
Abstract
Background
Mowat-Wilson syndrome (MWS) is a genetic condition characterized by distinctive facial features, moderate to severe intellectual disability, developmental delay and multiple congenital anomalies. MWS is caused by heterozygous mutations or deletions of the ZEB2 gene located on chromosome 2q22.3. At present, over 190 cases with mutations and deletions involving the ZEB2 gene have been reported, but triplication or duplication of reciprocal region of Mowat-Wilson syndrome has never been reported.
Case Presentation
Here we report a 2-year-2-month-old boy carrying a de novo 2.9 Mb complex copy number gain at 2q22.3 involving triplication of ZEB2 gene. The boy is characterized by intrauterine growth retardation, hypotonia, cognitive impairment, multiple congenital anomalies and behavioral abnormalities.
Conclusion
This case provides evidence that triplication of ZEB2 gene may be clinical significance and ZEB2 gene is likely to be a dosage sensitive gene.
Similar content being viewed by others


Background
Mowat-Wilson syndrome (MWS; OMIM# 235730) is an autosomal dominant genetic syndrome with multiple congenital anomalies. MWS is characterized by distinctive facial features, epilepsy, moderate to severe intellectual disability, global developmental delay, and congenital anomalies including agenesis of the corpus callosum, Hirschsprung disease, genitourinary anomalies, hypospadias, congenital heart disease, short stature and hypotonia [1–6]. MWS individuals display behavior problems including a happy affect and sociable demeanor, repetitive behaviors, pain insensitivity and a high rate of oral behaviors [7]. Eye abnormalities and craniosynostosis are rare features of this syndrome [8–10]. Eye abnormalities include iris/retinal colobomas, atrophy or absence of the optic nerve, hyphema, and deep refraction troubles, sometimes leading to severe visual consequences [8]. The syndrome is caused by heterozygous deletions or mutations of ZEB2 (OMIM# 605802) gene located on chromosome 2q22.3. So far, more than 190 individuals with MWS have been described, who result from more than 100 different mutations or deletions of ZEB2 gene. However, no obvious genotype-phenotype correlation was observed unless MWS patients carrying large deletions presented with more severe conditions, which may be the effect of continuous genes deletion [11–14]. Currently, no clinical presentations of patients with ZEB2 copy number gain have been reported. Here, we report the first case of a de novo 2.9 Mb copy number gain at 2q22.3 involving triplication of the entire ZEB2 gene detected by chromosomal microarray analysis (CMA). This case suggests that ZEB2 gene is likely to be a dosage sensitive gene.
Case presentation
The proband was the first child of healthy unrelated parents and family history was unremarkable. Intrauterine growth retardation was noticed by ultrasound examination at 7 months of pregnancy. He was born by vaginal delivery at 38 weeks of gestation. Birth weight was 3.0 kg (20.3 %), length 48 cm (8.5 %) and head circumference 32 cm (1.7 %). Apgar scores were all 9. He had severe hypotonia. No feeding difficulty was noted at all times. The development milestones were delayed: he raised his head at 4 months of age, sat alone at 8 months and walked without assistance at 1 year 8 months. Language development was significantly delayed.
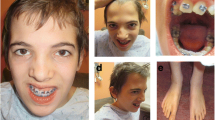
The patient was 2 years 2 months old at the time of molecular evaluation. His weight was 12.5 kg (39.7 %), height 86.2 cm (17.9 %) and head circumference 48.5 cm (46.3 %). He demonstrated catch-up growth but hypotonia persisted. His voice was low and he cried weakly. His receptive language was relative normal but he used body language to communicate. His cognitive competence was lower than his peers. On physical examination, his distinctive facial features included scaphocephaly, flat facial profile, auricle dysplasia, low-set and asymmetrical ears, small eyes, flat nose bridge, shallow philtrum, small mouth, teeth dysplasia, micrognathia, sparse eyebrows and hair. He had short hands and broad fingers (Fig. 1). Echocardiography revealed a small atrial septal defect. No genitourinary anomalies was noticed except for small testes. He had chronic and mild to moderate constipation, but no intestinal blockage and enlargement of the colon, and was not diagnosed with Hirschsprung disease. He always displayed a smiling, open-mouth expression and a happy, sociable demeanor as well as timid behavior. He never presented with epileptic seizures, and EEG was normal. The brain magnetic resonance imaging (MRI) showed normal corpus callosum and no other brain structural abnormalities. No additional abnormalities was noticed.

The proband at 2 years 2 months of age. Note scaphocephaly and flat facial profile, auricle dysplasia, low-set and asymmetrical ears, small eyes, flat nose bridge, shallow philtrum, small and open mouth, teeth dysplasia, micrognathia, sparse eyebrows and hair, short hands and broad fingers
Methods
Chromosomal microarray analysis
Chromosomal microarray analysis was performed for the proband and both parents using Affymetrix Cytoscan HD Array (Affymetrix, USA). Genomic DNA was extracted from peripheral blood using a commercial kit (Qiagen). The labeling and hybridization procedures were performed following manufacturer’s instructions. The raw data of chromosomal microarray was analyzed by Affymetrix Chromosome Analysis Suite Software.
Results
CMA test revealed a complex gain of copy number at 2q22.2q22.3, which involves a duplication (chr2:143,886,436-144,391,185) and a triplication (chr2:144,391,186-146,831,592) (Fig. 2). Parental tests were normal. Thus, the proband carried a de novo copy number variant.

Affymetrix cytoscan HD array analysis including weighted log2 ratio (upper), copy number state (middle) and allele peaks (lower) are shown for chromosome 2. The result shows copy number gain at 2q22.2q22.3 encompassing the entire ZEB2 gene. The genomic coordinates (hg19): chr2: 143,886,436-146,831,592. The copy number gain region is denoted by a black bar
Discussion
ZEB2 gene mutations or deletions cause Mowat-Wilson syndrome through a haploinsufficiency mechanism, but little is known about the clinical significance of ZEB2 copy number gain. In this study, we report a 2-year-2-month-old boy with global developmental delay, cognitive impairment, multiple congenital anomalies and behavior problems who carried a de novo 2.9 Mb triplication at 2q22.3 involving the entire ZEB2, GTDC1 and TEX41 genes and part of ARHGAP15 gene. No other clinical significant CNVs were detected. The patient’s clinical presentation was compared with the typical features of Mowat-Wilson syndrome (Table 1). Some of our patient’s clinical features overlapped with that of Mowat-Wilson syndrome, in particular, severe speech impairment with relative preservation of receptive language, open-mouth appearance and happy demeanor. However, his distinctive facial features were significantly different from that of MWS which included deep-set large and widely spaced eyes, upturned earlobes, saddle nose with rounded nasal tip, pointed chin, flaring eyebrows and elongated face. He had significant intrauterine growth retardation and severe hypotonia whereas he demonstrated postnatal catch-up growth but hypotonia persisted. No triplication at this locus had been reported in literature or described in database. We identified several cases with duplications of ZEB2 gene in DECIPHER and ISCA databases (Table 2 and Fig. 3). All these duplications were de novo except for ones without parental tests and no copy number gain including ZEB2 gene was reported in the DGV, which strongly suggested a pathogenic nature of these copy number gains.

The panel shows a genome view of all duplications or triplication cases (blue or black colored custom tracks) relative to the genomic coordinates and RefSeq genes at 2q22.3 region, extracted from Human Genome Build 37 (hg19). Red arrow pinpoints the ZEB2 gene. Blue: duplication; Black: triplication
There are four genes involved in the copy number gain at 2q22.3 of our patient: ZEB2, GTDC1 and TEX41 genes are triplicated, part of ARHGAP15 is duplicated. ARHGAP15, a member of the RHO GTPase-activating proteins (GAPs), regulates RHO GTPases (see ARHA; MIM 165390) which regulates diverse biologic processes [15]. GTDC1 is ubiquitous expressed at relatively high levels in lung, spleen, testis, and peripheral blood leukocytes, suggesting that it may have biochemical functions in these organs [16]. TEX41 is a non-protein coding gene. Currently, none of the three genes are known to have any clinical significance.
The protein encoded by ZEB2 gene is a member of δEF1/Zfh-1 family, containing a Smad-binding domain, a homeodomain-like sequence, and two separate clusters of zinc fingers at the amino and carboxy terminals [17]. The ZEB2 protein interacts with SMAD proteins and acts as a transcriptional repressor in response to TGF-β signaling [17]. The SMAD proteins are cytoplasmic mediators that are tightly controlled and play an important role in relaying TGF-ß signals from cell-surface receptors to the nucleus. The TGF-ß family exerts a wide range of biological functions in cell growth, differentiation, apoptosis and development of the embryo. ZEB2 gene is highly conserved among different species. The homologous alignment at amino acid levels reveals 97 % similarities between human and mouse, and 88 % between human and Xenopuslaevis. In addition, these proteins share the same amino acids in the zinc finger domain and certain similarities in their Smad binding domain (SBD). These findings suggest that the protein plays a similar role in vivo.
It was important to note that overexpression of Xenopus SIP1 (XSIP1) induced enlargement of neural tissue in anterior region, and some embryos failed to form eye vesicles and normal head phenotypes. Ectopic expression of XSIP1 induced anterior neural markers suggesting that XSIP1 played a role in early neurogenesis [18]. The animal model evidence shows that the ZEB2 gene is dosage sensitive and its precise regulation and expression is vital to embryonic neural and neural crest development.
Currently several genes have been known to be dosage sensitive genes, such as MECP2, NIPBL and NSD1 etc. For example, it is well known that haploinsufficiency of MECP2 gene typically results in Rett syndrome in females and severe neonatal encephalopathy or lethality in males [19]. Duplications overlapping the entire MECP2 gene are associated with MECP2 duplication syndrome characterized by global developmental delay, intellectual disability, autistic features, epilepsy and recurrent infections [20]. Patients with MECP2 triplications have also been reported with more severe phenotypes [21]. Cornelia de Lange syndrome is a multisystem congenital anomaly disorder and mutations or deletions of NIPBL gene is a major cause for this condition [22]. Conversely, NIPBL copy number gain is responsible for 5p13 duplication syndrome consisting of developmental delay, learning disability, distinctive facial features and behavior problems [23–25]. Similarly, haploinsufficiency of the NSD1 gene located on 5q35 is the major cause of Sotos syndrome recognized by intellectual disability, overgrowth, typical facial appearance, behavior problems and seizures [26], whereas reciprocal duplications of Sotos syndrome region overlapping the entire NSD1 gene present a reverse phenotype including delayed bone age, microcephaly, developmental delay and seizures [27, 28]. We believe more dosage sensitive genes exist in the human genome and are yet to be discovered. Here we provide the first evidence suggesting that ZEB2 gene is such a dosage sensitive gene similar to the aforementioned genes.
In conclusion, we first report a patient carrying a triplication at 2q22.3 involving the entire ZEB2 gene who presents overlapping features of Mowat-Wilson syndrome. Based on the clinical evidence from patients with de novo copy number gain involving the ZEB2 gene and the experimental evidence from Xenopus ZEB2 overexpression model, we propose that ZEB2 copy number gain is functionally and clinically significant.
Consent
Written informed consent was obtained from the patient for publication of this Case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
References
Mowat DR, Croaker GD, Cass DT, Kerr BA, Chaitow J, Adès LC, et al. Hirschsprung disease, microcephaly, mental retardation, and characteristic facial features: Delineation of a new syndrome and identification of a locus at chromosome 2q22–q23. J Med Genet. 1998;35(8):617–23.
Mowat DR, Wilson MJ, Goossens M. Mowat–Wilson syndrome. J Med Genet. 2003;40(5):305–10.
Wakamatsu N, Yamada Y, Yamada K, Ono T, Nomura N, Taniguchi H, et al. Mutations in SIP1, encoding Smad interacting protein-1, cause a form of Hirschsprung disease. Nat Genet. 2001;27(4):369–70.
Zweier C, Horn D, Kraus C, Rauch A. Atypical ZFHX1B mutation associated with a mild Mowat–Wilson syndrome phenotype. Am J Med Genet A. 2006;140(8):869–72.
Wilson M, Mowat D, Dastot-Le Moal F, Cacheux V, Kaariainen H, et al. Further delineation of the phenotype associated with heterozygous mutations in ZFHX1B. Am J Med Genet A. 2003;119A(3):257–65.
Garavelli L, Mainardi PC. Mowat–Wilson syndrome. Orphanet J Rare Dis. 2007;2:42.
Evans E, Einfeld S, Mowat D, Taffe J, Tonge B, Wilson M. The behavioral phenotype of Mowat-Wilson syndrome. Am J Med Genet A. 2012;158A(2):358–66.
Bourchany A, Giurgea I, Thevenon J, Goldenberg A, Morin G, Bremond-Gignac D, et al. Clinical spectrum of eye malformations in four patients with Mowat-Wilson syndrome. Am J Med Genet A. 2015;167(7):1587–92.
Wenger TL, Harr M, Ricciardi S, Bhoj E, Santani A, Adam MP, et al. CHARGE-like presentation, craniosynostosis and mild Mowat–Wilson syndrome diagnosed by recognition of the distinctive facial gestalt in a cohort of 28 new cases. Am J Med Genet A. 2014;164A(10):2557–66.
Hartill VL, Pendlebury M, Hobson E. Mowat-Wilson syndrome associated with craniosynostosis. Clin Dysmorphol. 2014;23(1):16–9.
Zweier C, Thiel CT, Dufke A, Crow YJ, Meinecke P, Suri M, et al. Clinical and mutational spectrum of Mowat–Wilson syndrome. Eur J Med Genet. 2005;48(2):97–111.
Dastot-Le Moal F, Wilson M, Mowat D, Collot N, Niel F, Goossens M. ZFHX1B mutations in patients with Mowat-Wilson syndrome. Hum Mutat. 2007;28(4):313–21.
Garavelli L, Zollino M, Mainardi PC, Gurrieri F, Rivieri F, Soli F, et al. Mowat–Wilson syndrome: facial phenotype changing with age: study of 19 Italian patients and review of the literature. Am J Med Genet Part A. 2009;149A(3):417–26.
Adam MP, Conta J, Bean LJH. Mowat-Wilson Syndrome. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2015. http://www.ncbi.nlm.nih.gov/pubmed/?term=Adam+MP%2C+Conta+J%2C+Bean+LJH.+Mowat-Wilson+Syndrome.
Seoh ML, Ng CH, Yong J, Lim L, Leung T. ArhGAP15, a novel human RacGAP protein with GTPase binding property. FEBS Lett. 2003;539(1–3):131–7.
Zhao E, Li Y, Fu X, Zhang JY, Zeng H, Zeng L, et al. Cloning and expression of human GTDC1 gene (glycosyltransferase-like domain containing 1) from human fetal library. DNA Cell Biol. 2004;23(3):183–7.
Verschueren K, Remacle JE, Collart C, Kraft H, Baker BS, Tylzanowski P, et al. SIP1, a novel zinc finger/homeodomain repressor, interacts with Smad proteins and binds to 5'-CACCT sequences in candidate target genes. J Biol Chem. 1999;274(29):20489–98.
Eisaki A, Kuroda H, Fukui A, Asashima M. XSIP1, a member of two-handed zinc finger proteins, induced anterior neural markers in Xenopuslaevis animal cap. Biochem Biophys Res Commun. 2000;271(1):151–7.
Amir RE, Van den Vewer IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23(2):185–8.
Ramocki MB, Tavyev YJ, Peters SU. The MECP2 duplication syndrome. Am J Med Genet A. 2010;152A(5):1079–88.
Wax JR, Pinette MG, Smith R, Chard R, Cartin A. Second-trimester prenasal and prefrontal skin thickening-association with MECP2 triplication syndrome. J Clin Ultrasound. 2013;41(7):434–7.
Boyle MI, Jespersgaard C, Brondum-Nielsen K, Bisgaard AM, Tumer Z. Cornelia de lange syndrome. Clin Genet. 2015;88(1):1–12.
Novara F, Alfei E, D'Arrigo S, Pantaleoni C, Beri S, Achille V, et al. 5p13 microduplication syndrome: a new case and better clinical definition of the syndrome. Eur J Med Genet. 2013;56(1):54–8.
Yan J, Zhang F, Brundage E, Scheuerle A, Lanpher B, Erickson RP, et al. Genomic duplication resulting in increased copy number of genes encoding the sister chromatid cohesion complex conveys clinical consequences distinct from Cornelia de Lange. J Med Genet. 2009;46(9):626–34.
Oexle K, Hempel M, Jauch A, Meitinger T, Rivera-Brugues N, Stengel-Rutkowski S, et al. 3.7 Mb tandem microduplication in chromosome 5p13.1-p13.2 associated with developmental delay, macrocephaly, obesity, and lymphedema. Further characterization of the dup(5p13) syndrome. Eur J Med Genet. 2011;54(3):225–30.
Tatton-Brown K, Cole TRP, Rahman N. Sotos Syndrome. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 2004. 1993–2015.
Novara F, Stanzial F, Rossi E, Benedicenti F, Inzana F, Di Gregorio E, et al. Defining the phenotype associated with microduplication reciprocal to Sotos syndrome microdeletion. Am J Med Genet A. 2014;164A(8):2084–90.
Rosenfeld JA, Kim KH, Angle B, Troxell R, Gorski JL, Westemeyer M, et al. Further Evidence of Contrasting Phenotypes Caused by Reciprocal Deletions and Duplications: Duplication of NSD1 Causes Growth Retardation and Microcephaly. Mol Syndromol. 2013;3(6):247–54.
Acknowledgments
We cherish our sincerest gratitude for Dr. Yiping Shen's guidance and selfless help who is employed by Boston Children’s Hospital. We would like to thank the family of the proband for their cooperation with this study.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interest.
Authors' contributions
HY carried out the cytogenetic studies and wrote the manuscript. LL made the clinical evaluation and collected clinical information of the patient in detail. LZ and ZM coordinated the clinical evaluation. MC and JZ carried out the cytogenetic studies. All the authors have read and approved the manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Yuan, H., Zhang, L., Chen, M. et al. A de novo triplication on 2q22.3 including the entire ZEB2 gene associated with global developmental delay, multiple congenital anomalies and behavioral abnormalities. Mol Cytogenet 8, 99 (2015). https://doi.org/10.1186/s13039-015-0206-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13039-015-0206-8