
Abstract
Background
5-Aminolevulinic acid (ALA) recently received much attention due to its potential application in many fields. In this study, an ALA production strain of Escherichia coli was constructed by rational metabolic engineering and stepwise improvement based on known regulatory and metabolic information and CRISPR/Cas9 mediated gene knockout.
Results
A metabolic strategy to produce ALA directly from glucose in this recombinant E. coli via the C5 pathway was applied herein. The rational metabolic engineering by gene knockouts significantly improved ALA production from 662.3 to 1601.7 mg/L. In addition, we managed to synergistically produce ALA via the C4 pathway in recombinant strain. The expression of a modified hemA gene, encoding an ALA synthase from Rhodobacter sphaeroides, improved ALA production from 1601.7 to 2099.7 mg/L. After 24 h cultivation, a yield of 0.210 g ALA per g glucose was achieved by constructed E. coli D5:FYABD-RSA.
Conclusion
Our study revealed that an industrially competitive strain can be efficiently developed by metabolic engineering based on combined rational modification and optimization of gene expression.
Similar content being viewed by others

Background
Synthetic biology plays a critical part in bio-based production of fuels, chemicals and materials from biomass. The sophistication of available genetic and biochemical tools has developed as synthetic biology applications have grown in complexity. In the past 20 years, a dramatic change in the scope and complexity of efforts within the space of synthetic biology has occurred [1]. Due to the great development of genetic engineering, accomplishments have progressed from simple reconstitution of biosynthetic pathways in heterologous hosts to complicated refactoring efforts [2,3,4] that can produce medicinally relevant compounds (strictosamide, taxadiene, etc.) [5, 6] or amino acids (L-valine, L-threonine, etc.) [7, 8] in a high titer.
Among many systems available for heterologous protein production, the Gram-negative bacterium Escherichia coli remains one of the most attractive organisms because of its ability to grow rapidly and at high density on inexpensive substrates. Well-characterized genetics and availability of a large number of cloning vectors and mutant host strains [9]. As a commonly used organism for expression and optimization of heterologous biosynthetic pathways, E. coli has been the most widely used prokaryotic system that produces heterologous proteins for industrial production of bacterial metabolites by batch and fed-batch operations [10, 11]. E. coli is a facultative anaerobic, non-sporulating bacterium that is considered as the workhorse of modern biotechnology in the microbial production of bio-fuels and biochemicals [12]. Recent advances in the fundamental understanding of transcription, translation and protein folding in E. coli, together with developments of improved genetic tools have made E. coli more valuable than ever [9].
5-Aminolevulinic acid (ALA) is a key intermediate involved in the biosynthesis of tetrapyrrole, which has attracted much attention for its potential applications in agriculture, cosmetics and cancer therapy. Due to its nontoxicity to crops, animals and humans, ALA is also used as selective biodegradable herbicide and insecticide in agriculture [13].
In living organisms, there are two major pathways described for ALA biosynthesis [13]. One is the C4 pathway, which occurs in mammals, birds, yeast and purple non-sulfur photosynthetic bacteria. In this pathway, ALA is formed through catalyzation of 5-aminolevulinate synthase, which condenses glycine and succinyl-CoA, an intermediate of the tricarboxylic acid (TCA) cycle [14]. The second pathway, C5, is present in higher plants, algae and many bacteria including E. coli [15]. In the C5 pathway, glutamate is the only substrate for biosynthesis of ALA.
In E. coli, ALA biosynthesis is through the C5 pathway and is tightly regulated by feedback inhibition of heme, the end product of the C5 pathway [16]. Earlier study has developed an available metabolic strategy of ALA biosynthesis via C5 pathway in E. coli [17]. Through this strategy, ALA can be synthesized in E. coli by over-expressing the key genes, hemA and hemL, in a modified minimal medium using glucose as the sole carbon source. In the research reported herein, we used a similar process for ALA biosynthesis. We managed to integrate T7 RNA Polymerase into the genome of E. coli BW25113 in our earlier work. This mutant strain was named E. coli BW25113-T7 [18]. In this mutant strain, the key genes, hemA, hemL and RhtA, were expressed efficiently and controlled by the T7 Expression System.
Our earlier work proved that this process for ALA biosynthesis in E. coli BW25113-T7 works. Through efficient and controllable protein expression using the T7-Lac promoter, E. coli BW25113-T7 accumulated 42.4% more ALA than E. coli BL21(DE3) did [18]. Here, we report the development of the genetically defined ALA overproducing E. coli strain by systems metabolic engineering. It was thus our goal to engineer E. coli to make a 5-Aminolevulinic acid overproducer with completely known genotype by systems-level metabolic engineering. In this study, rational metabolic engineering based on known regulatory and metabolic information was carried out to develop an E. coli strain capable of overproducing ALA.
Results
Available metabolic strategy of ALA production by overexpression of hemA, hemL and eamA
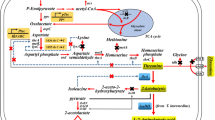
We constructed an E. coli strain from BW25113 which achieved high efficiency of protein expression through T7 expression system in our earlier work [18]. This mutant E. coli BW25113-T7 strain was rationally engineered to produce ALA by over-expression of key genes. The biosynthetic pathways of ALA in E. coli and the strategies for constructing ALA production strain are shown in Fig. 1. The key genes of ALA synthesis via the C5 pathway in this strategy are hemA and hemL. The hemA gene encodes a NADPH-dependent glutamyl-tRNA reductase which catalyzes the reduction of glutamyl-tRNA to glutamate-1-semialdehyde (GSA) [17]. The hemL gene encodes glutamate-1-semialdehyde aminotransferase which quickly converts GSA to ALA. The eamA gene encodes an O-acetylserine or cysteine exporter which is capable of translocating dipeptides and amino acid analogs from the cytosol to the periplasm.

The biosynthetic pathways of ALA in E. coli and the strategies for constructing the ALA producing strain. The shaded boxes indicate the genes that were knocked out. Red arrows indicate increased flux or activity by directly over-expressing the corresponding genes. Blue arrows indicate decreased flux or activity by knocking out the corresponding genes. Dotted boxes represent the corresponding metabolic pathways
Plasmid pET-ALA-LAA was constructed to over-express these three key genes. In this plasmid, these three genes can be tightly controlled and efficiently expressed by the T7-Lac promoter. Cells that harbored pET-ALA-LAA were cultured in M9YE medium. After 24 h of induced fermentation, an ALA titer (662.3 mg/L) was determined.
Overexpression of gltB, gltD and gltX genes involved in the C5 pathway in E. coli
Glutamate and L-glutamyl-tRNA are both precursors of ALA synthesis via the C5 pathway. To increase glutamate and L-glutamyl-tRNA amounts for ALA synthesis, we overexpressed gltB, gltD and gltX genes alone or in combination.
There are two pathways by which E. coli synthesizes glutamate from ammonia [19]. The pathway catalyzed by glutamate dehydrogenase (encoded by gdhA) is ATP-independent. The other is a cyclic and ATP-driven pathway (Shown in Fig. 2) which catalyzed by glutamate synthase (encoded by gltBD). Our modified minimal medium does not provide a high level of ammonia. In this case, the ATP-driven pathway functions (Fig. 1). Glutamate synthase is a tetramer of dimers, with each dimer having one large and one small subunit (gltB and gltD, respectively) [19]. Glutamate synthase catalyzes the single-step conversion of L-glutamine and alpha-ketoglutarate into two molecules of L-glutamate (Fig. 2). In doing so, glutamate synthase simultaneously operates as the major source of L-glutamate for the cell and as a key step in ammonia assimilation during nitrogen-limited growth [19,20,21]. The ammonia-dependent activity can be catalyzed very slowly by just the small subunit in the absence of the full complex [22]. Glutamate-tRNA ligase (GluRS) is a member of the family of aminoacyl-tRNA synthetases, which is encoded by gltX gene [23, 24]. GluRS charges GlutRNA for both protein and ALA synthesis [25].

The ATP-driven pathway of L-glutamate biosynthesis in E. coli
In this study, we constructed four extra plasmids to overexpress these three genes (Table 1). These four plasmids were electroporated into cells that harbored pET-ALA-LAA. The resulting strains were cultivated in modified minimal medium (see ‘‘Section Materials and Methods’’) supplied with 10 g/L glucose for ALA accumulation analysis. After 24 h of fermentation, ALA concentration was determined (Table 2 and Fig. 3A). Strain BW25113-T7 which only contained pET-ALA-LAA was set as the control.

ALA production in recombinant E. coli expressing various related genes. Cultivation was performed in 300 mL Erlenmeyer flask supplied with 30 mL modified minimal medium supplied with 10 g/L glucose for 24 h. Results are the average of three individual experiments. A final ALA accumulation result was measured. Adjusted P values were calculated using Dunnett's multiple comparisons test (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001)
Beyond our expectation, we found all strains that harbored extra plasmids exhibited decreased ALA accumulation with E. coli BDX having the greatest decrease in 5-ALA accumulation relative to the control (Table 2). We found that all strains harbored extra plasmids grew much more slowly than control strains (Table S1). These results indicate that overexpression of gltB, gltD and gltX genes may not have a positive effect on the ALA biosynthesis in E. coli. This finding suggests that the biosynthesis of L-glutamate and conversion of L-glutamate to GlutRNA are not rate-limiting steps of ALA biosynthesis.
Enhanced production of 5-ALA by metabolic pathway modification
To further improve the BW25113-T7 strain, following targeted genetic modifications were performed (Fig. 1).
HemF is a strictly aerobic enzyme that requires molecular oxygen as the electron acceptor and produces hydrogen peroxide. The knock-out of hemF gene would mightily repress the biosynthesis of protoporphyrinogen IX (PPG) resulting in reduced endogenous loss of 5-ALA. After knocking out the hemF gene in BW25113-T7 (named D1:F), yield of 5-ALA increased 2.06-fold compared to the original strain (named as D0). This result indicated that knock-out of the hemF gene has a positive effect on ALA biosynthesis.
L-glutamate is a precursor of ALA synthesis via the C5 pathway. Therefore, we attempted to increase ALA accumulation by reducing endogenous loss of L-glutamate. GdhA, which encodes glutamate dehydrogenase, catalyzes the ATP-independent amination of α-ketoglutarate to yield L-glutamate [26]. This reaction is reversible (Fig. 4A). YbdK catalyzes ATP-dependent ligation of glutamate with cysteine at a low catalytic rate [27] (Fig. 4B). GadB, a glutamate decarboxylase enzyme, catalyzes cleavage of L-glutamate into carbon dioxide and 4-aminobutanoate [28] (Fig. 4C). These three genes in the ALA biosynthetic pathway were knocked out (Fig. 1) individually or jointly using the E. coli D1:F strain. ALA accumulations of those mutant strains ranged from a reduction to a 2.30-fold increase in ALA accumulation (Table 3). Among the mutant strains, E. coli D3:FYB significantly increased ALA accumulation (2.30-fold) compared with E. coli D0.

The reactions catalyzed by corresponding genes. The reaction directions shown in accordance with the physiological direction of the reaction. A GdhA catalyzes the ATP-independent amination of α-ketoglutarate to yield L-glutamate. B YbdK catalyzes the ATP-dependent ligation of glutamate with cysteine. C GadB catalyzes the cleavage of L-glutamate into carbon dioxide and 4-aminobutanoate
The periplasmic binding proteins—mppA, the L-alanyl-g-D-glutamyl-meso-diaminopimelate binding protein, or dppA, the dipeptide binding protein—actively import ALA through an interaction with the dipeptide inner membrane ATP-binding cassette transporter, DppBCDF, in E. coli [29]. We theorized that inactivation of dppA and/or mppA genes would reduce ALA assimilation and therefore increase ALA accumulation in the medium. As expected, inactivation of dppA improved ALA production but mppA did not (Table 3). To further improve ALA production, we used the E. coli D4:FYAB strain to perform dppA and mppA gene knockouts individually or jointly. We found that mutant strain, E. coli D5:FYABD, showed the highest production of ALA which was 2.42 fold greater than D0 (Table 3).
Growth characteristics of mutant strains and selection of optimal mutant E. coli strain
After knocking out these genes, growth of mutant strains in different media were examined to assess whether CRISPR/Cas9-mediated gene knock-out affected metabolic characteristics of the bacteria. These strains all harbored pET-ALA-LAA for producing ALA.
These mutant strains were cultured in M9YE medium (see ‘‘Section Materials and Methods’’) or standard LB medium (Fig. 5). Growth rate among these strains in LB medium ranged from 0.25 to 0.3 (Table 4). When cultured in M9YE medium, growth rate of these strains varied from 0.3 to 0.5. Two mutant strains (D2:FA and D3:FYB) showed significant change in growth rate when culture in M9YE medium. These results reveal that knockouts of some genes may lead to growth retardation of bacteria.

Growth characteristic of mutant E. coli strains in different medium. Cells were cultured in M9YE (A) or LB (B) medium with 50 μg/mL Kanamycin. Bacteria contained pET28b-ALA-LAA. Data are means of three replicates
We easily selected D5:FYABD as the optimal mutant strain for ALA production based on its growth rate and ALA production (Fig. 6). The ALA production was increased 2.42-fold with similar growth rate when compared with the original-type (D0).

The results of all mutant strains with respect to ALA production and growth rates. The ALA production and growth rates of the control D0 strain harboring pET28b-ALA-LAA are also indicated for comparison
Increasing ALA accumulation via C4 pathway
In E. coli, primary ALA biosynthesis is through the C5 pathway. To further increase accumulation of ALA, we tried to synergistically produce ALA via the C4 pathway. The photosynthetic bacterium, Rhodobacter sphaeroides, can accumulate ALA under certain conditions or after mutagenesis [30,31,32]. Through metabolic engineering, recombinant E. coli was also able to produce ALA from the C4 pathway through biotransformation [33]. In this aspect, the gene encoding for ALA synthase from R. sphaeroides was introduced into E. coli through genetic engineering [33]. Biosynthesis of glycine and succinyl-CoA are regulated in E. coli. Consequently, glycine and succinate (the precursor of succinyl-CoA) must be added to the culture medium artificially to provide sufficient substrates for enhanced ALA biosynthesis. In the current study, we overexpressed a modified heterologous hemA from R. sphaeroides using the T7 Expression System (Fig. 7). The amino acid sequence of hemARS was modified for better expression in E. coli. Glycine (2 g/L) was added to the M9YE medium as a substrate for the C4 pathway. These modifications increased ALA accumulation from 1602.72 mg/L with E. coli D5:FYABD to 2099.7 mg/L for the mutant strain containing the hemARS gene (Table 2 and Fig. 3B). After 24 h cultivation, a yield of 0.210 g ALA per g glucose was achieved (Fig. 8).

Schematic presentation ALA of production in E. coli via both C4 and C5 pathways. Glycine was added to the culture medium as a substrate for the C4 pathway. G6P, glucose-6-phosphate; Pyr, pyruvate; Ket, α-ketoglutarate; Glu, glutamate; GlutRNA, Glutamyl-tRNA; GSA, glutamate 1-semialdehyde aminotransferase; ALA, 5-aminolevulinic acid; PBG, porphobilinogen

Fermentation of ALA in E. coli D5:FYABD-RSA. A 1% (v/v) inoculum from an overnight culture for 12 h was used. 10 g/L glucose and 2 g/L glycine were added initially. 0.1 mM IPTG was added as indicated. During the fermentation, the pH was controlled optimally at 6.5 ± 0.3
Discussion
Metabolic engineering provides a powerful tool for regulating metabolic pathway towards accumulation of desired compounds [34]. Many bio-based products are now produced in E. coli through metabolic engineering or synthetic biology [17, 35,36,37]. In the present study, we developed a metabolic strategy to produce ALA directly from glucose and constructed a well-defined genetically-engineered E. coli strain based on known metabolic and regulatory information and CRISPR-Cas9 mediated gene knock-outs. After systematic metabolic engineering of E. coli, we were able to achieve a high yield of 0.210 g of ALA per gram of glucose. More importantly, the engineered strain developed in this study can be further improved because all of the modifications are clearly defined.
We first constructed the ALA-producing E. coli base strain by amplifying three key genes. HemA encodes Glutamyl-tRNA reductase, which catalyzes the first step of porphyrin biosynthesis [38]. Glutamate-1-semialdehyde 2,1-aminomutase (hemL) catalyzes the pyridoxal 5'-phosphate-dependent transfer of the amino group from C2 of glutamate-1-semialdehyde (GSA) to C1, thereby forming ALA [39]. Synergistic functions of hemA and hemL efficiently converted glutamate to ALA. The eamA gene encoded an integral membrane protein which is implicated in exporting metabolites of the cysteine pathway [40]. Deletion of eamA in E. coli strain BL21 is associated with a decline in extracellular 5-aminolevulinate concentration [41] which suggests that eamA might be a transporter involved in export cysteine pathway metabolites. We overexpressed hemA, hemL and eamA genes using T7 system in E. coli BW25113-T7 and achieved a yield of 0.066 g of ALA per gram of glucose. Then several genes encoding those enzymes directly involved in Glutamate and L-glutamyl-tRNA biosynthesis were amplified. However, this modification did not increase accumulation of ALA. This result was similar to previous research by Kang [17]. In their study, amplifying gltX genes decreased ALA accumulation. Our result also suggested biosynthesis of L-glutamate and conversion of L-glutamate to GlutRNA are not rate-limiting steps in ALA biosynthesis. Overexpression of gltX, gltB and gltD genes may increase metabolic stress on cells which may lead to growth retardation and decreased ALA accumulation.
Further improvement of the ALA-producing strain was achieved to modify metabolic pathway rationally by using the metabolic and regulatory information available in the literature (Fig. 1). In E. coli, biosynthesis of ALA via the C5 pathway is tightly regulated by the end product heme [16]. Synthesis of heme leads to an endogenous depletion of ALA. Additionally, heme also makes hemA unstable which inhibits accumulation of ALA [42]. Therefore, we expected that down-regulation of heme biosynthesis might lead to high ALA production. However, knock-outs of most genes encoding enzymes directly involved in heme biosynthesis (hemBCDEGH) lead to severe growth limitation or even death. Oxidative decarboxylation of coproporphyrinogen III to protoporphyrinogen IX is catalyzed by two enzymes in E. coli. Under aerobic conditions, the hemF gene product is active. Under anaerobic conditions, HemN catalyzes the coproporphyrinogen III dehydrogenase reaction. Hence, we believed that knock-out of hemF will ultimately restrict biosynthesis of heme. This belief was confirmed with the observation that the mutant strain E. coli D1:F increased ALA yield from 662.25 to 1361.22 mg/L.
Our next step was to knock out the genes responsible for major competing pathways based on published metabolic and regulatory information (Fig. 1). The major competing pathways, including nitrogen metabolism, glutathione metabolism and butanoate metabolism, were suppressed in the present study. In E. coli, there are two pathways to synthesize glutamate from ammonia [19]. The pathway catalyzed by glutamate dehydrogenase (encoded by gdhA) directly from ammonia, α-ketoglutarate, and NADPH [26]. The other is a cyclic and ATP-driven pathway (Shown in Fig. 2) and is catalyzed by glutamate synthase (encoded by gltBD). Each turn of which utilizes one molecule each of ammonia, ATP, NADPH, and α-ketoglutarate to produce one molecule of glutamate [19]. When culture in our modified minimal medium (M9YE) which does not provide a high level of ammonia, the ATP-driven pathway functions (Fig. 1). As the reaction catalyzed by gdhA is reversible, we assumed that down-regulation of gdhA catalyzed reversible reaction is believed to reduce endogenous loss of L-glutamate and led to a raise of ALA production. The ybdK (encoding putative glutamate—cysteine ligase 2) and gadB (encoding glutamate decarboxylase B) genes were also knocked out to increase L-glutamate availability for ALA synthesis. In our study, all triple mutant strains (D3:FYA, D3:FYB and D3:FAB) yielded more ALA than double mutant strains (D2:FY, D2:FA and D2:FB). We propose that synergistical effects of knocking out these three genes (ybdK, gadB and gdhA) improves ALA yields because of the increased flux through L-glutamate. These observations led us to select mutant strain D4:FYAB for further modification even though this strain did not produce the highest yield of ALA.
To avoid ALA accumulation inside cells and to produce more ALA in the medium, inactivation of mppA and/or dppA genes was carried out. Cultivation results showed that inactivation of dppA (D1:D) improved ALA production while inactivation of mppA (D1:M) did not. The D1:D strain harboring pET28b-ALA-LAA accumulated 27.9% more ALA than the D0 strain did (Table 3). Further knockout of dppA and/or mppA genes in strain D4:FYAB showed a similar result. The additional knockout of the mppA gene in D4:FYAB did not increase ALA production. Double mutant of dppA and mppA in D4:FYAB strain even showed a 29.2% decrease in ALA production. These results suggest that silencing the dppA gene is more beneficial for ALA biosynthesis than knocking out the mppA gene.
According to metabolic and regulatory information available in the literature, we identified a mutant strain that allowed elevated ALA production with an acceptable growth rate. In the D5:FYABD strain, heme biosynthesis was limited and flux through glutamate was increased. Deletion of the dppA gene reduced ALA assimilation by E. coli which allowed increased ALA accumulation in the medium. The D5:FYABD strain harboring pET-ALA-LAA showed a 141.8% increase in ALA production compared with the D0 strain harboring the same plasmids. These results suggest that beneficial effects on ALA production of flux redistribution achieved in D5:FYABD strain could be further strengthened by engineering regulatory and export functions.
There are two major pathways described for ALA biosynthesis in living organisms [13]. We successfully measured production of ALA via the C5 pathway in this study and tried to develop a strategy to improve ALA production in recombinant E. coli via the C4 pathway. The ALA synthase from R. sphaeroides was introduced into E. coli to produce ALA from the C4 pathway. This modified hemARS was able to work synergistically with native hemA and hemL from E. coli (Fig. 7). In R. sphaeroides, glycine and succinyl-CoA can be catalyzed to form ALA by hemA. Since biosynthesis of glycine and succinyl-CoA was also regulated in E. coli, we added glycine in the medium artificially to provide more substrates for ALA biosynthesis. Through this strategy, ALA production in D5:FYABD was increased from 1601.7 mg/L to 2099.7 mg/L, a 31.1% increase in ALA accumulation.
Conclusion
In summary, rational metabolic engineering based on known metabolic and regulatory information, and co-expression of native key genes and modified heterologous gene, allowed development of an E. coli strain capable of efficiently producing ALA. An impressively high yield of 0.210 g of ALA per gram of glucose could be achieved using a metabolically engineered strain in flask culture. Further optimization of the fermentation process could improve ALA production to even higher levels. The approaches described in this study can also be applied more broadly to develop strains for efficient production of other metabolites.
Methods and materials
E. coli strains
Molecular cloning and manipulation of plasmids were done with E. coli DH5α (TransGen, Beijing). BW25113-T7 strains were used for CRISPR/Cas9-induced Double Strain Break and recombination. All E. coli strains were cultured routinely in standard LB medium when not mentioned otherwise.
Growth conditions
LB medium (10 g/L tryptone, 5 g/L yeast extract and 10 g/L NaCl, pH 7.2) was used in all DNA manipulations. During cultivation and fermentation, the modified M9 medium (M9YE) was used that contained 1 g/L NH4Cl, 0.5 g/L NaCl, 3 g/L KH2PO4, 17.1 g/L Na2HPO4·12H2O, 2 mM MgSO4, 0.1 mM CaCl2, 2 g/L yeast extract and 10 g/L glucose. Glycine (2 g/L) was added as indicated to serve as the substrate for the C4 pathway. Ampicillin (100 mg/mL), chloramphenicol (25 mg/mL) and kanamycin (50 mg/mL) were added to provide selective pressure for E. coli during cultivation when necessary. To induce expression of plasmid-borne genes, Isopropyl-β-D-thiogalactopyranoside (IPTG) was added to cultures which resulted in a final concentration of 0.1 mM. Considering cell growth and ALA stability, the pH was measured by a glass electrode and controlled at 6.5 ± 0.3 with 4 M NaOH.
Selection of integration site and design of homologous recombination
The function and detailed message of the hemF gene (and other genes) was verified in NCBI and BioCyc Database. Sequence of the hemF gene (and other genes) in the BW25113-T7 genome was confirmed in NCBI. As the recognition site for sgRNA, N20 site directs the Cas9 protein to enable site-specific induction of a DSB. The N20 site was found by BROAD international design tool, which is available at: http://www.broadinstitute.org/rnai/public/analysis-tools/sgrna-design.
Plasmid construction for CRISPR and preparation of linear donor dsDNA by PCR
All primer pairs we designed for gene cloning and intermediate plasmid construction are listed in Table S2. Plasmid pCas (Fig. S1A) and pTarget were prepared in our laboratory. Plasmid pTarget-gene was constructed by Reverse-PCR and T4-Ligation (T4 DNA Ligase, NEB, England) to replace the N20 fragment (Fig. S1B) with specific primers (Table S2). Plasmid pACYCD-gene and pACYCD-Donor-Gene were both constructed for preparing Donor DNA (Fig. S2) by In-Fusion® HD Cloning Kit (Takara, Japan). Donor DNA was cloned from pACYCD-Donor-Gene by Hi-Fi PCR (Phusion® High-Fidelity PCR Master Mix, NEB, England). All plasmids used in this research are listed in Table 1.
Electroporation, cell recovery, and plating
For transformation, the plasmid or linear DNA were electroporated into competent cells in the pre-chilled cuvette (0.1 cm) using Bio-Rad MicroPulser (1.8 kV, time constant > 5.0 ms). For selection, 25 μg/mL chloramphenicol (Chl) or 50 μg/mL kanamycin (Kan) were used alone or in combination. For induction of λ-Red proteins and lac operator, 1 mM arabinose and 1 mM IPTG were used.
To prepare cells harboring pCas, cells cultured at 37℃ (OD600 = 0.45–0.55) were made competent, mixed with pCas (100 ng) and subjected to electroporation, after which the cells were recovered in SOC medium (1 mL) for 1 h at 30℃, plated onto the Kan plate, and cultured at 30 ℃ for 18–24 h.
For CRISPR/Cas9-mediated homologous recombination, cells harboring pCas were cultured at 30℃ in medium containing Kan and Arabinose and made competent. After co-electroporation of Donor DNA (400 ng) with pTarget-Gene (100 ng), cells were recovered in SOC (1 mL) medium for 1 h at 30℃, plated onto Chl/Kan plate, and cultured at 30℃ for 18–24 h.
For elimination of pTarget-Gene, cells harboring both pCas and pTarget were cultured at 30℃ in medium containing Kan and IPTG for 2 h. Cells were plated onto Kan plates and cultured at 30℃ for 18–24 h.
For elimination of pCas, cells harboring pCas were cultured at 37℃ in the medium without any antibiotic for 12–16 h. Then the cells were plated onto non-antibiotic plates and cultured at 37℃ for 12–16 h.
Confirmation of CRISPR/Cas9-mediated gene knock-in in BW25113
The targeted genetic modifications were rationally engineered by CRISPR/Cas9 in E. coli BW25113-T7. The six targeted genes (hemF, ybdK, gadB, gdhA, dppA and mppA) were cloned from E. coli BW25113-T7 to prepare for Donor DNA. The process of hemF gene knock-out is shown here as example. The map of hemF gene knock-out in BW25113-T7 in ideal condition is shown in Fig. S3. The homologous left arm (HRL) and the homologous right arm (HRR) were set near site of N20. Both homologous arms were approximately 400 bp, which could have a high efficiency of recombination [43]. After CRISPR/Cas 9 mediated gene knock-out, it would make a deletion of 100 bp DNA in targeted gene.
To make CRISPR/Cas9-mediated homologous recombination in BW25113-T7, we electroporated pCas (encoding both Cas and λ-Red proteins) into E. coli BW25113-T7, followed by Arabinose (Ara) induction of pCas-encoded λ-Red proteins Gam, Bet and Exo. After preparing competent cells, pTarget-gene (such as pTarget-hemF) and Donor DNA were co-electroporated into cells (Fig. S4).
To verify deletion in target locus, all mutated bacterial strains were selected for colony PCR (Fig. S5). The primer pairs for colony PCR are shown in Table S2. Characteristics of those mutant strains are shown in Table 1.
Growth of bacteria in different medium
All E. coli strains were grown at 37℃ in conical flasks (250 mL) containing M9YE medium or LB medium with Kanamycin (50 μg/mL). Growth was measured by monitoring optical density at 600 nm (OD600) using a spectrophotometer.
The growth rate was fitted with Sigmoidal-4PL curves. The OD of the stationary phase (ODsp) and the time required to reach it are calculated from this fitted curve. The ratio of the ODsp to the time required to reach it was set as growth rate (hr-1) of the strain in medium.
Analytical procedures
Flask cultivations were carried out in 100 mL conical flasks supplied with 30 mL modified minimal medium at 37℃ with agitation of 200 rpm. A 1% (v/v) inoculum from an overnight culture (12) was used. Cells were cultured in M9YE at 37℃ until OD600 reached 0.7. Then IPTG with a final concentration of 0.1 mM was added to the induced group. To analyze ALA production, culture (30 mL) after inducing for 24 h was centrifuged (12,000 × g for 2 min at 4℃). The supernatant was analyzed for extracellular ALA concentration. ALA concentration was analyzed using modified Ehrlich’s reagent [44]. Specifically, standard or sample (2 ml after diluted) was mixed with 1 ml 1.0 M sodium acetate (pH 4.6) in a cuvette, and 0.5 ml acetylacetone (2,4-pentanedione) was added to each cuvette. Then the mixtures were heated to 100 ℃ for 15 min. After cooling for 15 min, the reaction mixture (1 ml) and freshly prepared modified Ehrlich's reagent (1 ml) were mixed together. After 30 min, the absorbance at 554 nm was measured. Standard plot for ALA measurement is shown in Fig. S6.
For analyzing glucose, 1 mL of culture was centrifuged (12,000 g for 2 min at 4 ℃) and the supernatant was then filtered through a 0.22 mm syringe filter for analysis. The HPLC system was equipped with a cation exchange column (HPX-87 H, BioRad Labs), and a differential refractive index (RI) detector (Shimadzu RID-10 A). A 0.5 mL/min mobile phase using 5 mM H2SO4 solution was applied to the column. The column was operated at 65 ℃.
Data analysis
Data for ALA production of were subjected to analysis of variance (ANOVA) by GraphPad Prism (version 7.00). Error bars indicate standard error of the mean (SEM). P values were calculated using Dunnett's multiple comparisons test (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001). The mean of each column was compared with the mean of a control column. BW25113-T7 (D0) was set as control column.
Availability of data and materials
The majority of data generated or analyzed during this study are included in this published article or in the supplementary information. Data not shown in this manuscript are available upon request from the corresponding author.
Abbreviations
- E. coli :
-
Escherichia coli
- ALA:
-
5-Aminolevulinic Acid
- GSA:
-
Glutamate-1-semialdehyde
- GluRS:
-
Glutamate-tRNA ligase
- IPTG:
-
Isopropyl-β-D-thiogalactopyranoside
- Kanr :
-
Kanamycin resistance gene
- Chlr :
-
Chloramphenicol resistance
- sgRNA:
-
Small-guide RNA
- HRL:
-
Homologous left arm
- HRR:
-
Homologous right arm
References
Phelan RM, Sachs D, Petkiewicz SJ, Barajas JF, Blake-Hedges JM, Thompson MG, Reider Apel A, Rasor BJ, Katz L, Keasling JD, et al. Development of Next Generation Synthetic Biology Tools for Use in Streptomyces venezuelae. ACS Synth Biol. 2017;6(1):159–66.
Yamanaka K, Reynolds KA, Kersten RD, Ryan KS, Gonzalez DJ, Nizet V, Dorrestein PC, Moore BS. Direct cloning and refactoring of a silent lipopeptide biosynthetic gene cluster yields the antibiotic taromycin A. Proc Nat Acad Sci USA. 2014;111(5):1957–62.
Shao Z, Rao G, Li C, Abil Z, Luo Y, Zhao H. Refactoring the Silent Spectinabilin Gene Cluster Using a Plug-and-Play Scaffold. ACS Synth Biol. 2013;2(11):662–9.
Temme K, Zhao D, Voigt CA. Refactoring the nitrogen fixation gene cluster from Klebsiella oxytoca. Proc Nat Acad Sci USA. 2012;109(18):7085–90.
Brown S, Clastre M, Courdavault V, O’Connor SE. De novo production of the plant-derived alkaloid strictosidine in yeast. Proc Nat Acad Sci USA. 2015;112(11):3205–10.
Soliman S, Tang Y. Natural and engineered production of taxadiene with taxadiene synthase. Biotechnol Bioeng. 2015;112(2):229–35.
Park JH, Lee KH, Kim TY, Lee SY. Metabolic Engineering of Escherichia coli for the Production of L-Valine Based on Transcriptome Analysis and in Silico Gene Knockout Simulation. Proc Nat Acad Sci USA. 2007;104(19):7797–802.
Lee SY, Lee KH, Park JH, Kim TY, Kim HU. Systems metabolic engineering of Escherichia coli for L -threonine production. Mol Syst Biol. 2007;3(1):149.
Baneyx F. Recombinant protein expression in Escherichia coli, vol. 10. England: Elsevier Ltd; 1999. p. 411–21.
Lee SY. High cell-density culture of Escherichia coli. Trends Biotechnol. 1996;14(3):98–105.
Yee L, Blanch HW. Recombinant Protein Expression in High Cell Density Fed-Batch Cultures of Escherichia Coli. BMC Biotechnol. 1992;10(12):1550–6.
Himmel ME, Karplus PA, Sakon J, Adney WS, Baker JO, Thomas SR. Polysaccharide hydrolase folds diversity of structure and convergence of function. Appl Biochem Biotech. 1997;63(1):315–25.
Sasaki K, Watanabe M, Tanaka T, Tanaka T. Biosynthesis, biotechnological production and applications of 5-aminolevulinic acid. Appl Microbiol Biot. 2002;58(1):23–9.
Burkovski A, Krämer R. Bacterial amino acid transport proteins: occurrence, functions, and significance for biotechnological applications. Appl Microbiol Biot. 2002;58(3):265–74.
Schön A, Krupp G, Gough S, Berry-Lowe S, Kannangara CG, Söll D. The RNA required in the first step of chlorophyll biosynthesis is a chloroplast glutamate tRNA. Nature. 1986;322(6076):281–4.
Woodard SI, Dailey HA. Regulation of Heme Biosynthesis in Escherichia coli. Arch Biochem Biophys. 1995;316(1):110–5.
Kang Z, Wang Y, Gu P, Wang Q, Qi Q. Engineering Escherichia coli for efficient production of 5-aminolevulinic acid from glucose. Metab Eng. 2011;13(5):492–8.
Ye C, Chen X, Yang M, Zeng X, Qiao S. CRISPR/Cas9 mediated T7 RNA polymerase gene knock-in in E. coli BW25113 makes T7 expression system work efficiently. J Biol Eng. 2021;15(1):1–22.
Miller RE, Stadtman ER. Glutamate synthase from Escherichia coli. An iron-sulfide flavoprotein. J Biol Chem. 1972;247(22):7407–19.
Berberich MA. A glutamate-dependent phenotype in E. coli K12: the result of two mutations. Biochem Bioph Res Co. 1972;47(6):1498–503.
Lozoya E, Sanchez-Pescador R, Covarrubias A, Vichido I, Bolivar F. Tight linkage of genes that encode the two glutamate synthase subunits of Escherichia coli K-12. J Bacteriol. 1980;144(2):616–21.
Mantsala P, Zalkin H. Active Subunits of Escherichia coli Glutamate Synthase. J Bacteriol. 1976;126(1):539–41.
Eriani G, Delarue M, Poch O, Gangloff J, Moras D. Partition of tRNA synthetases into two classes based on mutually exclusive sets of sequence motifs. Nature. 1990;347(6289):203–6.
Landès C, Perona JJ, Brunie S, Rould MA, Zelwer C, Steitz TA, Risler JL. A structure-based multiple sequence alignment of all class I aminoacyl-tRNA synthetases. Biochimie. 1995;77(3):194–203.
Friesen JD. Escherichia coli and Salmonella typhimurium: Cellular and Molecular Biology. In: vol 240. American Association for the Advancement of Science. Washington, D.C: Academic; 1988. p. 1678.
Veronese FM, Boccu E, Conventi L. Glutamate dehydrogenase from Escherichia coli: induction, purification and properties of the enzyme. Biochim Biophys Acta. 1975;377(2):217.
Lehmann C, Doseeva V, Pullalarevu S, Krajewski W, Howard A, Herzberg O. YbdK is a carboxylate-amine ligase with a γ-glutamyl: Cysteine ligase activity: Crystal structure and enzymatic assays. Proteins. 2004;56(2):376–83.
Moreau PL. The Lysine Decarboxylase CadA Protects Escherichia coli Starved of Phosphate against Fermentation Acids. J Bacteriol. 2007;189(6):2249–61.
Letoffe S, Delepelaire P, Wandersman C. The Housekeeping Dipeptide Permease Is the Escherichia coli Heme Transporter and Functions with Two Optional Peptide Binding Proteins. Proc Nat Acad Sci USA. 2006;103(34):12891–6.
Tangprasittipap A, Prasertsan P, Choorit W, Sasaki K. Biosynthesis of intracellular 5-aminolevulinic acid by a newly identified halotolerant Rhodobacter sphaeroides. Biotechnol Lett. 2007;29(5):773–8.
Nishikawa S, Watanabe K, Tanaka T, Miyachi N, Hotta Y, Murooka Y. Rhodobacter sphaeroides mutants which accumulate 5-aminolevulinic acid under aerobic and dark conditions. J Biosci Bioeng. 1999;87(6):798–804.
Neidle EL, Kaplan S. Expression of the Rhodobacter sphaeroides hemA and hemT genes, encoding two 5-aminolevulinic acid synthase isozymes. J Bacteriol. 1993;175(8):2292–303.
Van Der Werf MJ, Zeikus JG. 5-Aminolevulinate production by Escherichia coli containing the Rhodobacter sphaeroides hemA gene. Appl Environ Microb. 1996;62(10):3560–6.
Stephanopoulos G. Metabolic Fluxes and Metabolic Engineering. Metab Eng. 1999;1(1):1–11.
Ajikumar PK, Xiao W-H, Tyo KEJ, Wang Y, Simeon F, Leonard E, Mucha O, Phon TH, Pfeifer B, Stephanopoulos G. Isoprenoid pathway optimization for Taxol precursor overproduction in Escherichia coli. Science. 2010;330(6000):70.
Liao JC, Atsumi S, Hanai T. Non-fermentative pathways for synthesis of branched-chain higher alcohols as biofuels. Nature. 2008;451(7174):86–9.
Xia X-X, Qian Z-G, Ki CS, Park YH, Kaplan DL, Lee SY. Native-sized recombinant spider silk protein produced in metabolically engineered Escherichia coli results in a strong fiber. Proc Nat Acad Sci USA. 2010;107(32):14059–63.
Schauer S, Chaturvedi S, Randau L, Moser J, Kitabatake M, Lorenz S, Verkamp E, Schubert W-D, Nakayashiki T, Murai M, et al. Escherichia coli glutamyl-tRNA reductase. Trapping the thioester intermediate. J Biol Chem. 2002;277(50):48657–63.
Ilag LL, Jahn D, Eggertsson G, Soll D. The Escherichia coli hemL gene encodes glutamate 1-semialdehyde aminotransferase. J Bacteriol. 1991;173(11):3408–13.
Daßler T, Maier T, Winterhalter C, Böck A. Identification of a major facilitator protein from Escherichia coli involved in efflux of metabolites of the cysteine pathway. Mol Microbiol. 2000;36(5):1101–12.
Feng C, Pan M, Tang L. 5-Aminolevulinic acid level and dye-decolorizing peroxidase expression regulate heme synthesis in Escherichia coli. Biotechnol Lett. 2021;44(2):271–7.
Wang L, Elliott M, Elliott T. Conditional stability of the HemA protein (glutamyl-tRNA reductase) regulates heme biosynthesis in Salmonella typhimurium. J Bacteriol. 1999;181(4):1211–9.
Pyne ME, Moo-Young M, Chung DA, Chou CP. Coupling the CRISPR/Cas9 System with Lambda Red Recombineering Enables Simplified Chromosomal Gene Replacement in Escherichia coli. Appl Environ Microbiol. 2015;81(15):5103–14.
Burnham BF. δ-Aminolevulinic acid synthase (Rhodopseudomonas sphaeroides). Method Enzymol. 1970;17A:195–204.
Acknowledgements
Not applicable.
Funding
The work presented in this manuscript was supported by the Beijing Swine Innovation Team of Modern Agriculture Industry Technological System.
Author information
Authors and Affiliations
Contributions
QSY and YCC designed the study, wrote and edited the manuscript. YCC conducted all experiments reported herein. YCC, CX, YLJ, YMJ and HX performed experiments and analyzed experimental results. QSY and ZXF supervised the study. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Fig S1.
Map of plasmids which were constructed for CRISPR. A) Map of pCas, which harbored the temperature sensitive oriR101 with repA101ts, kanamycin resistance gene, the λ-Red operon encoding Gam, Bet, and Exo proteins under the control of arabinose-inducible promoter ParaB, S. pyogenes-derived cas9 driven by endogenous promoters and sgRNA guided to ori-p15a which is under the control of lac operator. B) Map of pTarget-gene, which harbored Chloramphenicol resistance, ori-p15a and sgRNA guided to E. coli BW25113-T7 targeted gene.
Additional file 2: Fig S2.
Construct of intermediate cloning vectors for preparing Donor DNA. (A) Fragment A cloned from BW25113-T7 was concatenated to pACYCD-Blank to assemble pACYCD-gene. (B) Reverse-PCR to constructed pACYCD-Donor. (C) The map of Donor DNA, which contains HRL and HRR.
Additional file 3: Fig S3.
Gene map for targeted Cas9-mediated gene Knock-In. (A) The knock-out site of hemF in BW25113-T7. (B) map of hemF knock-out in ideal condition and the location of PCR product (899 bp) for sequencing.
Additional file 4: Fig S4.
Schematic illustration of DSB induction and homologous recombination. After preparing competent cells, the pTarget-gene and Donor DNA which harbored homology arms (HRR and HRL) that targeted a chromosomal locus spanning the middle of targeted gene and the DSB site were electroporated into cells.
Additional file 5: Fig S5.
Detection of successful gene knock-outs of BW25113-T7.
Additional file 6: Fig S6.
Standard Plot for ALA measurement. x: the absorbance at 554 nm; y: ALA concentration of sample after diluted (mg/L).
Additional file 7: Table S1.
Growth rate of strain expressing various related genes in different medium.
Additional file 8: Table S2.
Primers for Plasmid constructions and Testing.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ye, C., Yang, Y., Chen, X. et al. Metabolic engineering of Escherichia coli BW25113 for the production of 5-Aminolevulinic Acid based on CRISPR/Cas9 mediated gene knockout and metabolic pathway modification. J Biol Eng 16, 26 (2022). https://doi.org/10.1186/s13036-022-00307-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13036-022-00307-7