
Abstract
Owing to the thermal instability and low affinity of BlaR-CTD to some β-lactams, the receptor assay based on BlaR-CTD is limited in the detection of abundant variety of drugs and the result is often unstable. In this study, the three-dimensional structure of BlaR-CTD from Bacillus licheniformis ATCC14580 was constructed by homologous modeling based on the crystal structure of BlaR-CTD from B. licheniformis 749/I, and the binding sites of this protein to 40 β-lactams were also obtained by molecular docking. To improve the stability and affinity of the protein, 23 mutant proteins were designed based on docking and homologous alignment results as well as by inserting disulfide bond and building the salt bridge. The mutation was rationality evaluated by SIFT and PloyPhen2 software. The heterologous expressed and purified mutant proteins were then subjected to the activity and stability assay. It was shown that among all mutant proteins, I188K/S19C/G24C, A138E/R50C/Q147C and S190Y/E183C/I188K respectively exhibited a higher affinity to 33, 22 and 21 β-lactams than the wild-type protein, while I188K/S19C/G24C exhibited the best stability. This may due to that the conformation of the active site in mutant protein I188K/S19C/G24C changed, and the random coli in the surface of protein activity increased. Our study suggests a possible structure-function relationship on the stability and affinity of BlaR-CTD, which provides new insights into protein rational design study and lays a solid foundation for establishing the receptor-based screening assay for the detection of β-lactam residues.
Similar content being viewed by others


Introduction
The penicillin binding protein (PBP) BlaR is a signal transduction membrane protein which induces the synthesis of β-lactamase. The C-terminal domain of BlaR (BlaR-CTD) protein is located in the extracellular region which acts as a drug binding site [19]. BlaR-CTD, a penicillin-binding protein. BlaR-CTD is firstly the sensor domain of a penicillin receptor that is acylated by penicillin. This protein can identify and bind to a variety of β-lactams [8, 12]. In the active site of the protein, STYK, serine was a key amino acid (AA) that can participate in the binding of β-lactams [31].
BlaR-CTD protein has been used to detect the β-lactam residues in the receptor-based screening assay, such as a biological fluid method using colloidal gold labeled receptor protein [7] and an receptor-based enzyme linked immune-sorbent assay developed by our lab in which BlaR-CTD from B. licheniformis ATCC14580 was immobilized on the plate [25]. The limits of detection for 11 β-lactams were lower than the maximum residue limits regulated by European Union. However, the study showed that BlaR-CTD protein had a poor affinity for some β-lactams, e.g. cephalexin and cefadroxil. In addition, the thermal stability of the protein was poor that the protein activity only remained 70% at − 20 °C after 1 month, and lost fast at 4 °C [25].
Presently, site-directed mutagenesis was an effective means for the structural modification of protein [14], such as, improving the enzymatic activity and stability [9, 15]. Contreras-Martel et al. obtained a wild-type and the mutant PBP2b from Streptococcus pneumoniae by heterologous expression in E. coli, and the structure analysis showed that the mutation of AAs around the active site could affect the distribution of charge and polarity, preventing the entry of substrate into the active site, thus results in a bacterial resistance against β-lactam drugs [4]. Powell et al. mutated the AAs around the active site of the PBP2 from Neisseria gonorrhoeae, and the results showed that the acylation efficiency for penicillin G was reduced [26]. Therefore, the mutation site should avoid the active site and active site of the protein when the structure of BlaR-CTD protein was reconstructed. Molecular docking can quickly simulates the binding mode between a protein and a ligand, and analyze the key AAs involved in the binding, such as, AAs forming a hydrogen bond(s) with the drug [33]. Moreover, disulfide bond played an important role in the structural stability of proteins. It was demonstrated that the protein expression and enzyme activity of β-mannanase from Aspergillus niger BK01 (ManBK) were reduced when the conserved C171-C174 disulfide bond was missing, indicating that disulfide bond has a crucial in the stability of [15]. Through the site-directed mutagenesis of PBP3 from Streptococcus pneumoniae R6, the thermal stability of the mutant A353C/E393C was increased while the affinity of the mutant did not change [32].
In this study, 23 mutant proteins of BlaR-CTD from Bacillus licheniformis ATCC14580 were designed by homologous modeling, molecular docking, and prediction of mutation site in software, disulfide bond inserting and salt bridge building. The mutant proteins were obtained by site-directed mutagenesis and heterologous expression in E. coli. Based on the results of activity and stability tests, I188K/S19C/G24C showed equal or higher affinity to 33 β-lactams and better stability as compared to the wild-type protein. This study for the first time modified BlaR-CTD protein by rational design and site-directed mutagenesis, and the mutant protein I188K/S19C/G24C exhibited not only the improvement in stability but also higher affinity than the wild-type. Current study offers a powerful base for establishing the screening method for the detection of β-lactam residues.
Materials and methods
Chemicals
Penicillin G, nafcillin, dicloxacillin, ceftiofur, cefquinome, cefuroxim, carbenicillin and flucloxacillin were bought from Dr. Ehrenstorfer (Deisenhofen, Germany). Ampicillin, amoxicillin, oxacillin, azlocillin, penicillin V, cloxacillin, cefalotin, cefoperazone, cefazolin, cefalexin, ceftriaxone, cefadroxil, cefepime, cefradin, latamoxef, cefixim, ceftazidim, cefoxitin, sulbenicillin, ticarcillin and aztreonam were supplied by the National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China). Piperacillin and cefalonium were from Toronto Research Chemicals (Toronto, Canada). Cefapirin was from Witega (Berlin, Germany). Benzylpenicillin was from European Directorate for the Quality of Medicines & HealthCare (Strasbourg, France). Kanamycin was obtained from TianYuan (Wuhan, China). 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC) and N-hydroxysuccinimide (NHS) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Bovine serum albumin (BSA) was obtained from Bovogen (East Keilor, VIC, Australia). β-isopropyl-d-thiogalactopyranoside (IPTG) and horseradish peroxidase (HRP) were purchased from Solarbio (Beijing, China). 3,3′,5,5′-tetramethylbenzidine (TMB) and Ni2+ Sepharose Fastflow were from Thermo Fisher Scientific (Waltham, MA, USA). Luria-Bertani (LB) broth and agar were purchased form Qingdao Hope Bio-Technology (Qingdao, China). All other reagents were purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China) and were all of analytical grade.
Bacteria and plasmid
E. coli BL21 (DE3) and E. coli DH5α were purchased from TransGen Biotech (Wuhan, China). Recombinant plasmid pET-28a(+)-BlaR-CTD was obtained from the National Reference Laboratory of Veterinary Drug Residues (HZAU), Wuhan, China [25].
Homologous modeling and molecular docking
By sequence alignment using NCBI-BLAST (Basic Local Alignment Search Tool), the BlaR-CTD protein from B. licheniformis 749/I was chosen as a template to build the three dimensional (3D) structure model of BlaR-CTD protein from B. licheniformis ATCC14580 (the AA sequence identity is 91%). Using homologous modelling motif of Sybyl-X2.0 software (Tripos, USA), the 3D structure of BlaR-CTD protein was obtained based on the crystal structure of BlaR-CTD protein from B. licheniformis 749/I (PDB ID: 1NRF) [16]. Sybyl-X2.0 was also used for molecular docking between BlaR-CTD protein and 40 β-lactams, and the key AAs involved in the formation of hydrogen bonds between the protein and each drug were obtained. The AAs within 5 Å of the active pocket were considered as AAs that may be associated with affinity.
The 3D structure of mutant protein was also obtained by homologous modelling and the key AAs involved in the formation of hydrogen bonds between mutant protein and each β-lactam were obtained by molecular docking using Sybyl-X2.0 software, as described above. Structural model was visualized by PYMOL (www.pymol.org).
Design of mutational sites
SIFT (https://sift.bii.a-star.edu.sg) was used to predict whether an AA substitution effects on protein function based on the degree of conservation of AA residues in a sequence alignments derived from closely related sequences [21]. PloyPhen2 (http://genetics.bwh.harvard.edu/pph2/) was used to predict the possible impact of an AA substitution on the structure and function of a protein based on a number of features comprising the sequence, phylogenetic and structural information characterizing the substitution [1]. The higher score of SIFT and the lower score of PloyPhen2 indicated that the mutation was more reasonable and rational [23]. To design the mutational sites, first, the AAs formed hydrogen bond with β-lactam molecules were removed from the list of AAs within 5 Å of the active pocket from docking results. Then, the mutations of the remaining AAs were predicted by SIFT software, and the mutational sites with score of 1.0 were screened out. These sites were taken into the PloyPhen2 analysis to select the mutational sites with low scores. Through the sequence alignment of active sites, the AAs involved in a protein binding with drugs also can be applied to mutation.
In addition, basic AAs are easy to form salt bridges with acidic AAs, which can improve the protein stability [5, 20]. Therefore, site-directed mutagenesis can be carried out by analyzing the physicochemical properties of AAs. As BlaR-CTD protein did not contain free cysteine and disulfide bonds, disulfide bond were introduced in the flexible region of the protein to improve the stability by using the software Disulfide by Design 2 (http://cptweb.cpt.wayne.edu/DbD2/). The sites where disulfide bonds could be inserted were based on the predicted energy and B-Factors [11]. The ΣB-factor, which is related to the protein stability, indicates the smearing of atomic electron densities regarding to their equilibrium positions on account of thermal motion and positional disorder [24]. The mutational sites were selected according to the principle of small bonding energy and large ΣB-factor. χ3 torsion angle is defined by rotation of the two Cβ atoms about the Sγ-Sγ bond [6].
Construction of mutants
The site-directed mutagenesis was used for the structural modification of BlaR-CTD. Mutant plasmids were obtained by one step overlap extension polymerase chain reaction [29], using a recombinant plasmid pET-28a(+)-BlaR-CTD as the template and the primers for the mutation were shown in Table 1. The obtained 0.7-kb DNA fragments were digested with restriction enzymes of Dpn I, and the mutant plasmids were transformed into E.coli DH5α. After confirming by sequencing analysis, the mutant plasmids were transformed into E.coli BL21 (DE3) for the recombinant expression.
Preparation of mutant proteins
E. coli BL21 (DE3) harboring mutant plasmid were cultured overnight in 1 L of LB broth containing 50 μg·mL− 1 kanamycin at 37 °C in shaker. The culture was diluted 100-fold into a fresh LB broth containing 50 μg·mL− 1 kanamycin and incubated with vigorous shaking at 37 °C until the OD600 reached 0.6. Then 1 mM IPTG was added and the culture was incubated at 18 °C for 12 h. Bacteria were collected by centrifugation at 10,000 rpm for 10 min, and were washed 3 times with ice cold phosphate-buffered saline (PBS) (8 g NaCl, 2.9 g Na2HPO4·12H2O, 0.2 g KCl, 0.2 g KH2PO4 per liter, pH 7.4). Then, 50 mL binding buffer (7.6 g Na3PO4, 29.22 g NaCl, 0.68 g imidazole per liter, pH 7.4) was added into the precipitate, and ultrasonic fragmentation was carried out after resuspension of the precipitate. The suspension was centrifuged at 10,000 rpm for 40 min at 4 °C, and the soluble fraction was loaded onto a 2 mL Ni2+-charged chelating sepharose resin column. After washing by different concentrations of imidazole eluent, the protein was eluted at imidazole concentration of 60, 100 and 200 mM. The purified mutant proteins were dialyzed against PBS, and were confirmed by SDS-PAGE with Coomassie Brilliant Blue staining and western blot. The protein concentration was determined by the method of Bradford [3].
Activity assay
The activity assay was based on directly competitive inhibition of binding of horseradish peroxidase-labeled ampicillin (HRP-AMP) to the immobilized BlaR-CTD or its mutants by β-lactams. HRP-AMP was prepared according to the previous study [25]. PBS was used as the coating buffer to dilute the wild-type and mutant proteins, and 100 μL protein (1 μg·mL− 1) were coated on the microtiter plates overnight at 4 °C. The plate was washed 3 time with 250 μL of PBST (PBS containing 0.5% Tween-20), followed by blocking with 1% (w/v) BSA in PBS for 12 h at 4 °C. After washing 3 times with PBST, 50 μl of the standard drug (100 μg·L− 1) and 50 μl of HRP-AMP solution were added successively to each well and incubated at 30 °C for 30 min. After washing 3 times with PBST, 100 μl of enzyme substrate TMB was added and incubated at 37 °C for 15 min, and the reaction was stopped by adding 50 μl of 2 M H2SO4. Then, the absorbance values at 450 nm were measured with microplate reader (Tecan, Seestrasse, Schweiz). The inhibition rate was used to indicate the recognition ability of protein to drugs. The smaller the inhibitions rate the stronger recognition ability.
Inhibition rate (%) = OD value(drug)/OD value(no drug) × 100%
Statistical analyses
Descriptive statistical parameters such as average (AVG) and standard deviation (SD) were calculated. Statistical analysis of the data was performed using Microsoft Excel 2003.
Results
Homologous model of BlaR-CTD and docking with β-lactams
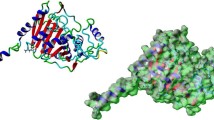
The 3D structure of BlaR-CTD protein from B. licheniformis ATCC14580 was shown in Fig. 1a. It was mainly composed of two domains. One was the α/β domain, which was covered by two α helices (α1 in N-terminal and α10 in C-terminal) on one side of a seven parallel β sheets (β1-β7), with the other side covered by α3 and α8 helix. The other domain was mainly composed of four helices (α5, α6, α7 and α9) encircling the hydrophobic α4 helix. The active pocket of the BlaR-CTD was composed of S(55)TYK(58), S(103)AT(105) and K(192)TG(194) [19].

3D structure of BlaR-CTD protein (a) and interaction with cefquinome (b). a The 3D structure of BlaR-CTD was represented as a cartoon, with α-helices colored in cyan, β-strands in magenta, and loops in salmon. The active sites of Ser55, Ser103, Thr105 and Thr195 were colored in yellow. b The amino acid residues involved in the interaction of BlaR-CTD with cefquinome. The hydrogen bonds were represented as black dotted lines. The cefquinome molecule was colored in gray. The AAs of the active site were colored in yellow
The docking result of cefquinome with BlaR-CTD was shown in Fig. 1b. Nine hydrogen bonds were formed between the protein and cefquinome in the active pocket. Among the hydrogen bond forming AAs, Ser55, Ser103, Thr105 and Thr193 were reported in the previous literature [19], in which one hydrogen bond was respectively formed with Ser55, Ser103 and Thr105, and three hydrogen bonds were formed with Thr193. In addition, two hydrogen bonds between cefquinome and Tyr87, and one hydrogen bond between the drug and Thr195 were also formed.
Molecular docking between BlaR-CTD protein and other 39 β-lactams was also carried out by Sybyl-X2.0, and 65 key AAs existed within 5 Å around active pocket (Additional file 1: Table S1). The key AAs forming hydrogen bonds to each β-lactam were shown in Table 2, including Ser55, Ser101, Ser102, Ser103, Thr105, Thr193, Thr195, Try87, Glu89, Arg229 and Ser233. When the score was less than 4, the binding ability between protein and drug was poor, and when the score was greater than 7, the binding ability was stronger.
Determination of mutational sites
PBP3 from Streptococcus pneumoniae R6 had a high binding ability with cefadroxil, and the docking results of PBP3 and cefadroxil showed that D224 in the active site K(239)TGTTD(244) and cefadroxil formed a hydrogen bond [32]. The sequence alignment demonstrated that K(192)TGTSV(197) in BlaR-CTD activity pocket corresponded to K(239)TGTTD(244) in PBP3. Therefore, the mutation of V197D was selected in order to improve the affinity.
Mutation prediction and evaluation was carried out according to SIFT and Polyphen 2. The mutational sites with a SIFT score of 1.0 were obtained as shown in Additional file 1: Table S2. In order not to affect the structure and function of the protein, the selection of mutational sites of protein design should avoid the active site, especially the hydrogen bond-forming AAs of the protein [4]. Twelve AAs from the list of AAs in the active pocket but exclude the active site and hydrogen bond-forming AAs (Additional file 1: Table S1) showed a SIFT score of 1.0 (Additional file 1: Table S2), and the single point mutations of A138E, Q147K and S190Y with the lowest Polyphen scores were screened out (Additional file 1: Table S3).
In addition, salt bridge and disulfide bond were inserted into the protein flexible region to improve the stability of the protein. Through the analysis of protein structure, I188K mutation could form a salt bridge (2.92 Å) with Glu212 in β6 sheet of BlaR-CTD. According to the prediction by software Disulfide by Design 2.0, the mutant proteins S19C/G24C, R50C/Q147C, S76C/L96C, S135C/S145C, E183C/I188C were selected according to the principle of smaller binding energy value and larger ΣB-factor value as shown in Table 3. In addition, the single point mutation was combined with the disulfide bond insertion mutation when necessary to improve the affinity and stability simultaneously.
Expression and purification of BlaR-CTD mutant proteins
The optimized induction condition for the expression of protein was IPTG at final concentration of 1 mM and culturing for 12 h at 18 °C (Additional file 1: Figure S1). As shown in Fig. 2, each single point (Fig. 2a) or combined mutant protein (Fig. 2b) were purified with purity above 95%, and the bands for each mutant were in good agreement with the expected size of 26 kDa. After determination of protein concentration, 600 μg·mL− 1 of each purified mutant protein was stored at − 20 °C for further use.

Purification of single point mutants and disulfide bond mutants (a) and multipoint mutants of BlaR-CTD protein (b). a Lanes 1–11 were the purified proteins of BlaR-CTD, A138E, Q147K, I188K, S190Y, V197D, S19C/G24C, R50C/Q147C, S76C/L96C, S135C/S145C, and E183C/I188C, respectively. b Lanes 1–13 were the purified proteins of A138E/S19C/G24C, Q147K/S19C/G24C, I188K/S19C/G24C, S190Y/S19C/G24C, V197D/S19C/G24C, A138E/R50C/Q147C, I188K/R50C/Q147C, S190Y/R50C/Q147C, V197D/R50C/Q147C, A138E/E183C/I188C, Q147K/E183C/I188C, S190Y/E183C/I188C, and V197D/E183C/I188C, respectively. M, protein mass marker
The formation of disulfide bond in mutant proteins (S19C/G24C, R50C/Q147C, S76C/L96C, S135C/S145C, E183C/I188C) were detected by DTNB colorimetry. Standard curve was established according to different absorption values of cysteine at 412 nm (Additional file 1: Figure S2). The absorbance value of mutant protein was measured and the content of cysteine was calculated. The results showed that no free sulfhydryl group was detected in wild type and mutant proteins, indicating the formation of disulfide bond (Additional file 1: Figure S2).
Activity identification of BlaR-CTD mutant proteins
The binding activities of 5 single point mutation proteins and 5 disulfide bond inserting mutant proteins were shown in Additional file 1: Table S5. The OD values of no free drug competition for mutant proteins S76C/L96C and S135C/S145C were low, indicating that the binding ability of β-lactams of these two mutant proteins was poor, so they were not observed for the following studies.
The inhibition rate, which was inversely proportional to the protein activity, was used to indicate the recognition ability of the protein to the drug compared between each mutant and the wild-type. The results showed that compared with the wild-type protein, the binding ability of each mutant protein to the drug varied, and there was no obvious advantage (Table 4). So, the three active disulfide bond inserting mutants, S19C/G24C, R50C/Q147C and E183C/I188C, and the single point mutations, I188K, V197D, A138E, Q147K, and S190Y, were combined in order to get the superposition effect.
Among the multipoint mutation proteins, three mutant proteins with highest affinity, I188K/S19C/G24C, A138E/R50C/Q147C, and S190Y/E183C/I188C, exhibited significantly lower inhibition rate to 33, 22 and 21 drugs respectively than the wild-type protein (Table 5).
Stability analysis of BlaR-CTD mutant proteins
By comparing the stability of the mutant and wild-type protein, the stability of I188K/S19C/G24C, A138E/R50C/Q147C and S190Y/E183C/I188C were higher than that of wild-type protein and single point mutant protein I188K at − 20 °C in 6 months (Fig. 3). At 4 °C, I188K/S19C/G24C was more stable than wild-type protein and mutant proteins I188K, A138E/R50C/Q147C and S190Y/E183C/I188C for 6 months. At 25 °C and 37 °C, the stability of mutant protein I188K/S19C/G24C was higher than that of wild-type protein and mutant proteins I188K, A138E/R50C/Q147C and S190Y/E183C/I188C in 30 days. When the mutant proteins A138E/R50C/Q147C and S190Y/E183C/I188C were stored at 4 °C, 25 °C and 37 °C for 1 month, the protein activity decreased to about 20%, and these two mutant proteins only has a good stability at − 20 °C.The activity of I188K/S19C/G24C could maintain over 90% in 6 months at − 20 °C and 4 °C, and over 90 and 60% at 25 °C and 37 °C respectively in 30 days (Fig. 3).

Stability of BlaR-CTD and mutant proteins. The wild-type BlaR-CTD protein and its mutants, I188K I188K/S19C/G24C, A138E/R50C/Q147C and S190Y/E183C/I188C were respectively stored at − 20 °C and − 4 °C for 6 months and at 25 °C and 37 °C for 30 days. At indicated times, each protein was taken to react with HRP-AMP. Residual activity (%) = OD value (after storage)/ OD value (before storage) × 100%
Comparison of protein structure and binding mode of I188K/S19C/G24C with the wild-type BlaR-CTD protein
According to the 3D structure of BlaR-CTD protein obtained earlier (Fig. 1a),
the 3D structure of mutant protein I188K/S19C/G24C was obtained by homologous modelling of Sybyl-X2.0 software (Fig. 4a). The β5 sheet in the α/β domain of the wild-type became two β sheets, β5 and β6, in the mutant protein I188K/S19C/G24C because of I188K mutation. The active site, K(192)TG(194), changed from β sheet in the wild-type to random coil in the mutant protein, which may have effect on the affinity of the protein. Sybyl-X2.0 was used for the molecular docking between mutant protein I188K/S19C/G24C and β-lactams. As shown in Fig. 4b, five hydrogen bonds were formed between Ser55, Ser103, Thr195 in the mutant protein and cefquinome, among which two hydrogen bonds formed with Ser55 and Thr195 respectively, and one hydrogen bond formed with Ser103. Hydrogen bond is the strongest non-covalent interactions, and the more hydrogen bonds, the stronger the binding ability of the protein to the ligand [28]. However, compared with the wild-type protein (Fig. 1b), the number of hydrogen bonds was decreased 44.4% in I188K/S19C/G24C docked with cefquinome (from 9 hydrogen bonds in the wild-type to 5 hydrogen bonds in I188K/S19C/G24C) (Fig. 4b) while the binding activity improved more than 3 fold (Table 5). The key AAs involved in the formation of hydrogen bonds between protein and 40 β-lactam drug were obtained (Additional file 1: Table S6). In a comparison of the wild type protein result (Table 2), it was found that the total score of the mutant protein was improved as a whole.

3D structure of mutant protein I188K/S19C/G24C (a) and interaction with cefquinome (b). a The 3D structure of BlaR-CTD was represented as a cartoon, with α-helices colored in cyan, β-strands in magenta, and loops in salmon. The mutational sites of I188K, S19C and G24C were colored in yellow. b The AA residues involved in the interaction of protein with cefquinome. The hydrogen bonds were represented as black dotted lines. The cefquinome molecule was colored in gray. The AAs of the mutational sites were colored in yellow
Discussion
Evaluation of the affinity of BlaR-CTD mutant proteins
The recombinant BlaR-CTD protein originating from B. licheniformis ATCC 14580 and heterologously expressed in E. coli contained a total of 288 residues, corresponding to a theoretical molecular mass of 32,427 Da, since it contained an N-terminal extension sequence originating from pET28 vector (Additional file 1: Figure S3, amino acid residue numbers from − 34 to − 1). However, the molecular weight of the recombinant BlaR-CTD protein estimated from SDS-PAGE was only 26 kDa (Fig. 2). We attribute this discrepancy to the fact that due to the low isoelectric point of BlaR-CTD (pI = 5.24) in the alkaline separation gel the denatured protein might be more negatively charged than average proteins, accelerating its mobility. Similarly, in the study of a related 262 residues protein (Met346-Arg601 C-terminal domain of the BlaR sensory-transducer protein of B. licheniformis with a hexapeptide extension), the recombinant protein with 29.3 kDa theoretical molecular mass and pI 5.72 migrated with 26 kDa on SDS-PAGE [17].
The affinity of BlaR-CTD to each β-lactam is different, due to the different chemical structures of drugs, the interaction and binding modes of BlaR-CTD protein with drugs are also different. There are some difficulties in selecting mutation sites because it was uncertain which amino acids in proteins interact directly with drugs. Molecular docking was used to find the key amino acids that may interact with each drug (e.g., those amino acids forming hydrogen bonds with the drug). Through the results of molecular docking between protein and β-lactam antibiotics, the amino acid sites involved in the hydrogen bonding between protein and drug were obtained. These key amino acids should be avoided in the selection of mutation sites, which provided a reference for the selection of subsequent mutation sites.
Mutation of V197D in BlaR-CTD was designed based on the sequence alignment with PBP3 in which D224 formed a hydrogen bond with cefadroxil [32]. The inhibition rate of V197D to cefadroxil was significantly decreased in this study (Table 4), indicating that V197D had a higher affinity for cefadroxil, which was consistent with the result of PBP3 to cefadroxil [32]. This suggests that it is reliable to select mutation through AA sequence as compared to active sites. The inhibition rate of mutant protein Q147K on 17 drugs increased significantly. The mutational site Gln147 is located in the flexible region between α7 and α8, and the distance between Arg50 and Gln147 (4.38 Å) is close in 3D structure. When the polar Gln147 with no charge was changed into the alkaline Lys147 with positive charge, it was easy to produce the repulsion effect since both Arg50 and Lys147 had a positive charge and the side chain was large, making the active pocket become smaller, thus leading to the decrease of protein affinity.
Among the disulfide bond inserting mutants, S76C/L96C and S135C/S145C exhibited nearly no activity (Additional file 1: Table S5). Since the mutational site Leu96 in the mutant protein S76C/L96C was located within 5 Å of the active pocket (Additional file 1: Table S1), its mutation may influence the protein activity pocket, resulting in the reduced activity of mutant protein.
Compared with S19C-G24C mutant protein (Table 4), the inhibition rate of mutant protein A138E/S19C/G24C and I188K/S19C/G24C decreased significantly (Table 5), indicating a superposition effect. But the inhibition rate of other combined mutant proteins (Q147K/S19C/G24C, S190Y/S19C/G24C, V197D/S19C/G24C) was not quite different from that of single point mutation. It might be due to the mutation of hydrophobic AAs to polar AAs, which increased the affinity of the proteins. The mutation of V197D was also from hydrophobic AA to polar AA, but Asp197 was an acid AA, and it was easy to form a salt bridge with the alkaline residue Arg229, which was close to it, thus making the active pocket smaller and reducing the protein activity. Compared with R50C-Q147C (Table 4), the inhibition rate of the A138E/R50C/Q147C to 21 drugs significantly decreased (Table 5). It probably due to Glu138 and Cys147 were located in the flexible region of Ω-loop behind α7, and the spatial locations are relatively close (13.35 Å), resulting in the change in the spatial structure of the protein and the increase of the affinity. The inhibition rates of combined mutations of A138E, Q147K, S190Y, and V197D with E183C-I188C to penicillins were significantly reduced, while the inhibition rates to most of the cephalosporins were significantly increased (Table 5). Cys183 was located on β4, Cys188 was located on β5, and they were located between adjacent β sheets (the distance is 4.29 Å), which may lead to a smaller protein active pocket. The β-lactam ring of cephalosporins is larger than that of penicillin [27]. When cephalosporins are entering into mutant proteins, the binding ability of the proteins may be reduced due to the small space of active pocket. The inhibitory rate of the mutant protein S190Y/E183C/I188C on the 21 drugs decreased significantly (Table 5). The spatial distances between Ser190, Glu183 and Ile188 were very close. The protein structure changed correspondingly after the mutation, since Tyr is larger than Ser, and the affinity of the protein was improved. Moreover, it is interesting to note that Tyr190 and Cys188 are located in the β5, implicating that this region is important for the binding ability of protein to drug.
The total score of molecular docking which predicted by software represented the binding affinity between protein and drug, and the score was expressed as binding constant, pKd [13]. A high value of total score indicates a good affinity of the drug with protein. Among the docking results for 40 drugs, the docking scores of I188K/S19C/G24C with 23 drugs were increased compared with those of the wild-type (Additional file 1: Table S6). In the 33 drugs involved in the activity test, the docking scores between the mutant protein and 18 drugs were increased, but the mutant protein showed a higher affinity to all of the 33 drugs (Table 5). Since the molecular docking result is only a software simulation, the binding ability of protein to drugs should still confirmed by activity identification. Nevertheless, our study shows that the software simulation had some reference value, but there still has some discrepancy.
Evaluation of the stability of BlaR-CTD mutant proteins
In a comparison of wild-type protein and single point mutation (I188K), it has been observed that disulfide bond mutation in I188K/S19C/G24C had a better stability and the activity of protein remained longer (Fig. 3), indicating that the disulfide bond played an important role in the stability of protein [10, 18, 22, 30]. I188K/S19C/G24C was more stable than A138E/R50C/Q147C and S190Y/E183C/I188C at 4 °C, 25 °C and 37 °C (Fig. 3). The disulfide bond of mutant protein I188K/S19C/G24C was formed in a flexible region, which is between the last AA of β2 and before the flexible region of α1 of the protein. However, the disulfide bonds of mutant proteins A138E/R50C/Q147C and S190Y/E183C/I188C formed in a more rigid region, which was behind the flexible region of α3 and α7, and between β4 and β5, respectively (Table 3). The different positions of disulfide bond may cause a different effect on the protein stability. The disulfide bonds formed in the flexible region of the protein would prevent the production of extra tension and was beneficial to the stability of the protein [2].
Moreover, when a single point mutation was combined with the introduction of disulfide bonds, it produced the superposition effect in the enhancement of protein affinity (Tables 4 and 5) and stability (Fig. 3). In mutant protein I188K/S19C/G24C, the mutational site I188K was located on the fragment of β5, which was close to the active pocket of BlaR-CTD protein. It might affect the affinity of protein, made the mutation bind to the various β-lactams and enhances the binding affinity of β-lactams. S19C/G24C, located between α1 helix and the flexible region in front of the β2 fragment, were far away from the active site of BlaR-CTD protein and would not change the structure of the active pocket, thus not affect the affinity of the protein.
Conclusions
To our best knowledge, our study is a new attempt on the structural modification of protein which may be used in the receptor assay for detecting β-lactam antibiotics residues. In this study, the structure of BlaR-CTD protein from Bacillus licheniformis ATCC14580 was modified by site-directed mutagenesis based on the combination use of computer simulations, including homologous modeling, molecular docking, disulfide bond inserting and salt bridge building as well as prediction and evaluation of mutation site by software. Mutant protein I188K/S19C/G24C was screened out from 23 mutant proteins for the best stability and higher affinity to 33 β-lactams. As expected, the study results of I188K/S19C/G24C showed that single point mutation combined with disulfide bond insertions could simultaneously increase the recognition ability and stability of the protein. In subsequent studies, mutant proteins I188K/S19C/G24C can be applied to establish a fast and effective receptor method for the screening of β-lactam antibiotic residues.
Abbreviations
- 3D:
-
Three-dimensional
- AA:
-
Amino acid
- BlaR-CTD:
-
The C-terminal domain of BlaR
- BSA:
-
Bovine serum albumin
- HRP-AMP:
-
Horseradish peroxidase-labeled ampicillin
- IPTG:
-
Isopropy-β-d-thiogalactoside
- LB:
-
Luria–Bertani
- PBP:
-
Penicillin binding protein
- PBS:
-
Phosphate-buffered saline
- TMB:
-
3,3′,5,5′-tetramethylbenzidine
References
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248.
Bhattacharyya R, Pal D, Chakrabarti P. Disulfide bonds, their stereospecific environment and conservation in protein structures. Protein Eng Des Sel. 2004;17(11):795–808.
Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72(1):248–54.
Contreras-Martel C, Dahout-Gonzalez CA, Martins Ados S, Kotnik M, Dessen A. PBP active site flexibility as the key mechanism for beta-lactam resistance in pneumococci. J Mol Biol. 2009;387(4):899–909.
Coquelle N, Fioravanti E, Weik M, Vellieux F, Madern D. Activity, stability and structural studies of lactate dehydrogenases adapted to extreme thermal environments. J Mol Biol. 2007;374(2):547–62.
Craig DB, Dombkowski AA. Disulfide by design 2.0: a web-based tool for disulfide engineering in proteins. BMC Bioinformatics. 2013;14(1):346.
Degelaen J, Granier B, Frere JM, Joris B. Process for determining antibiotics containing a β-lactam ring in a biological fluid. US, Patent; 2003.
Duval V, Swinnen M, Lepage S, Brans A, Granier B, Franssen C, Frere JM, Joris B. The kinetic properties of the carboxy terminal domain of the Bacillus licheniformis 749/I BlaR penicillin-receptor shed a new light on the derepression of beta-lactamase synthesis. Mol Microbiol. 2003;48(6):1553–64.
Gao C, Lan D, Liu L, Zhang H, Yang B, Wang Y. Site-directed mutagenesis studies of the aromatic residues at the active site of a lipase from Malassezia globosa. Biochimie. 2014;102(7):29–36.
Gasymov OK, Abduragimov AR, Glasgow BJ. Restoration of structural stability and ligand binding after removal of the conserved disulfide bond in tear lipocalin. Biochem Biophys Res Commun. 2014;452(4):1004–8.
Ge L, Li D, Wu T, Zhao L, Ding G, Wang Z, Xiao W. B-factor-saturation mutagenesis as a strategy to increase the thermostability of α-L-rhamnosidase from Aspergillus terreus. J Biotechnol. 2018;275:17–23.
Golemi-Kotra D, Cha JY, Meroueh SO, Vakulenko SB, Mobashery S. Resistance to beta-lactam antibiotics and its mediation by the sensor domain of the transmembrane BlaR signaling pathway in Staphylococcus aureus. J Biol Chem. 2003;278(20):18419–25.
Gupta SD, Bommaka MK, Mazaira GI, Galigniana MD, Subrahmanyam CVS, Gowrishankar NL, Raghavendra NM. Molecular docking study, synthesis and biological evaluation of Mannich bases as Hsp90 inhibitors. Int J Biol Macromol. 2015;80:253–9.
Hsieh PC, Vaisvila R. Protein engineering: single or multiple site-directed mutagenesis. Methods Mol Biol. 2013;978:173.
Huang JW, Chen CC, Huang CH, Huang TY, Wu TH, Cheng YS, Ko TP, Lin CY, Liu JR, Guo RT. Improving the specific activity of β-mannanase from Aspergillus Niger BK01 by structure-based rational design. Biochim Biophys Acta. 2014;1844(3):663.
Jensen K, Osmani SA, Hamann T, Naur P, Møller BL. Homology modeling of the three membrane proteins of the dhurrin metabolon: catalytic sites, membrane surface association and protein-protein interactions. Phytochemistry. 2011;72(17):2113–23.
Joris B, Ledent P, Kobayashi T, Lampen JO, Ghuysen JM. Expression in escherichia coli of the carboxy terminal domain of the blar sensory-transducer protein of bacillus licheniformis as a water-soluble mr 26 000 penicillin-binding protein. FEMS Microbiol Lett. 1990;70(1):107–13.
Kawade R, Akiba H, Entzminger K, Maruyama T, Okumura CJ, Tsumoto K. Roles of the disulfide bond between the variable and the constant domains of rabbit immunoglobulin kappa chains in thermal stability and affinity. Protein Eng Des Sel. 2018;31:243–7.
Kerff F, Charlier P, Colombo ML, Sauvage E, Brans A, Frere JM, Joris B, Fonze E. Crystal structure of the sensor domain of the BlaR penicillin receptor from Bacillus licheniformis. Biochemistry. 2003;42(44):12835–43.
Makhatadze GI, Loladze VV, Ermolenko DN, Chen X, Thomas ST. Contribution of surface salt bridges to protein stability: guidelines for protein engineering. J Mol Biol. 2003;327(5):1135–48.
Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31(13):3812–4.
Ning L, Guo J, Jin N, Liu H, Yao X. The role of Cys179-Cys214 disulfide bond in the stability and folding of prion protein: insights from molecular dynamics simulations. J Mol Model. 2014;20(2):2106.
Nissen PH, Christensen SE, Ladefoged SA, Brixen K, Heickendorff L, Mosekilde L. Identification of rare and frequent variants of the CASR gene by high-resolution melting. Clin Chim Acta. 2012;413(5):605–11.
Parthasarathy S, Murthy MR. Protein thermal stability: insights from atomic displacement parameters (B values). Protein Eng. 2000;13(1):9–13.
Peng J, Cheng G, Huang L, Wang Y, Hao H, Peng D, Liu Z, Yuan Z. Development of a direct ELISA based on carboxy-terminal of penicillin-binding protein BlaR for the detection of beta-lactam antibiotics in foods. Anal Bioanal Chem. 2013;405(27):8925–33.
Powell AJ, Tomberg J, Deacon AM, Nicholas RA, Davies C. Crystal structures of penicillin-binding protein 2 from penicillin-susceptible and -resistant strains of Neisseria gonorrhoeae reveal an unexpectedly subtle mechanism for antibiotic resistance. J Biol Chem. 2009;284(2):1202–12.
Tahlan K, Jensen SE. Origins of the β-lactam rings in natural products. J Antibiot. 2013;66:401.
Wang J, Papke RL, Stokes C, Horenstein NA. Potential state-selective hydrogen bond formation can modulate activation and desensitization of the alpha7 nicotinic acetylcholine receptor. J Biol Chem. 2012;287(26):21957–69.
Wei H, Hu J, Wang L, Xu F, Wang S. Rapid gene splicing and multi-sited mutagenesis by one-step overlap extension polymerase chain reaction. Anal Biochem. 2012;429(1):76–8.
Zabetakis D, Olson MA, Anderson GP, Legler PM, Goldman ER. Evaluation of disulfide bond position to enhance the thermal stability of a highly stable single domain antibody. PLoS One. 2014;9(12):e115405.
Zapun A, Contrerasmartel C, Vernet T. Penicillin-binding proteins and beta-lactam resistance. FEMS Microbiol Rev. 2008;32(2):361–85.
Zhang J, Wang Z, Wen K, Liang X, Shen J. Penicillin-binding protein 3 of Streptococcus pneumoniae and its application in screening of beta-lactams in milk. Anal Biochem. 2013;442(2):158–65.
Zhou C, Liu L, Zhuang J, Wei J, Zhang T, Gao C, Liu C, Li H, Si H, Sun C. A systems biology-based approach to uncovering molecular mechanisms underlying effects of traditional Chinese medicine Qingdai in chronic myelogenous leukemia, involving integration of network pharmacology and molecular docking technology. Med Sci Monit. 2018;24:4305–16.
Acknowledgments
The authors thank the National Key R&D Program of China for funding this research.
Funding
This work was supported by the National Key R&D Program of China (2018YFD0500300 and 2017YFC1600100), the Natural Science Foundation of Hubei Province (2017CFB445), the National Program for Risk Assessment of Quality and Safety of Livestock and Poultry Products (GJFP2019007) and National Program for Risk Assessment of Quality and Safety of Milk (GJFP201800804).
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Author information
Authors and Affiliations
Contributions
ZHY and GYC conceived the project, obtained grant support, designed research, and analyzed and interpreted data. JNN, SA and TC performed the experiments, analysis, interpretation, and statistic. JNN and SA wrote the paper and ZHY, GYC, YLW and DPP revised the manuscript. All authors edited and approved the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0) which permits copy and redistribute the material just in non-commercial usages, provided the original work is properly cited.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Table S1. Amino acid residues within 5 Å in the active pocket. Table S2. The mutational sites with score of 1.0 predicted by SIFT. Table S3. Scoring results by PolyPhen software. Table S4. Determination of free sulfhydryl group in mutant protein. Table S5. Acitivity identification of BlaR-CTD wild-type and mutant protein using HRP-AMP. Table S6. Binding sites of I188K/S19C/G24C to β-lactam antibiotics. Figure S1. Expression of BlaR-CTD protein. Figure S2. Standard curve of cysteine based on DTNB. Figure S3. The DNA and amino acid sequences of recombinant wildtype BlaR-CTD protein. (DOCX 120 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Ning, J., Ahmed, S., Cheng, G. et al. Analysis of the stability and affinity of BlaR-CTD protein to β-lactam antibiotics based on docking and mutagenesis studies. J Biol Eng 13, 27 (2019). https://doi.org/10.1186/s13036-019-0157-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13036-019-0157-4