Long-term analysis of the IFM 99 trials for myeloma: cytogenetic abnormalities [t(4;14), del(17p), 1q gains] play a major role in defining long-term survival. J Clin Oncol. 2012;30:1949–52.
Avet-Loiseau H, Attal M, Moreau P, Charbonnel C, Garban F, Hulin C, et al. Genetic abnormalities and survival in multiple myeloma: the experience of the Intergroupe Francophone du Myelome. Blood. 2007;109:3489–95.
An G, Xu Y, Shi L, Shizhen Z, Deng S, Xie Z, et al. Chromosome 1q21 gains confer inferior outcomes in multiple myeloma treated with bortezomib but copy number variation and percentage of plasma cells involved have no additional prognostic value. Haematologica. 2014;99:353–9.
An G, Li Z, Tai YT, Acharya C, Li Q, Qin X, et al. The impact of clone size on the prognostic value of chromosome aberrations by fluorescence in situ hybridization in multiple myeloma. Clin Cancer Res. 2015;21:2148–56.
Lee CK, Ma ES, Shek TW, Lam CC, Au WY, Wan TS, et al. Plasmablastic transformation of multiple myeloma. Hum Pathol. 2003;34:710–4.
Hartmann L, Biggerstaff JS, Chapman DB, Scott JM, Johnson KR, Ghirardelli KM, et al. Detection of genomic abnormalities in multiple myeloma: the application of FISH analysis in combination with various plasma cell enrichment techniques. Am J Clin Pathol. 2011;136:712–20.
Ma SK, Wan TS, Cheuk AT, Fung LF, Chan GC, Chan SY, et al. Characterization of additional genetic events in childhood acute lymphoblastic leukemia with TEL/AML1 gene fusion: a molecular cytogenetics study. Leukemia. 2001;15:1442–7.
Wan TS, Ma ES. Molecular cytogenetics: an indispensable tool for cancer diagnosis. Chang Gung Med J. 2012;35:96–110.
Dimopoulos M, Kyle R, Fermand JP, Rajkumar SV, San MJ, Chanan-Khan A, et al. Consensus recommendations for standard investigative workup: report of the International Myeloma Workshop Consensus Panel 3. Blood. 2011;117:4701–5.
Sewify EM, Afifi OA, Mosad E, Zaki AH, El Gammal SA. Cyclin D1 amplification in multiple myeloma is associated with multidrug resistance expression. Clin Lymphoma Myeloma Leuk. 2014;14:215–22.
Acknowledgement
The authors thank the clinicians who submitted myeloma samples for Target FISH study.
Funding
No research grant or external funding is used to support this study.
Availability of data and materials
The dataset supporting the conclusion of this article is included within the article.
Authors’ contributions
ESKM designed the study, analyzed the results and wrote the manuscript. CLNW, ATCW and GC performed the Target FISH experiments. TLC supervised the Target FISH experiments and interpreted the data. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Ma, E.S.K., Wang, C.L.N., Wong, A.T.C. et al. Target fluorescence in-situ hybridization (Target FISH) for plasma cell enrichment in myeloma. Mol Cytogenet 9, 63 (2016). https://doi.org/10.1186/s13039-016-0263-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13039-016-0263-7