
Abstract
Background
Although long non-coding RNA (lncRNA) NCK1-AS1 plays important roles in human cancer, its function in atherosclerosis (AS) remains unclear.
Method
The expression of NCK1-AS1 in AS blood samples was detected by qRT-PCR. Oxidized low-density lipoprotein (ox-LDL) was used to construct the AS cell model, and quantitative real-time polymerase chain reaction (qRT-PCR) assay was used to evaluate NCK1-AS1 level. Cell phenotypes including proliferation and apoptosis were assessed by Cell Counting Kit-8 (CCK-8) assay and flow cytometer, respectively. The malondialdehyde level was measured to evaluate oxidative stress. The expression of apoptosis-related proteins was evaluated by western blot. The expression of inflammatory cytokines (IL-1β, IL-6 and TNK-α) was measured by qRT-PCR and ELISA assays. The relationship among NCK1-AS1, miR-1197 and COX10 was determined by bioinformatic analysis and luciferase reporter assay.
Results
NCK1-AS1 was significantly upregulated in AS blood samples and ox-LDL stimulated vascular smooth muscle cells (VSMCs). Knockdown of NCK1-AS1 increased cell viability, reduced cell apoptosis and MDA level, and also inhibited the expression of inflammatory cytokines (IL-1β, IL-6 and TNK-α) in ox-LDL stimulated VSMCs. NCK1-AS1 could positively regulate COX10 expression by directly sponging miR-1197. Moreover, co-transfection of sh-NCK1-AS1 and miR-1197 inhibitor, or co-transfection of sh-NCK1-AS1 and pc-COX10 (COX10 overexpressing plasmid) obviously reduced cell viability, promoted cell apoptosis, and increased MDA level in VSMCs followed by ox-LDL treatment for 24 h compared to that in sh-NCK1-AS1 transfected VSMCs.
Conclusion
Our study revealed that knockdown of NCK1-AS1 attenuated the development of AS by regulating miR-1197/COX10 axis, suggesting that this lncRNA might be a potential therapeutic target for AS.
Similar content being viewed by others

Introduction
Atherosclerosis (AS) is a common leading cause of disability and death worldwide, which is closely involved in inflammation [1]. The major reason for this disease is the accumulation of low-density lipoprotein (LDL) in focal areas of medium and large arteries [2]. Recently, oxidized low-density lipoprotein (LDL) was used to construct the AS cell model in vitro [3]. Hence, more mechanisms involved in ox-LDL induced vascular smooth muscle cell (VSMC) injury is helpful to develop novel therapeutic targets for AS.
Long non-coding RNAs (lncRNAs) are a group of non-coding RNAs (> 200 nucleotide), and can participate in diverse biological processes [4]. Increasing evidences revealed that lncRNAs played essential roles in AS development through regulating the proliferation, apoptosis and inflammation of smooth muscle and endothelial cells [5, 6]. LncRNA NCK1 antisense RNA 1 (NCK1-AS1) is a newly identified lncRNA, and encodes a noncoding RNA with 1.4 kb in length [7]. Recently, NCK1-AS1 has been reported to play important roles in human cancers such as gastric cancer [8], lung squamous cell carcinoma [9], non-small cell lung cancer [10], urinary bladder cancer [11], ovarian cancer [12], and so on. However, the role of NCK1-AS1 in AS progression remains unclear.
MicroRNAs (miRNAs), is an another key non-coding RNA mediator that participated in biological pathways including AS [13]. MiRNAs have been identified to regulate the expression of downstream genes by binding to their 3′UTR and then inhibiting their expression [14]. Although the functions of miR-1197 in various biological processes including cell survival and apoptosis have been well studied [15], its mechanisms in ox-LDL-stimulated VSMCs remains unknown.
Cytochrome c oxidase assembly protein (COX10), a farnesyl transferase and is necessary for the maturation and stability of COX1, one of important cytokines with a pro-inflammatory function [16, 17]. It has been reported that high level of COX1 is positively correlated to the stronger inflammatory response during AS development [18]. In this study, we studied the role of NCK1-AS1/miR-1197/COX10 in cell proliferation, apoptosis and inflammatory response in ox-LDL stimulated VSMCs, and suggested that NCK1-AS1 might be a potential therapeutic target for AS.
Materials and methods
Clinical samples
Total 32 AS patients and 32 healthy volunteers were recruited at General Hospital of Ningxia Medical University between 2015.5 to 2020.9. Inclusion criteria: all patients were diagnosed with carotid atherosclerosis by angiography. Exclusion criteria: patients accompanied with cancers and autoimmune or inflammatory disease. The clinical features of participants were shown in Table 1. All participants have signed the written informed consent forms. The blood samples of all participants were collected and stored at − 80 °C immediately. This study was approved by the human Ethics Committee of General Hospital of Ningxia Medical University.
Cell culture and treatment
Human vascular smooth muscle cells (VSMCs) were purchased from American type culture collection (ATCC; Manassas, VA, USA). and cultured in Dulbecco’s Modified Eagles Medium (DMEM, Invitrogen, California, USA) containing 10% fetal calf serum (FBS, Gibco, USA) in a 37 °C incubator with 5% CO2. To mimic the atherosclerosis in condition in vitro, VSMCs were stimulated by 25, 50 and 100 μg/ml of ox-LDL (UnionBiol, Beijing, China) for 24 h, or 50 μg/ml ox-LDL for 6, 12, 24 and 48 h. To investigate the function of NCK1-AS1, transfected VSMCs were treated with 50 μg/ml ox-LDL for 24 h, and cells were used to perform subsequent experiments.
Cell transfection
The short hairpin RNA targeting NCK1-AS1 (sh-NCK1-AS1), negative control sh-NC, miR-1197 mimics (miR mimics), mimics NC, miR-1197 inhibitor (miR inhibitor) and inhibitor NC were purchased from GenePharma (Shanghai, China). sh-NCK1-AS1: GAAUGUCAUCCCAGCCGAAT, and sh-NC: UUCUCCGAACGUGUCACGUTT. To overexpress COX10, pcDNA3.1-based recombinant-overexpressing plasmid specific to COX10 was also constructed by GenePharma (Shanghai, China), and the empty vector as the negative control. 30 nM sh-RNA/mimics/inhibitor and 100 ng recombinant-overexpressing vector were transfected into VSMCs by Lipofectamine 2000 (Invitrogen). After 48 h of transfection, cells were used for subsequent experiments.
Cell proliferation
After transfection, VSMCs (2 × 105 cells/well) were stimulated by 50 μg/ml ox-LDL for 24 h. Cell viability was assessed by incubating with 10 μl of Cell Counting Kit-8 (CCK-8, Dojindo Molecular Technologies) reagent for 4 h. The absorbance at 450 nm was detected by a microplate reader. The assay was performed in triplicates of biological replicates.
Apoptosis analysis
After transfection, VSMCs (2 × 105 cells/well) were stimulated by 50 μg/ml ox-LDL for 24 h, harvested, washed in PBS and suspended to 100 μl 1× binding buffer, followed by the incubated with 5 μl of fluorescein isothiocyanate (FITC)-labeled Annexin-V and 5 μl of propidium iodide (PI) for 15 min in the dark according to the manufacturer’s instruction (Cat. No. 556419, KeyGen Biotech). The apoptotic cells were analyzed by flow cytometer (BD Biosciences, Cowley, UK). The assay was performed in triplicates of biological replicates.
Quantitative real time-polymerase chain reaction (qRT-PCR)
Total RNA was extracted by using TRIzol reagent. The cDNA was reverse transcribed from total RNAs by PrimeScript™ RT Master Mix (TaKaRa, Beijing, China) and the reverse transcription reactions were conducted on a LightCycler 480 (Roche Diagnostic, Sussex, UK) using 2 x SYBR Green qPCR Master Mix (Absource, Munich, Germany). The assay was performed in triplicates of biological replicates. The relative expression of target genes was calculated by the 2−ΔΔCt method, with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and U6 small nuclear RNA (U6) as the internal reference. The primers were listed in Table 2.
Western blot
Total protein was extracted by using RIPA lysis buffer (Sigma-Aldrich). Approximately equal amounts of protein were separated by 10% SDS-PAGE gels and then transferred into PVDF membranes (Roche, Basel, Switzerland). After blocking with 5% non-fat dry milk for 1 h, the membranes were incubated with primary antibodies against COX10 (1: 1000, ab228734), caspase 3 (1:1000, ab32351), cleaved-caspase 3 (1:1000, ab32042), Bax (1:1000, ab32503), Bcl-2 (1:1000, ab32124), and GAPDH (1:10000, ab181602) overnight at 4 °C. Then the membranes were incubated with HRP-conjugated secondary antibodies (1:10,000) for 2 h. All antibodies were purchased from Abcam. Protein bands were detected by enhanced chemiluminescence kit. The proteins were quantified by Quantity One software (Bio-Rad Laboratories, Inc., Hercules, CA, USA). The assay was performed in triplicates of biological replicates.
Target prediction
The binding sites among NCK1-AS1, miR-1197 and COX10 were predicted by Starbase (http://starbase.sysu.edu.cn/index.php) and TargetScan (http://www.targetscan.org/), respectively as previously described [19].
Luciferase reporter assay
The full fragment of NCK1-AS1 and 3′UTR of COX10 containing the wild type (WT) or mutant type (MUT) miR-1197 binding site were synthesized and cloned into pmirGLO vector (Promega, Mannheim, Germany) to generate recombinant luciferase vectors (NCK1-AS1 WT, NCK1-AS1 MUT, COX10 WT and COX10 MUT). The recombinant luciferase reporter vector was co-transfected singly with miR mimics or mimics NC into VSMCs using Lipofectamine 2000. Finally, a dual luciferase reporter assay system (Promega, USA) was used to detect the relative luciferase activities. The relative firefly luciferase activity was calculated by normalizing to renilla luciferase activity. The assay was performed in triplicates of biological replicates.
Malondialdehyde (MDA) content
To evaluate the oxidative stress, transfected VSMCs (2 × 105 cells/well) were stimulated by 50 μg/ml ox-LDL for 24 h. Subsequently, cells were harvested, lysed and the malondialdehyde (MDA) content was measured by using Lipid Peroxidation MDA Assay kit (cat. no. S0131, Beyotime) according to the manufacturer’s instructions. The oxidative stress was presented as the percentage of control cells without any treatments. The assay was performed in triplicates of biological replicates.
ELISA assay
To evaluate the effect of NCK1-AS1 on inflammatory response, transfected VSMCs (2 × 105 cells/well) were treated with 50 μg/ml ox-LDL for 24 h. Cells were lysed, and the production of inflammatory cytokines (IL-1β, IL-6 and TNK-α) were detected by using the commercial detection kits (Thermo Fisher Scientific, USA), respectively. The assay was performed in triplicates of biological replicates.
Statistical analysis
All data were presented as mean ± standard deviation (SD), and statistical analysis was performed using SPSS 18.0 software. Difference was tested by using student’s t test (two groups) and one-way analysis of variance (ANOVA) (multiple groups). P < 0.05 was considered statistically significant.
Results
NCK1-AS1 was highly expressed during AS development
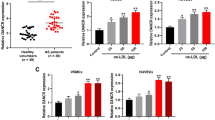
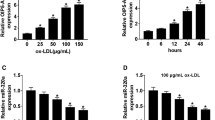
To investigate the role of NCK1-AS1 in AS, we firstly detected the expression of NCK1-AS1 and miR-1197 in the blood samples of AS patients and ox-LDL treated VSMCs. The results showed that NCK1-AS1 level increased 1.66 fold in the blood samples of AS patients compared to that in healthy volunteers (Fig. 1A), also upregulated 1.51 fold and 2.08 fold in 50 μg/ml ox-LDL treated VSMCs and 100 μg/ml ox-LDL VSMCs for 24 h compared to that in control cells, respectively (Fig. 1B). In addition, NCKA-AS1 level was also regulated 1.41 fold, 1.79 fold and 1.78 fold in 50 μg/ml ox-LDL treated VSMCs for 12 h, 24 h and 48 h, respectively (Fig. 1C). For miR-1197 expression, is was downregulated 2.13 fold in the blood samples of AS patients compared to that in healthy volunteers (Fig. 1D). Also, miR-1197 level was downregulated 1.42 fold and 2.13 fold in 50 μg/ml ox-LDL treated VSMCs and 100 μg/ml ox-LDL VSMCs for 24 h compared to that in control cells, respectively (Fig. 1E). MiR-1197 level was also downregulated 1.53 fold and 1.64 fold in 50 μg/ml ox-LDL treated VSMCs for 24 h and 48 h, respectively (Fig. 1F). Then 50 μg/ml ox-LDL for 24 h was used for the subsequent experiments.

The expression of NCK1-AS1 and miR-1197 during AS progression. A The expression of NCK1-AS1 in blood samples of AS patients was detected by qRT-PCR (n = 32). B and C VSMCs were stimulated by 25, 50 and 50 μg/ml ox-LDL for 24 h B or 50 μg/ml ox-LDL for 6, 12, 24 and 48 h C. The expression of NCK1-AS1 was detected by qRT-PCR. D The expression of miR-1197 in blood samples of AS patients was detected by qRT-PCR (n = 32). (E and F) VSMCs were stimulated by 25, 50 and 50 μg/ml ox-LDL for 24 h E or 50 μg/ml ox-LDL for 6, 12, 24 and 48 h F. The expression of miR-1197 was detected by qRT-PCR. * p < 0.05, ** p < 0.01 and *** p < 0.001
Knockdown of NCK1-AS1 increased cell viability, reduced oxidative stress and promoted cell apoptosis in ox-LDL stimulated VSMCs
To evaluate the role of NCK1-AS1, sh-NCL-AS1 was transfected into VSMCs to knockdown it. The results of qRT-PCR showed that sh-NCK1-AS1 reduced the level of NCK1-AS1 (1.62 fold) in ox-LDL treated VSMCs compared with sh-NC group and NC group (Fig. 2A). ox-LDL treatment decreased cell viability (2.16 fold), increased MDA content (1.67 fold) and promoted cell apoptosis (4.15 fold) of VSMCs compared to that in control group, and knockdown of NCK1-AS1 increased cell viability (1.45 fold), reduced MDA content (1.61 fold) and cell apoptosis (1.42 fold) in ox-LDL stimulated VSMCs compared with sh-NC group and NC group (Fig. 2B-D). In addition, ox-LDL increased the protein expression of apoptosis-related marker cleaved caspase 3 and pro-apoptotic factor Bax, while reduced anti-apoptotic factor Bcl-2 level in VSMCs compared with control group, and knockdown of NCK1-AS1 reduced the expression of cleaved caspase 3 and Bax, while increased Bcl-2 expression compared with sh-NC and NC group in ox-LDL treated VSMCs (Fig. 2E). The results suggested that knockdown of NCK1-AS1 inhibited AS development in vitro.

Knockdown of NCK1-AS1 attenuated ox-LDL induced cell injury in VSMCs. VSMCs were transfected with sh-NCK1-AS1 or sh-NC, and then treated with 50 μg/ml ox-LDL for 24 h. A The expression of NCK1-AS1 was detected by qRT-PCR. B CCK-8 assay. C MDA content was measured by specific detection kit. D Flow cytometry for cell apoptosis. E The expression of cleaved caspase3, Bax and Bcl-2 was measured by western blot. Control group is that VSMCs without any treatment, NC means that VSMCs received ox-LDL treatment. * p < 0.05, ** p < 0.01 and *** p < 0.001
NCK1-AS1 regulated COX10 by sponging miR-1197
Due to the negative correlation between the level of NCK1-AS1 and miR-1197 in the blood samples of AS patients (Fig. 3A), we thought that NCK1-AS1 might serve as the sponge of miR-1197. Starbase prediction showed that there was a putative binding site between NCK1-AS1 and miR-1197, and a mutant sequence of NCK1-AS1 against miR-1197 binding site was constructed (Fig. 3B). Overexpression of miR-1197 increased the expression of miR-1197 (3.47 fold) compared with mimics NC group, and miR-1197 inhibitor reduced miR-1197 expression (2.24 fold) compared with inhibitor NC group (Fig. 3C). Luciferase reporter assay indicated that miR-1197 mimics reduced the luciferase activity of NCK1-AS1 WT (1.64 fold) compared with miR-NC group (Fig. 3D). Meanwhile, the results of Targetscan prediction suggested that COX10 was a target of miR-1197, and a mutant sequence of 3′UTR COX10 against miR-1197 binding site was constructed (Fig. 3E). Luciferase reporter assay also confirmed the binding relationship between miR-1197 and 3′UTR of COX10 (1.58 fold) (Fig. 3F). Moreover, ox-LDL caused a significant downregulation of miR-1197 compared with control group (2.17 fold), and knockdown of NCK1-AS1 increased miR-1197 expression (1.46 fold) compared with sh-NC in ox-LDL treated VSMCs (Fig. 3G). In addition, COX10 was notably upregulated (2.34 fold) in ox-LDL treated VSMCs, and knockdown of NCK-1AS1 reduced the expression of COX10 (1.33 fold) compared with sh-NC in ox-LDL treated VSMCs (Fig. 3H and I). These results indicated that NCK1-AS1 could regulate COX10 by sponging miR-1197 in ox-LDL treated VSMCs.

NCK1-AS1 regulated COX10 by sponging miR-1197. A Correlation between NCK1-AS1 and miR-135a-5p level in AS blood samples. B Schematic model of miR-1197 binding site within NCK1-AS1 predicted by Starbase and the construction of mutant miR-1197 binding site. C VSMCs were transfected with miR-1197 mimics or inhibitor, and qRT-PCR for miR-1197 level. D VSMCs were transfected with miR-1197 mimics or mimics NC, and the relative luciferase activity. E Schematic model of miR-1197 binding site within 3′UTR COX10 predicted by Targetscan and the construction of mutant miR-1197 binding site. F VSMCs were transfected with miR-1197 mimics or mimics NC, and the relative luciferase activity. G - I VSMCs were transfected with sh-NCK1-AS1, and then treated with 50 μg/ml ox-LDL for 24 h. QRT-PCR for miR-1197 level G, COX10 level H, and western blot for COX10 level I. * p < 0.05, ** p < 0.01, and *** p < 0.001
Knockdown of NCK1-AS1 attenuated ox-LDL-induced VSMCs injury through regulating miR-1197/COX10 axis
To determine the function of NCK1-AS1/miR-1197/COX10 axis in AS, the rescue experiments were performed. Co-transfection of miR-1197 inhibitor and sh-NCK1-AS1 attenuated the elevation of cell viability (1.25 fold) and the reduction of cell apoptosis (1.39 fold) in sh-NCK1-AS1 transfected VSMCs followed by ox-LDL treatment for 24 h (Fig. 4A and B). Meanwhile, co-transfection of pc-COX10 (COX10 overexpressing plasmid) and sh-NCK1-AS1 attenuated the elevation of cell viability (1.67 fold) and the reduction of cell apoptosis (1.34 fold) in sh-NCK1-AS1 transfected VSMCs followed by ox-LDL treatment for 24 h (Fig. 4C and D). In addition, both co-transfection of sh-NCK1-AS1 and miR-1197 inhibitor, as well as co-transfection of sh-NCK1-AS1 and pc-COX10 notably increased MDA content in sh-NCK1-AS1 transfected VSMCs followed by ox-LDL treatment for 24 h (Fig. 4E and F). These findings revealed that knockdown of NCK1-AS1 attenuated ox-LDL-induced VSMCs injury through regulating miR-1197/COX10 axis.

Knockdown of NCK1-AS1 attenuated ox-LDL-induced VSMCs injury through regulating miR-1197/COX10 axis. A and B CCK-8 assay for cell viability A and flow cytometry for cell apoptosis B after co-transfection of sh-NCK1-AS1 and miR-1197 inhibitor in VSMCs followed by 50 μg/ml ox-LDL for 24 h. C and D CCK-8 assay for cell viability C and flow cytometry for cell apoptosis D after co-transfection of sh-NCK1-AS1 and pc-COX10 (COX10 overexpressing plasmid) in VSMCs followed by 50 μg/ml ox-LDL for 24 h. E and F VSMCs were co-transfected with sh-NCK1-AS1 and miR-1197 inhibitor E, or sh-NCK1-AS1 and pc-COX10 F, and then treated by 50 μg/ml ox-LDL for 24 h. The MDA content was measured by specific detection kit. * p < 0.05, ** p < 0.01, and *** p < 0.001
Knockdown of NCK1-AS1 attenuated inflammatory response in ox-LDL-induced VSMCs
Finally, we evaluated the impacts of NCK1-AS1 on inflammatory response in AS, and found that knockdown of NCK1-AS1 reduced the expression of inflammatory cytokines (IL-1β, 2.04 fold; IL-6, 1.19 fold and TNK-α, 1.17 fold) compared with sh-NC in ox-LDL stimulated VSMCs (Fig. 5A). In addition, knockdown of NCK1-AS1 reduced the production of inflammatory cytokines (IL-1β, 1.53 fold; IL-6, 1.71 fold and TNK-α, 1.62 fold) compared with sh-NC in ox-LDL stimulated VSMCs (Fig. 5B). These data further confirmed the inhibitory action of NCK1-AS1 knockdown on inflammatory response in AS progression.

Knockdown of NCK1-AS1 attenuated inflammatory response in ox-LDL-induced VSMCs. VSMCs were transfected with sh-NCK1-AS1 or sh-NC, and then treated with 50 μg/ml ox-LDL for 24 h. The expression of inflammatory cytokines (IL-1β, IL-6 and TNK-α) was evaluated by qRT-PCR A and ELISA assay B. *** p < 0.001
Discussion
In this study, we investigated the regulatory mechanism of NCK1-AS1/miR-1197/COX10 axis in the progression of AS, and our results constructed a AS cell model using ox-LDL to stimulate VSMCs, and found that ox-LDL notably reduced cell viability, increased cell apoptosis and inflammatory response, which was consistent with previous findings [20, 21]. These data confirmed our successful AS cell model, which was subsequently used to investigate the role of NCK1-AS1 during AS development.
Interestingly, we found that NCK1-AS1 was significantly upregulated in AS blood samples and ox-LDL treated VSMCs. Moreover, knockdown of NCK1-AS1 obviously increased cell viability, reduced cell apoptosis and inflammatory response in ox-LDL treated VSMCs. Previous studies have reported the essential functions of NCK1-AS1 in other human diseases. For example, inhibition of NCK1-AS1 prevents the migratory and invasive capacities in nasopharyngeal carcinoma cells [22]. Knockdown of NCK1-AS1 inhibits cell proliferative rate and migratory ability via the suppression of miR-134 in cervical cancer [23]. These reports indicated that NCK1-AS1 played important functions in cell survival and apoptosis. In this study, we revealed the essential role of NCK1-AS1 in cell proliferation, apoptosis and inflammatory response in ox-LDL stimulated VSMCs, suggesting that NCK1-AS1 might be a novel diagnostic and therapeutic biomarker for AS.
More previous studies revealed that lncRNAs can function as the sponge of miRNAs to participate in the various pathogenic processes [24]. For instance, SNHG7–003 suppresses cell proliferation and promoted apoptosis of VSMCs by targeting miR-1306-5p [25]. Kcnq1ot1 exacerbates the lipid accumulation and promotes AS progression through functioning as the competing endogenous RNAs (ceRNAs) of miR-452-3p [26]. Here, we identified that miR-1197 was negatively regulated by NCK1-AS1, and luciferase reporter assay and function assays confirmed this interaction relationship. Moreover, downregulation of miR-1197 obviously reversed the protective impacts of NCK1-AS1 knockdown in ox-LDL stimulated VSMCs injury. However, there were other previously identified miRNA targeted by NCK1-AS1 including miR-137 [27], miR-22-3p [8], miR-22-3p [28], and more attentions should be focused on these potential NCK1-AS1-downstream miRNAs regulatory axis during AS development.
In addition, our results also identified that COX10 was a direct target of miR-1197, and miR-1197 could directly bind to its 3′UTR and then inhibit COX10 expression. It has been reported that miRNAs can regulate the progression of AS by targeting downstream target genes [29]. Although inflammatory response always occurs during AS progression, the function of COX10 in AS remains unclear. In this study, we found that knockdown of NCK1-AS1 significantly reduced the expression of COX10 in ox-LDL treated VSMCs, and overexpression of COX10 obviously eliminated the protective roles of NCK1-AS1 knockdown on cell proliferation, apoptosis and inflammatory response. These results further determined that COX10 participated in the regulatory function of NCK1-AS1/miR-1197 in AS development. Similarly, miRNAs can regulate a series of downstream target genes in human diseases. For example, miR-1197 controls the progression of non-small-cell lung carcinoma through regulating MADD [30], and also promotes cell proliferation by upregulating HOXC11 [31]. Hence, there might be other potential target genes that mediated the function of miR-1197 in AS development, and this conjecture needed to be further investigated in the subsequent experiments.
In conclusion, our study demonstrated that knockdown of NCK1-AS1 could efficiently attenuate ox-LDL induced VSMCs injury through regulating miR-1197 and COX10, providing that NCK1-AS1 might be a potential therapeutic target for AS.
Availability of data and materials
The data that support the findings of this study are available on request from the corresponding author.
References
Roth GA, Mensah GA, Johnson CO, Addolorato G, Ammirati E, Baddour LM, et al. Global burden of cardiovascular diseases and risk factors, 1990-2019: update from the GBD 2019 study. J Am Coll Cardiol. 2020;76(25):2982–3021.
Soehnlein O. Libby P. Targeting inflammation in atherosclerosis - from experimental insights to the clinic. 2021:1–22.
Tiliwaldi H, Tursun A, Tohti A, Mamatzunun M, Wu Z. Circ_0000345 protects endothelial cells from oxidized low-density lipoprotein-induced injury by miR-129-5p/ten-eleven translocation Axis. J Cardiovasc Pharmacol. 2021;77(5):603–13.
Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol Cell. 2011;43(6):904–14.
Zhou T, Ding JW, Wang XA, Zheng XX. Long noncoding RNAs and atherosclerosis. Atherosclerosis. 2016;248:51–61.
Li H, Zhu H, Ge J. Long Noncoding RNA: Recent updates in atherosclerosis. Int J Biol Sci 2016;12(7):898–910.
Li H, Jia Y, Cheng J, Liu G, Song F. LncRNA NCK1-AS1 promotes proliferation and induces cell cycle progression by crosstalk NCK1-AS1/miR-6857/CDK1 pathway. Cell Death Dis. 2018;9(2):198.
Guan B, Ma J, Yang Z, Yu F, Yao J. LncRNA NCK1-AS1 exerts oncogenic property in gastric cancer by targeting the miR-22-3p/BCL9 axis to activate the Wnt/β-catenin signaling; 2021.
Zhou X, Bao W, Zhang D, Yang Y, Du X, Qiu G. NCK1-AS1 promotes the progression of lung squamous cell carcinoma through transcriptionally upregulating NCK1 via interacting with MYC. Cancer Biol Ther. 2021;22(3):196–203.
Zha LF, Zhang LD, Pan HM, Ma HD. Upregulation of lncRNA NCK1-AS1 predicts poor prognosis and contributes to non-small cell lung cancer proliferation by regulating CDK1. Eur Rev Med Pharmacol Sci. 2021;25(3):1351–7.
Qiao Z, Dai H, Zhang Y, Li Q, Zhao M, Yue T. LncRNA NCK1-AS1 promotes Cancer cell proliferation and increase cell Stemness in urinary bladder Cancer patients by downregulating miR-143. Cancer Manag Res. 2020;12:1661–8.
Chang H, Li B, Zhang X, Meng X. NCK1-AS1 promotes NCK1 expression to facilitate tumorigenesis and chemo-resistance in ovarian cancer. Biochem Biophys Res Commun. 2020;522(2):292–9.
Madrigal-Matute J, Rotllan N, Aranda JF, Fernández-Hernando C. MicroRNAs and atherosclerosis. Curr Atheroscler Rep. 2013;15(5):322.
Lu TX, Rothenberg ME. MicroRNA. J Allergy Clin Immunol. 2018;141(4):1202–7.
Zhang GM, An SY, El-Samahy MA, Zhang YL, Wan YJ, Wang ZY, et al. Suppression of miR-1197-3p attenuates H (2) O (2)-induced apoptosis of goat luteinized granulosa cells via targeting PPARGC1A. Theriogenology. 2019;132:72–82.
Fukui H, Diaz F, Garcia S, Moraes CT. Cytochrome c oxidase deficiency in neurons decreases both oxidative stress and amyloid formation in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2007;104(35):14163–8.
Mazhar D, Gillmore R, Waxman J. COX and cancer. QJM. 2005;98(10):711–8.
El-Sheakh AR, Ghoneim HA, Suddek GM, el SM A. Antioxidant and anti-inflammatory effects of flavocoxid in high-cholesterol-fed rabbits. Naunyn Schmiedebergs Arch Pharmacol. 2015;388(12):1333–44.
Jiang J, Bi Y, Liu XP, Yu D, Yan X. Yao J, et al. To construct a ceRNA regulatory network as prognostic biomarkers for bladder cancer. 2020;24(9):5375–86.
Hong D, Bai YP, Gao HC, Wang X, Li LF, Zhang GG, et al. Ox-LDL induces endothelial cell apoptosis via the LOX-1-dependent endoplasmic reticulum stress pathway. Atherosclerosis. 2014;235(2):310–7.
Zhang YL, Cao YJ, You SJ, Li RX, Liu HH, Liu CF. Protective effects of autophagy against oxidized LDL-induced injury in endothelial cells. Zhonghua Yi Xue Za Zhi. 2010;90(39):2792–6.
Hu H, Li H, Feng X. Downregulation of lncRNA NCK1-AS1 inhibits Cancer cell migration and invasion in nasopharyngeal carcinoma by upregulating miR-135a. Cancer Manag Res. 2019;11:10531–7.
Huang L, Gan X, He L, Wang L, Yu J. Silencing of long non-coding RNA NCK1-AS1 inhibits cell proliferation and migration via inhibition of microRNA-134 in cervical cancer. Exp Ther Med. 2019;18(3):2314–22.
Cheng DL, Xiang YY, Ji LJ, Lu XJ. Competing endogenous RNA interplay in cancer: mechanism, methodology, and perspectives. Tumour Biol J Int Soc Oncodev Biol Med. 2015;36(2):479–88.
Zheng J, Tan Q, Chen H, Chen K, Wang H, Chen Z, et al. lncRNA-SNHG7-003 inhibits the proliferation, migration and invasion of vascular smooth muscle cells by targeting the miR-1306-5p/SIRT7 signaling pathway. Int J Mol Med. 2021;47(2):741–50.
Yu XH, Deng WY, Chen JJ, Xu XD. LncRNA kcnq1ot1 promotes lipid accumulation and accelerates atherosclerosis via functioning as a ceRNA through the miR-452-3p/HDAC3/ABCA1 axis. Cell Death Dis. 2020;11(12):1043.
Chen M, Cheng Y, Yuan Z, Wang F, Yang L, Zhao H. NCK1-AS1 increases drug resistance of glioma cells to Temozolomide by modulating miR-137/TRIM24. Cancer Biother Radiopharm. 2020;35(2):101–8.
Wang B, Wang K, Jin T, Xu Q, He Y, Cui B, et al. NCK1-AS1 enhances glioma cell proliferation, radioresistance and chemoresistance via miR-22-3p/IGF1R ceRNA pathway. Biomed Pharmacother. 2020;129:110395.
Fernández-Tussy P, Ruz-Maldonado I. Fernández-Hernando C. MicroRNAs Circ RNAs Lipoprotein Metab. 2021;23(7):33.
Wang JY, Zhang F, Hong L, Wei SJ. CircRNA_0000429 regulates development of NSCLC by acting as a sponge of miR-1197 to control MADD. Cancer Manag Res. 2021;13:861–70.
Sun B, Hua J, Cui H, Liu H, Zhang K, Zhou H. MicroRNA-1197 downregulation inhibits proliferation and migration in human non- small cell lung cancer cells by upregulating HOXC11. Biomed Pharmacother Biomed Pharmacotherapie. 2019:117–109041.
Acknowledgements
Not Applicable.
Funding
Not Applicable.
Author information
Authors and Affiliations
Contributions
Bin Zhang, Jinfang Liu: study concepts, literature research, clinical studies, data analysis, experimental studies, manuscript writing and review; Juncheng Wang: study design, literature research, experimental studies and manuscript editing; Lei Du: definition of intellectual content, clinical studies, data acquisition and statistical analysis; Lufei Shao, Yourui Zou: data acquisition, manuscript preparation and data analysis; Haibo Liu: data acquisition and statistical analysis. All authors have read and approve the submission of the manuscript.
Corresponding author
Ethics declarations
Ethical approval and consent to participate
All patients signed the written informed consent. All procedures were approved by General Hospital of Ningxia Medical University Ethics Committee. Procedures operated in this research were completed in keeping with the standards set out in the Announcement of Helsinki and laboratory guidelines of research in China.
Consent for Publication
Not applicable.
Competing interests
All other authors have no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, B., Wang, J., Du, L. et al. Knockdown of NCK1-AS1 inhibits the development of atherosclerosis by targeting miR-1197/COX10 axis. J Biol Eng 16, 2 (2022). https://doi.org/10.1186/s13036-021-00281-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13036-021-00281-6