
Abstract
Human Papillomavirus is the major etiological agent in the development of cervical cancer but not a sufficient cause. Despite significant research, the underlying mechanisms of progression from a low-grade squamous intraepithelial lesion to high grade squamous intraepithelial lesion are yet to be understood. Deregulation of viral gene expression and host genomic instability play a central role in virus-mediated carcinogenesis. Key events such as viral integration and epigenetic modifications may lead to the deregulation of viral and host gene expression. This review has summarized the available literature to describe the possible mechanism and role of viral integration in mediating carcinogenesis. HPV integration begins with DNA damage or double strand break induced either by oxidative stress or HPV proteins and the subsequent steps are driven by the DNA damage responses. Inflammation and oxidative stress could be considered as cofactors in stimulating viral integration and deregulation of cellular and viral oncogenes during the progression of cervical carcinoma. All these events together with the host and viral genetic and epigenetic modifications in neoplastic progression have also been reviewed which may be relevant in identifying a new preventive therapeutic strategy. In the absence of therapeutic intervention for HPV-infected individuals, future research focus should be directed towards preventing and reversing of HPV integration. DNA damage response, knocking out integrated HPV sequences, siRNA approach, modulating the selection mechanism of cells harboring integrated genomes and epigenetic modifiers are the possible therapeutic targets.
Similar content being viewed by others
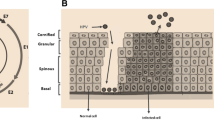


Background
Cervical cancer is the fourth most common cancer in women worldwide [1]. It is well known that high-risk HPV is the main etiological agent for this infectious viral carcinoma. Human papillomaviruses are small (50 nm) double-stranded DNA viruses composed of a genome of 8kilobase pair, enclosed inside a non-enveloped capsid protein. The genome includes three portions: (a) early genes (E1, E2, E4, E5, E6, E7) those regulate the vegetative and productive phase of viral life cycle;(b) late genes (L1, L2) which encode the capsid protein and (c) a non-coding regulatory region called long control region (LCR) involved in the regulation of viral replication and transcription [2].
Among 184 different HPV genotypes, only 40 diverse types can infect anogenital region which can be classified into 3 classes based on their oncogenic potential. HPV16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, 68, 73 and 82 are included in high-risk group while HPV6, 11, 40, 42, 43, 44, 54, 61, 70, 72 and 81 are included in low-risk group whereas HPV 26, 53 and 66 belong to the group of intermediate risk [3, 4]. Compelling evidence supports the high prevalence of HPV16 and 18 in the high-grade cervical lesion and considers these types to be the most potent carcinogenic viruses [5].
Based on the histopathological features cervical cancers are classified as squamous cell carcinoma (SCC), adenocarcinoma and adenosquamous carcinoma. Squamous cell carcinoma is the most common type of cervical cancer. The precancerous lesions which progress to SCC are called cervical intraepithelial neoplasia (CIN) or squamous intraepithelial lesion (SIL) which is classified according to the grade of the lesion [6]. A productive HR-HPV infection may develop into low-grade SILs (LSILs) which are nonmalignant bearing the low risk of progression to malignancy and corresponding to CIN1 [7]. The high-grade SILs (HSILs) comprise abortive virus infections in which there is deregulated expression of HPV early genes in basal epithelial cells, a greater risk of progression to invasive disease and corresponding to CIN2/3 [6]. Most of the HPV infections are subclinical and only a small fraction of HR-HPV infections produces early epithelial lesions [8, 9] and a more modest fraction of those lesions progress to higher grade lesion and invasive cancer. The mechanisms behind the progression of neoplastic lesions have not clearly understood. However, several viral and host factors and their interactions with each other have been proposed as potential candidates of carcinogenesis. In this review, we take a comprehensive look at the current understanding of molecular mechanisms behind the process of HPV-induced carcinogenesis with relevance to cervical cancer progression.
Entry of HPV and life cycle in cervical epithelial cell
HPV, an epitheliotropic virus, infects basal epithelial cells of the squamous-columnar junction of the cervix. The virus makes its entry into the basal epithelial cells through micro-wounds or micro-abrasions. Hence early age first sexual debut, coarse sex and other sexually transmitted infections (STIs) promote virus entry and infection [10]. Heparan sulfate proteoglycans (HSPG) found in the extracellular matrix (ECM) on the cell surface are thought to be the initial receptors of HPV VLP [11, 12]. α6-integrin and laminin-5 play an important role as co-receptors for an efficient viral infection [13]. Initial attachment of HSPG moieties to L1 facilitates the conformational changes in L2 [14, 15]. Subsequently, the L2 protein is cleaved by furin on the cell surface at a consensus cleavage site that is conserved among all papillomaviruses [16]. Entry of virus into a host is very slow (up to 12 h), possibly due to conformational changes in capsid and receptors [17].
After a successful binding to the receptor, virus is internalized into the cell by clathrin or caveolae-mediated endocytosis [18, 19]. The viral genome enters into the nucleus through nuclear envelope breaks instead of nuclear pore. Then it localizes the ND10 bodies inside the nucleus [20–22]. Once entered the nucleus, HPV replicates to a low copy number (10–200 copy number/cell) during the initial amplification and establishment of infection [23]. As the proliferating cells (harboring HPV genomes) undergo transition through differentiation, the mode of viral genome replication switches to support productive viral genome amplification concomitant with increased levels of the E1 and E2 replication proteins [24]. In the terminally differentiated layer of epithelium L1 and L2 capsid proteins are expressed and viral particles are assembled. The virions are sloughed off with the dead squamous cells of the host epithelium for further transmission [25].
Mechanism of HPV integration
Although integration is not a part of the normal HPV life cycle, high-risk HPV (HR-HPV) DNA is often integrated into the human genome in cervical SSC tissue sample [26–30]. It has been proposed that integration can be an early event associated with LSIL to HSIL progression [31–34], so expected to be a biomarker in cancer progression. However, the current understanding of the mechanism of integration is unclear.
Unlike retroviruses in which protein integrase facilitates their integration into the host genome, HPV encodes no such protein for this purpose. The site of integration is distributed throughout the genome as chromosomal fragile sites where DNA double strand breaks are failed to repair [35–37]. Integration hotspots in various genomic regions such as 3q28, 17q21, 13q22.1, 8q24.21, and 4q13.3 are reported by various researchers [36]. DNA damage is often induced by oxidative molecules such as reactive Oxygen species, reactive nitrogen species [38, 39] and HPV proteins E1, E6, &E7 [40–43].
Cell evolved with a special pathway to repair the continuous exogenous and endogenous damage in DNA named as DNA damage response (DDR). DDR plays a crucial role in preparing the cell to continue cell division. Once the damage is repaired, the cell cycle checkpoints are mitigated and cell continues to divide. If unrepaired cell undergoes apoptosis. However, unrepaired break points are the prerequisites for integration to occur.
Viral oncoproteins play a role in combating the downstream consequences of DNA damage response (DDR) in various ways. This favors in maintaining the breakpoints in host chromosomes required for integration. E6 and E7 oncoproteins disrupt cell cycle checkpoint control by inhibiting CDKs inhibitors [P21, P27] and degrading P53 [44–46]. HPV-16 E7 oncoprotein attenuates the DNA damage checkpoint response by accelerating the proteolytic turnover of claspin, a critical regulator of the ATR/CHK1 signaling axis and DNA damage checkpoint recovery in the G2 phase of the cell cycle [47]. HPV oncoproteins relax G1-S checkpoint control to induce unscheduled entry into S-phase and promote S-phase–like milieu conducive for viral genome replication in differentiated human keratinocytes [48, 49]. P53 is degraded by E6 oncoprotein and it is required for sensing base excision repair machinery and repairing of oxidative damage [50]. Thus, damage response fails to pose apoptotic threats to cell and allows replication to produce rearranged host genome with multiple breakpoints.
Interestingly, HPV takes advantage of this damage response pathway for its own replication and produces an ample number of episomal HPV which possibly increase the availability of more HPV DNA for integration into the host DNA. The DNA break-induced DDR triggers the accumulation of the factors for replication factories at the replication foci [43, 51]. Importantly, in the differentiated cells, this can occur in the G2 phase of the cell cycle where there is no competition from host DNA synthesis which is an additional advantage.
DNA damage response acts as a driving force throughout viral replication. Presence of ND10 at DNA damage region indicates the link between initial amplification of viral DNA with DDR [52]. It showed that during the initial amplification of viral DNA, the ATR-dependent DNA damage response engages at the HPV 18 replication centers [53]. Besides the initial amplification, DDR plays an important role to maintain the viral replication [54, 55]. The implication of DDR machinery in maintaining replication could be presumed by considering the association of CFS with ERD4 which helps to tether the viral genome with host DNA [56, 57].
The occasional association of Head-to-tail tandem repeats in the chromosome of cervical cancer cells [58, 59] leads to a plausible explanation that a linear concatemeric HPV genome is synthesized in cells by a rolling circle mechanism of replication and integrates into the host chromosome [60]. Upon induction of differentiation, genome switched to rolling circle mechanism of replication whereas in undifferentiated cells bidirectional replication occurs. The homologous recombination machinery is recruited to the regions of double strand breaks only and it is generated through collapsed replication fork during replication of viral genome [61] which might be induced by HPV E1. Double strand break in viral genome may be induced by HPV E1 during replication and homologous recombination [43, 51, 53, 61]. It is reported that DDR is involved in maintaining the double strand break in the viral genome [62, 63]. Failure to re-circularize the breaks produces linear HPV DNA with the double strand break which possibly enhances the scope for integration.
Proximity between host and HPV genome is required so that the fusion between virus and host could be completed. During the HPV life cycle, HPV genome tethers to host chromatin by HPVE2-BRD4 complex for partitioning of genomes to daughter cells [57]. E2-BRD4 complexes with acylated histones are observed at CFS region [56]. The association of BRD4 with the chromosomal fragile region (common site of integration) or the region of DNA damage leads to the presumption that BRD4 may play an important role in increasing the mechanistic feasibility of integration by promoting tethering of viral DNA with host genome at the region of DNA damage. However, there is no literature available to explain the role of BRD4 in HPV integration or any association of genome tethering with viral integration.
To complete the process of integration, fusion between host and HPV genome with DSB is expected to be accomplished through a recombination directed repair mechanism. Initially, nonhomologous end joining (NHEJ) is thought to be implicated in HPV integration [64]. According to a recent report, the presence of microhomologous sequences near the integration breakpoints indicate a homology-mediated DNA repair pathway during the fusion of human and viral DNA [65]. Although sequence homology is observed at the integration site, it is not perceived to be a prerequisite. There are two types of integration events [58]. In type-1 integration, a single copy of the viral genome gets integrated while in type-2, a concatamer of viral genome integrates into the host chromosome.
Summarizing the above literature, the integration is started with DNA damage, induced either by oxidative stress or HPV protein and the subsequent steps are driven by the DNA damage responses (Fig. 1). Virus uses the DDR machinery to promote viral amplification while the viral oncoproteins render the cells to overcome the downstream consequences of damage response. Breaks in HPV DNA are introduced possibly during the replication of virus which might be induced by E1 and these breaks fail to get repaired. Availability of ample viral episomes to the host genome with multiple DSB enhances the possibility of integration. Proximity between viral and host genome by E2-BRD4-mediated tethering could possibly increase the mechanistic feasibility of viral integration. Finally, the fusion between both the genome via either homologous or nonhomologous recombination is regulated by the DNA damage response pathway (ATM/ATR and DNA-PK pathways) [66].

Conceptual model of viral integration. 1. Induction of DSB by ROS/NOS/Viral protein 2. DNA damage induces DDR (DNA damage response), ATM/ATR and P53 get activated to repair the damage. 3. HPV oncogenes deactivate the normal function of DDR molecules and DNA damage failed to be recognized. E7 degrades claspins and attenuate DNA damage checkpoints, while E6 degrades p53 and base excision repair gets suppressed so that the genomic DNA remains unrepaired and cell cycle proceeds. 4. Virus utilizes the DDR machinery for its replication which increases the availability of episomal DNA for integration. Breaks in the circular viral DNA may occur due to replication stress. 5. Virus and host genome come to a close proximity mediated by the BRD4-E2 complex. 6. Fusion between host and viral genome is accomplished either by Nonhomologous mediated end joining or homologous recombination repair pathway
Role of HPV integration in carcinogenesis
Integration of HPV influences both viral and host genome. Viral genes such as E1, E2, E5, and L2 region get disrupted by integration and consequently E6 and E7 oncogenes overexpressed in the absence of E2 repressor protein [67]. Overexpression of E6/E7 results in cell cycle deregulation and promotes several other pathways leading to carcinogenesis. Integrated HPV DNA gives a selective growth advantage to the cell as compared to the episomal counterparts and it is attributed to the presence of host polyadenylation tail and loss of apoptotic protein E2 [68]. Episomal loss due to IFN response is considered to be a determinant for the selection of integrant is being supported by many studies [69–71].
Genomic instability is the hallmark of cancer. A recent study reported a direct association between HPV integration and host genomic instability. It showed HPV integration driven chromosomal rearrangements which include deletions, translocations and inversion in the genomic regions flanking HPV integrants. The study suggested that integration is a possible insertional mutagenesis event which could lead to a specific change in gene expression at the site of integration [72]. This hypothesis is strengthened by another study showing a high level of host gene expression at HPV integration sites [73] as compared to the expression of the same genes in tumors without viral integration at the same sites [73]. Genes nearer to the integration sites with high expression level are reported to be MYC, ERBB2, GLI2, TNIK, NR4A2, PROX1, EIF2C2, FAM179B, and SERPINB4, RPS6KB1, MAFA, PARN, EGFL7, SNIP1, POC1B, and BCL11B [73]. Integration impacts the host genome by amplification of oncogenes and disruption of tumor suppressor genes as well as driving inter- and intra-chromosomal rearrangements.
Inflammation and oxidation play role as cofactors in neoplastic progression
Inflammation contributes to carcinogenesis via promoting oxidative damage, cell proliferation, invasion, and metastasis, inhibiting apoptosis and secreting immune suppressors [74, 75]. Chronic inflammation may lead to DNA damage by enhancing cellular ROS, NOS and foster the progress of low-grade to the high-grade lesion.
The interplay of viral oncoproteins and inflammatory cytokines may lead to develop persistent infection by continuous immune evasion which promotes progression of the lesion and ultimately leads to malignancy. Being double standard DNA, HPV genome is recognized by TLR-9 present in the DCs, macrophages, and NK cells. Interferon responses possibly facilitate selection of integrants and overexpression of E6/E7 through enhancing the depletion of episomal DNA [76, 77]. Once the early genes E6/E7 are expressed, TLR9 downregulated and IFN response impaired resulting in a conducive milieu for immune evasion and persistent infection [78–81]. Impairment of interferon pathway leads to down-regulation of MHC I expression on a cell surface. As a result, CD8-cytotoxic T cell can’t get activated and unable to initiate T cell response. This process of continuous immune evasion may ultimately lead to the persistence infection and carcinogenesis. Overexpression of E6 and E7 in cell imparts resistance to TNF- α induced apoptosis and antiproliferative effect [82–84]. NF-κB, a key modulator for chronic inflammation, is activated by PDZ binding domain of HPV E6. Diminished cell proliferation and rapid apoptosis observed in cells infected with mutant PDZ binding domain [83] which suggests that NF-κB involve in E6/E7 mediated carcinogenesis.
An increased Oxidative stress in cell attributes to inflammation, chemical stress, UV exposure, oxidative phosphorylation etc. Oxidative stress leads to DNA damage and viral integration which have been described earlier. Besides, DNA damage, proteins are also prone to oxidative modifications such as carbonyls and nitrotyrosine adducts, as observed in conditions like aging and cancer [85, 86]. Accumulation of oxidative proteins may lead to the deregulation of important pathways leading to carcinogenesis. Increased levels of carbonyl in cytokeratin-6, actins, cornulin, retinal dehydrogenase and GAPDH have been observed in HPV16 dysplasia and neoplastic tissue [87]. However, the effect of protein oxidation on cellular transformation and carcinogenesis is not yet been clearly understood. This phenomenon has a greater implication in improving clinical protocol for screening and prognostic evaluation of cervical cancer. The role of HPV proteins to generate oxidative stress is yet to be explored. However, there is limited data demonstrating the role of E6 and E2. No experimental data is available on the impact of OS on the regulation and function of E4, E5, L1 and L2. The impact of viral mediated oxidative stress on viral life cycle and carcinogenesis needs to be explored.
Inflammation and oxidative stress could be the cofactors in stimulating viral integration and deregulation of cellular and viral oncogenes during the progress of cervical carcinoma.
Epigenetic modification in cervical cancer
Genetic and epigenetic modifications at regulatory region of HPV may contribute to the overexpression of viral oncogene at episomal state [88–91]. Association of cervical carcinoma progression with the methylation of L1, L2, E5, E2 and L2 region of different HR-HPV genotypes is supported by many epidemiological and in vitro studies [92–94]. It is important to consider the methylation in the long control region (LCR) as it regulates the transcription and replication of HPV genes. The LCR is the region of the E2 binding site which possesses CpG sites for potential methylation, resulting in inhibition of E2 function. It was initially reported that HPV16 LCR methylation in the promoter and enhancer region decreases with severity of lesion [95]. Methylation is found to be more common in invasive cervical carcinoma and cervical intraepithelial neoplasia (CIN) III than in CIN I-II (84.6% and 46.2% vs. 29.4%, respectively) as reported by Hong et al. in 2008(97). In a later study, it is revealed that CpG hypermethylation of the HPV16 LCR increase with the severity of the cervical neoplasia [low grade squamous intraepithelial lesion (LSIL): 5.9%; high-grade squamous intraepithelial lesion (HSIL): 33.3%; squamous cell carcinoma (SCC): 53.3%] [96]. LCR methylation is also reported in 71.4% of asymptomatic infection cases. However, a clear association of LCR methylation with neoplastic progression has not yet been established.
The important histone modifications such as methylation and acetylation at H3 lysine 27, H3 lysine 9 and H4 lysine 20 positions significantly contribute to the regulation of viral gene expression [97, 98]. In the context of the neoplastic progression, acetylation of H3 increases as the cell progresses phenotypically from normal to SCC in both episomal and integration mediated carcinogenesis. H3 lysine 27 trimethylation and H3 lysine 9 trimethylation markers decrease with the neoplastic progression in integration mediated carcinogenesis [99, 100].
Like other cancers, epigenetic modification is a prominent feature of cervical cancer. A wide range of host genes involved in cell cycle regulation, apoptosis, DNA repair and WNT pathway often undergo epigenetic modification in cervical cancer. DcR1 and DcR2 are the apoptotic genes reported being hypermethylated in invasive cancer [101] suggesting that cervical cells get a growth advantage by downregulating decoy receptor expression [102]. Other examples include the downregulation of p73 in cervical cancer due to promoter hypermethylation [103] and correlation of hypomethylated hTERT with disease prognosis [104]. RASSF1 is a key gene involved in the apoptotic signaling pathway which is downregulated in cervical cancer via methylation is being reported in many studies [105, 106]. Downregulation of CADM1 gene leads to metastasis and cancer progression. Promoter hypermethylation is associated with decreased expression of CADM1 in high¬grade CIN and SCC [107]. Frequency and density of CADM1 methylation increase with severity of dysplasia [107]. Hence CADM1 is suggested to be a possible epigenetic diagnostic marker in predicting the risk of cancer progression. WIF1, APC, and CDH1 are significantly hypermethylated in cervical cancers [108] which are the key regulators of Wnt/β-catenin pathway. Increased methylation in DAPK1, RARB, TIMP3, CCNA, and FHIT is associated with cervical cancer and low or no methylation is observed in LSHL lesion and normal cytology [109–111]. Host genes are reported as methylation marker to distinguish abnormal lesion from the normal one. CCNA1 could be a methylation marker to distinguish normal lesion from high-grade lesion while CCNA1, hTERT1, hTERT2 and TWIST1 could distinguish cervical cancer from normal and precancerous stage [112].
Apart from demographic and lifestyle factors [113], methylation of HPV and host gene are likely to be influenced by HPV integration and multiple genotypes. Methylation is higher in single type as compared to infection with multiple HPV genotypes [113]. Moreover, host methylation pattern is reported to be integration dependent [114].
Association of miRNAs in carcinogenesis
miRNAs are the noncoding RNAs that regulate expression of the target mRNAs at the post-transcriptional level. miRNAs play a significant role in initiating and promoting the process of carcinogenesis by inducing cellular proliferation, apoptosis and genomic instability [115].
Carcinogenesis process is influenced by both up regulation and down regulation of miRNAs. Increased expression of certain mi-RNAs (viz., miR-886-5p, miR-10a, miR-141, miR-21, miR-135b, miR-148a, miR-214 and miR-106b) plays vital role in cervical cancer progression as they are involved in regulation of cell proliferation, apoptotic pathway or cell adhesion [116–122]. Down regulation of let-7c, miR-124, miR-126, miR-143, and miR-145 regulates the expression of oncogenes.
The role of miRNAs via E6 and E7 oncogenes in regulating cell cycle progression, senescence, and apoptosis have been reported by several studies [123]. In cervical cancer, p53 is the most important target of E6 oncogene. Hence, expression of all the miRNAs regulated by P53 is thought to be regulated by E6 oncogene function. For example, miR-23b, miR-34a, and miR-218 are down-regulated by E6 oncogene via degradation of p53 [124, 125].
E7 induced overexpression of miR-15/16 via E2F1 deactivation resulting down-regulation of c-myc or c-myb. E7 enhances the expression of miR-15a/miR-16-1 which inhibits cell proliferation, survival, and invasion. miR-203 is downregulated by E7 via MAPK/PKC pathway [125].
Considering the significant changes in miRNA expression during the progress of normal cervical epithelium to high-grade CIN lesions and ultimately to SCC or AdCAs [126], the microRNA expression profiling may help in preparing a specific genomic and/or transcriptomic signature of cervical tissue. It could be used as possible diagnostic and prognostic biomarkers for cervical carcinoma. However, more study is required for its proper validation in clinical samples.
Association of HPV variants in cervix cancer pathogenesis
Sequence variations in HPV genome would lead to the evolution of variants with differences in infectivity and pathogenicity. The attribution of HPV variants to cancer development could be due to the difference in the capacity to cause persistence infection and regulation of viral oncogene expression. The association of HPV16, HPV31, HPV52 and HPV 58 variants with high oncogenicity, persistence, and progression of infection is reported in many studies [127–130].
Several epidemiological data from the regions with high prevalence of cervical carcinomas such as Latin America, Africa, and Asia have a high prevalence of sublineages AA and Af [131]. Non-European HPV16 variants such as Af1 and AA were found at an increased frequency in invasive lesions while it is low in high-grade lesion [131]. A Recent study in South Mexican population suggests that oncogenic HPV 16 AA-a strain carries a higher risk to progress cervical cancer. The association of sub-lineage AA with persistent infection and the risk of cancer development are higher than that of EUR sublineage [130, 132, 133]. These data collectively shows a strong association of HPV 16 AA variants with persistent infection and development of cancer while other non-European variants have a lesser degree of association. Hence HPV16AA variant is considered to be a potential carcinogenic strain. Only three amino acid changes within the E6 of HPV16 AA are connected to this augmented carcinogenic ability [129]. Enhanced ability of AA E6 variants in promoting cellular immortalization, migration, invasiveness and ability to undergo transformation to resilient phenotypes have been demonstrated using in vitro model of retrovirally transduced primary human foreskin keratinocytes mimicking persistently infected and post-integrated keratinocyte system [134, 135]. Tumorigenic potential of full-length HPV 16 AA E6 variant has also been demonstrated in organotypic tissue culture model [136].
Besides HPV16, the variants of other genotypes (HPV 18, HPV 31, HPV 52 and HPV 58) have also been reported to be associated with the risk of persistent infection and development of cervical cancer. Xi et al. demonstrated that HPV 18 E/AsAi variants had a 2-fold risk of developing CIN3 as compared to the African variants [137]. Studies also suggest that the HPV 18 AsAi lineage (A1/A2) is 4-fold more common in Adenocarcinoma than the E lineage [138].
Literature shows conflicting data regarding the association of HPV 31 variants with the risk of persistence infection and progression. It is reported that HPV 31 lineage C is more persistent than that of A and B lineage [139, 140]. However, other literature reflects that A and B lineage are mostly associated with persistent infection and progression of diseases. More particularly lineage B was associated with CIN 3 as compared to C or B [139, 141].
In Case of HPV 52 variants, it is reported that B lineage is associated with a 7.6 fold risk [95% CI: 1.3–44] as compared to lineage C, in women with HPV52 as a single infection [142]. However, A1 lineage is more common in CIN 2/3. For HPV 58, A lineage is associated with CIN3+ as compared to B, C and D. Geographical distribution of HPV variants, epidemiological risk factors and complex interaction of viral variants with host genetics need to be considered while designing epidemiological studies with HPV variants and cancer pathogenesis.
Conclusion
This review has described the important molecular mechanisms of HPV mediated carcinogenesis in human cells. After a successful persistent infection of HR-HPV variants, neoplastic progression is accomplished through several routes (Fig. 2). Events of fundamental importance leading to neoplastic changes during the progression of cervical carcinoma are persistent infection, overexpression of viral oncogene and host genomic instability. These events could be possibly driven by host immune responses, viral integration, and host/viral epigenetic modifications.

HPV-driven carcinogenesis: a multistep molecular mechanism of host-viral interaction. The initial outcome of carcinogenesis is modulated by both viral (high-risk versus low-risk HPV types, HPV integration) and host factors (inflammatory response, oxidative stress). Inflammatory response upon initial infection such as IFN response plays role in reducing episomal HPV resulting clearance of infection. Integration of HPV is initiated with DNA damage. The IFN induced loss of episomal HPV and down-regulation of E2 leads to the selection of cells with integrated HPV genomes expressing higher levels of E6 and E7. Once the early genes E6 &E7 are expressed, TLR9 downregulated and IFN response impaired, resulting a conducive milieu for immune evasion and persistent infection. Upregulation of E6/E7 increases genetic instability and chromosomal rearrangements that increase the risk of integration. Overexpression of E6/E7 leads to deregulation of the cell cycle via p53 & Rb degradation, deregulation of oncogenes and miRNAs expression. Epigenetic and genetic modification in viral and host genome leads to the deregulation of E6 &E7 oncogenes, and host tumor suppressor genes that lead to carcinogenesis. Oxidative modification of TFs also leads to altered gene expression and carcinogenesis
Current HPV vaccination strategy targets at preventing the infection of few restricted HPV genotypes in uninfected individuals. There is also a lack of treatment strategy for clearance of viral infection. In this scenario future research focusing prevention and reversal of HPV integration and epigenetic modifications may provide a way to arrest neoplastic progression. A clear understanding of the role of DNA damage response in viral integration may consider DDR as a target to prevent HPV integration. Knocking out integrated HPV DNA sequences by gene editing and SiRNA approach to reducing viral oncogene transcripts and modulating the mechanisms of selection of cells harboring integrated HPV genome may be considered in this regard. The implication of epigenetic modifiers as a therapeutic approach could be a major future strategy.
Abbreviations
- AP-1:
-
Activator protein 1
- ATM:
-
Ataxia telangiectasia mutated
- ATR:
-
Ataxia telangiectasia and Rad3-related protein
- Bax:
-
BCL2-associated X protein
- BRCA1:
-
Breast cancer type1
- BRD4:
-
Bromodomain-containing protein4
- CADM1:
-
Cell adhesion molecule 1
- CCNA1:
-
Cyclin-A1
- CDKs:
-
Cyclin-dependent kinases
- CEBPB:
-
CCAAT/enhancer-binding protein beta i
- CHK1:
-
Checkpoint kinase 1
- CIN:
-
Cervical intraepithelial neoplasia
- DAPK1:
-
Death-associated protein kinase 1
- DC:
-
Dendritic cells
- DcR1:
-
Death receptor 1
- DDR:
-
DNA damage response
- DNA-PK:
-
DNA-dependent protein kinase
- DNMT1:
-
DNA (cytosine-5)-methyltransferase 1
- DSB:
-
Double strand break
- FHIT:
-
Fragile histidine triad protein
- GADPH:
-
Glyceraldehyde 3-phosphate dehydrogenase
- GRE:
-
Glucocorticoid response element
- HDAC:
-
Histone deacetylases
- HPV E/As/Ai variant:
-
HPV European/Asian/American Indian variant
- HPV:
-
Human papillomavirus
- HSIL:
-
High-grade squamous epithelial lesion
- HSPG:
-
Heparan sulfate proteoglycans
- hTERT:
-
Human telomerase reverse transcriptase
- IFN:
-
Interferon
- LCR:
-
Long control region
- LSIL:
-
Lowgrade squamous epithelial lesion
- MCP-1:
-
Monocyte chemoattractants protein
- MRN:
-
Mre II, Rad50, NbsI
- ND10:
-
Nuclear bodies 10
- NF-1:
-
Neurofibromatosis type-1
- NF-κB:
-
Nuclear factor kappa-light-chain-enhancer of activated B cells
- NGS:
-
Next generation sequencing
- NHEJ:
-
Nonhomologous end joining
- NK:
-
Natural killer cells
- Oct-1:
-
Octamer transcription factor-1
- OS:
-
Oxidative stress
- P53:
-
53 Kilodalton phosphoprotein
- PRB:
-
Retinoblastoma protein
- RARB:
-
Retinoic acid receptor, beta
- RASSF1:
-
Ras association domain-containing protein 1
- SiRNA:
-
Small interfering RNA
- TFs:
-
Transcription factors
- TIMP3:
-
Metalloproteinase inhibitor 3
- TLR:
-
Toll-like receptor
- TOPBP1:
-
DNA Topoisomerase-2 binding protein 1
- TWIST1:
-
Twist-related protein 1
- URR:
-
Upstream regulatory region
- VLP:
-
Virus-like particle
- WIF1:
-
Wnt inhibitory factor 1
- WNT:
-
Wingless-related integration site
- YY-1:
-
Yin Yang 1
References
Cervical cancer: estimated incidence mortality and prevalence worldwide in 2012. Globacan 2012. “http://globocan.iarc.fr/old/FactSheets/cancers/cervix-new.asp. Accessed 5 Dec 2015.
Howley PM, Lowry DR. Papillomaviruses. In: Knipe DM and Howley PM editors Fields virology. Philadelphia: Lippincott, Williams and Wilkins; 2007. p. 2299-354
Trottier H, Burchell AN. Epidemiology of mucosal human papillomavirus infection and associated diseases. Public Health Genomics. 2009;12:291–307.
Bouvard V, Baan R, Straif K, Grosse Y, Secretan B, et al. A review of human carcinogens–Part B: biological agents. Lancet Oncol. 2009;10:321–2.
Guan P, Howell-Jones R, Li N, Bruni L, et al. Human papillomavirus types in 115,789 HPV-positive women: A meta-analysis from cervical infection to cancer Int. J Cancer. 2012;131:2349–59.
Baldwin P, Laskey R, Coleman N. Translational approaches to improving cervical screening. Nat Rev Cancer. 2003;3:217–26.
Middleton K, Peh W, Southern S, et al. Organization of human papillomavirus productive cycle during neoplastic progression provide a basis for selection of diagnostic markers. J Virol. 2003;77:10186–201.
Liaw KL, Hildesheim A, Burk RD, et al. A prospective study of human papillomavirus (HPV) type 16 DNA detection by polymerase chain reaction and its association with acquisition and persistence of other HPV types. J Infect Dis. 2001;183:8–15.
Woodman CB, Collins S, Winter H, et al. Natural history of cervical human papillomavirus infection in young women: a longitudinal cohort study. Lancet. 2001;357:1831–6.
Stanley M. HPV-immune response to infection and vaccination. Infect Agent Cancer. 2010;5:19.
Mudhakir D, Harashima H. Learning from the viral journey: how to enter cells and how to overcome intracellular barriers to reach the nucleus. AAPS J. 2009;11:65–77.
Johnson KM, Kines RC, Roberts JN, Lowy DR, Schiller JT, Day PM. Role of heparan sulfate in attachment to and infection of the murine female genital tract by human papillomavirus. J Virol. 2009;83:2067–74.
De Marco F. Oxidative Stress and HPV Carcinogenesis. Viruses. 2013;2013(5):708–31.
Day PM, Lowy DR, Schiller JT. Heparan sulfate-independent cell binding and infection with furin-precleaved papillomavirus capsids. J Virol. 2008;82:12565–8.
Horvath CAJ, Boulet GAV, Renoux VM, Delvenne PO, Bogers J. Mechanisms of cell entry by human papillomaviruses: an overview Caroline AJ. Virol J. 2010;7:11.
Richards RM, Lowy DR, Schiller JT, Day PM. Cleavage of the papillomavirus minor capsid protein, L2, at a furin consensus site is necessary for infection. Proc Natl Acad Sci U S A. 2006;103:1522–7.
Sapp M, Day PM. Structure, attachment and entry of polyoma -and papillomaviruses. Virology. 2009;384:400–9.
Day PM, Lowy DR, Schiller JT. Papillomaviruses infect cells via a clathrindependent pathway. Virology. 2003;307:1–11.
Smith JL, Campos SK, Ozbun MA. Human papillomavirus type 31 uses a caveolin 1- and dynamin 2-mediated entry pathway for infection of human keratinocytes. J Virol. 2007;81:9922–31.
Pyeon D, Pearce SM, Lank SM, Ahlquist P, Lambert PF. Establishment of human papillomavirus infection requires cell cycle progression. PLoS Pathog. 2009;5:e1000318.
Day PM, Baker CC, Lowy DR, Schiller JT. Establishment of papillomavirus infection is enhanced by promyelocytic leukemia protein (PML) expression. Proc Natl Acad Sci U S A. 2004;101:14252–7.
Aydin I, Weber S, Snijder B, Samperio VP, Kuhbacher A, Becker M, Day PM, Schiller JT, Kann M, Pelkmans L, et al. Large scale RNAi reveals the requirement of nuclear envelope breakdown for nuclear import of human papillomaviruses. PLoS Pathog. 2014;10:e1004162.
Doorbar J, Griffin H. Intrabody strategies for the treatment of human papillomavirus-associated disease. Expert Opin Biol Ther. 2007;7:677–89.
McKinney CC. Hussmann KL and McBride A A. The Role of the DNA Damage Response throughout the Papillomavirus Life Cycle Viruses. 2015;7:2450–69.
Stanley M. Prophylactic HPV vaccines: prospects for eliminating ano-genital cancer. Br J Cancer. 2007;96(9):1320–3.
Cullen AP, Reid R, Campion M, et al. Analysis of the physical state of different human papillomavirus DNAs in intraepithelial and invasive cervical neoplasm. J Virol. 1991;65:606–12.
Pirami L, Giache V, Becciolini A. Analysis of HPV16, 18, 31 and 35 DNA in pre-invasive and invasive lesions of the uterine cervix. J Clin Pathol. 1997;50:600–4.
Badaracco G, Venuti A, Sedati A, et al. HPV16 and HPV18 in genital tumors: significantly different levels of viral integration and correlation to tumor invasiveness. J Med Virol. 2002;67:574–82.
Vinokurova S, Wentzensen N, Kraus I, et al. Type-dependent integration frequency of human papillomavirus genomes in cervical lesions. Cancer Res. 2008;68:307–13.
Kalantari M, Blennow E, Hagmar B, et al. Physical state of HPV16 and chromosomal mapping of the integrated form in cervical carcinomas. Diagn Mol Pathol. 2001;10:46–54.
Daniel B, Mukherjee G, Seshadri L, et al. Changes in the physical state and expression of human papillomavirus type 16 in the progression of cervical intraepithelial neoplasia lesions analysed by PCR. J Gen Virol. 1995;76:2589–93.
Peitsaro P, Johansson B, Syrjanen S. Integrated human papillomavirus type 16 is frequently found in cervical cancer precursors as demonstrated by a novel quantitative real-time PCR technique. J Clin Microbiol. 2002;40:886–91.
Hudelist G, Manavi M, Pischinger KI, et al. Physical state and expression of HPV DNA in benign and dysplastic cervical tissue: different levels of viral integration are correlated with lesion grade. Gynecol Oncol. 2004;92:873–80.
Kulmala SM, Syrjanen SM, Gyllensten UB, et al. Early integration of high copy HPV16 detectable in women with normal and low grade cervical cytology and histology. J Clin Pathol. 2006;59:513–7.
Dall KL, Scarpini CG, Roberts I, et al. Characterization of naturally occurring HPV16 integration sites isolated from cervical keratinocytes under noncompetitive conditions. Cancer Res. 2008;68:8249–59.
Wentzensen N, Vinokurova S, von Knebel Doeberitz M. Systematic review of genomic integration sites of human of the female lower genital tract. Cancer Res. 2004;64:3878–84.
Thorland EC, Myers SL, Gostout BS, Smith DI. Common fragile sites are preferential targets for HPV16 integrations in cervical tumors. Oncogene. 2003;22:1225–37.
Williams VM, Filippova M, Soto U, Duerksen-Hughes PJ. HPV-DNA integration and carcinogenesis: putative roles for inflammation and oxidative stress. Futur Virol. 2011;6:45–57.
Beckman KB, Ames BN. Oxidative decay of DNA. J Biol Chem. 1997;272:19633–6.
Kessis TD, Connolly DC, Hedrick L, Cho KR. Expression of HPV16 E6 or E7 increases integration of foreign DNA. Oncogene. 1996;13:427–31.
Yu T, Ferber MJ, Cheung TH, Chung TK, Wong YF, Smith DI. The role of viral integration in the development of cervical cancer. Cancer Genet Cytogenet. 2005;158:27–34.
Williams Vonetta M, Filippova M, Filippov V, Payne Kimberly J, Duerksen-Hughes P. Human Papillomavirus Type 16 E6* Induces Oxidative Stress and DNA Damage. J Virol. 2014;88(12):6751–61.
Sakakibara N, Mitra R, McBride AA. The papillomavirus E1 helicase activates a cellular DNA damage response in viral replication foci. J Virol. 2011;85:8981–95.
Longworth MS, Laimins LA. Pathogenesis of human papillomaviruses in differentiating epithelia. Microbiol Mol Biol Rev. 2004;68:362–72.
Klingelhutz AJ, Foster SA, McDougall JK. Telomerase activation by the E6 gene product of human papillomavirus type 16. Nature. 1996;380:79–82.
Bishop B, Dasgupta J, Chen XS. Structure-based engineering of papillomavirus major capsid L1: controlling particle assembly. Virol J. 2007; 4:3.
Spardy N, Covella K, Cha E, Elizabeth E. Hoskins, Susanne I. Wells et al. Human Papillomavirus 16 E7 Oncoprotein Attenuates DNA Damage Checkpoint Control by Increasing the Proteolytic Turnover of Claspin. Cancer Res. 2009;69(17):7022–9.
Longworth MS, Laimins LA. The binding of histone deacetylases and the integrity of zinc finger-like motifs of the E7 protein are essential for the life cycle of human papillomavirus type 31. Virology. 2004;78:3533–41.
DiMaio D, Liao JB. Human papillomaviruses and cervical cancer. Adv Virus Res. 2006;66:125–59.
Achanta G, Huang P. Role of p53 in sensing oxidative DNA damage in response to reactive oxygen species-generating agents. Cancer Res. 2004;64:6233–9.
Fradet-Turcotte A, Bergeron-Labrecque F, Moody CA, Lehoux M, Laimins LA, Archambault J. Nuclear accumulation of the papillomavirus E1 helicase blocks S-phase progression and triggers an ATM-dependent DNA damage response. J Virol. 2011;85:8996–9012.
Everett RD. Interactions between DNA viruses, ND10 and the DNA damage response. Cell Microbiol. 2006;8:365–74.
Reinson T, Toots M, Kadaja M. Pipitch, Allik M, et al. Ustava Engagement of the ATR-Dependent DNA Damage Response at the Human Papillomavirus 18 Replication Centers during the InitialAmplification. J Virol. 2013;87(2):951–64.
Edwards TG, Vidmar TJ, Koeller K, Bashkin JK, Fisher C. DNA damage repair genes controlling human papillomavirus (HPV) episome levels under conditions of stability and extreme instability. PLoS One. 2013;8:e75406.
Brimer N, Vande Pol SB. Papillomavirus E6 PDZ interactions can be replaced by repression of p53 to promote episomal human papillomavirus genome maintenance. J Virol. 2014;88:3027–30.
Jang MK, Shen K, McBride AA. Papillomavirus genomes associate with BRD4 to replicate at fragile sites in the host genome. PLoSPathog. 2014;10:e1004117.
Jang MK, Anderson DE, van Doorslaer K, McBride AA. A Proteomic approach to discover and compare interacting partners of Papillomavirus E2 proteins from diverse phylogenetic groups. Proteomics. 2015;15(12):2030–50.
Jeon S, Allen-Hoffmann BL, Lambert PF. Integration of human papillomavirus type 16 into the human genome correlates with a selective growth advantage of cells. J Virol. 1995;69:2989–97.
Pett M, Coleman N. Integration of high-risk human papillomavirus: a key event in cervical carcinogenesis? J Pathol. 2007;212:356–67.
Rika K-M, Tadahito K, Iwao K. Rolling circle replication of human papillomavirus type 16 DNA in epithelial cell extracts. Genes Cells. 2011;16:23–33.
Li X, Heyer WD. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008;18(1):99–113.
Kenric G, Mehta Kavi P, Laimins LA, Moodya Cary A. Human Papillomaviruses Recruit Cellular DNA Repair and Homologous Recombination Factors to Viral Replication Centers. J Virol. 2012;86(17):9520–6.
Sakakibara N, Chen D, McBride AA. Papillomaviruses Use Recombination-Dependent Replication to Vegetatively Amplify Their Genomes in Differentiated Cells. PLoS Pathog. 2013;9(7):e1003321.
Ziegert C, Wentzensen N, Vinokurova S, Kisseljov F, Einenkel J, Hoeckel M, et al. A comprehensive analysis of HPV integration loci in anogenital lesions combining transcript and genome-basedamplification techniques. Oncogene. 2003;22:3977–84.
Hu Z, Zhu D, Wang W, Li W, Jia W, Zeng X, et al. Genome-wide profiling of HPV integration in cervical cancer identifies clustered genomic hot spots and a potential microhomology-mediated integration mechanism. Nat Genet. 2015;47:158–63.
Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–8.
Thierry F, Howley PM. Functional analysis of E2-mediated repression of the HPV18 P1o5 promoter. New Biologist. 1991;3:90–100.
Matsukura T, Kanda T, Furuno A, Yoshikawa H, Kawana T, Yoshiike K. Cloning of monomeric human papillomavirus type 16 DNA integrated within cell DNA from a cervical carcinoma. J Virol. 1986;58:979–82.
Tonon SA, Picconi MA, Bos PD, Zinovich JB, Galuppo J, Alonio LV, et al. Physical status of the E2 human papilloma virus 16 viral gene in cervical preneoplastic and neoplastic lesions. J Clin Virol. 2001;21:129–34.
Hopman AH, Smedts F, Dignef W, Ummelen M, Sonke G, Mravunac M, et al. Transition of high-grade cervical intraepithelialneoplasia to micro-invasive carcinoma is characterized by integration of HPV 16/18 and numerical chromosome abnormalities. J Pathol. 2004;202:23–33.
Boccardo E, Lepique Ana P, Villa LL. The role of inflammation in HPV carcinogenesis. Carcinogenesis. 2010;31(11):1905–12.
Akagi K, Jingfeng L, Tatevik R, Broutian HP-N, et al. Genome-wide analysis of HPV integration in human cancers reveals recurrent, focal genomic instability. Genome Res. 2014;24:185–99.
Ojesina AI, Lichtenstein L, Freeman SS, Pedamallu CS, et al. Landscape of genomic alterations in cervical carcinomas. Nature. 2014;506(7488):371–5.
Peebles KA, Lee JM, Mao JT, Hazra S, Reckamp KL, Krysan K, Dohadwala M, Heinrich EL, Walser TC, Cui X, et al. Inflammation and lung carcinogenesis: Applying findings in prevention and treatment. Expert Rev Anticancer Ther. 2007;7:1405–21.
Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–7.
Pett M et al. Acquisition of high-level chromosomal instability is associated with integration of human papillomavirus type 16 in cervical keratinocytes. Cancer Res. 2004;64(4):1359-68.
Pett M, et al. Selection of cervical keratinocytes containing integrated HPV16 associates with episome loss and an endogenous antiviral response. Proc Natl Acad Sci U S A. 2006;103:3822–7.
Barnard P, et al. The human papillomavirus E7 oncoprotein abrogates signaling mediated by interferon-alpha. Virology. 1999;259:305–13.
Barnard P, et al. The human papillomavirus E7 protein is able to inhibit the antiviral and anti-growth functions of interferon-alpha. Virology. 2000;277:411–9.
Cordano P, et al. The E6E7 oncoproteins of cutaneous human papillomavirus type 38 interfere with the interferon pathway. Virology. 2008;377:408–18.
Hasan UA, Bates E, Takeshita F, et al. TLR9 expression and function is abolished by the cervical cancer-associated human papillomavirus type 16. J Immunol. 2007;178(5):3186–97.
Filippova M, et al. The human papillomavirus 16 E6 protein can either protect or further sensitize cells to TNF: effect of dose. Cell Death Differ. 2005;12:1622–35.
James MA, et al. Human papillomavirus type 16 E6 activates NFkappaB, induces cIAP-2 expression, and protects against apoptosis in a PDZ binding motif-dependent manner. J Virol. 2006;80:5301–7.
Boccardo E, et al. HPV-18 confers resistance to TNF-alpha in organotypic cultures of human keratinocytes. Virology. 2004;328:233–43.
Butterfield DA. Keller JN Antioxidants and antioxidant treatment in disease. Biochem Biophys Acta. 1822;2012:615.
Butterfield DA. Oxidative stress in neurodegenerative disorders. Antioxid Redox Signal. 2006;2006(8):1971–3.
De Marco F, Bucaj E, Foppoli C, Fiorini A, Blarzino C, Filipi K, Giorgi A, Schininà ME, et al. Oxidative stress in HPV-driven viral carcinogenesis: redox proteomics analysis of HPV-16 dysplastic and neoplastic tissues. PLoS One. 2012;7:e34366.
Dong XP, Stubenrauch F, Beyer-Finkler E, et al. Prevalence of deletions of YY1-binding sites in episomal HPV 16 DNA from cervical cancers. Int J Cancer. 1994;58:803–8.
May M, Dong XP, Beyer-Finkler E, et al. The E6/E7 promoter of extrachromosomal HPV16 DNA in cervical cancers escapes from cellular repression by mutation of target sequences for YY1. EMBO J. 1994;13:1460–6.
Veress G, Szarka K, Dong XP, et al. Functional significance of sequence variation in the E2 gene and the long control region of human papillomavirus type 16. J Gen Virol. 1999;80:1035–43.
Lace MJ, Isacson C, Anson JR, et al. Upstream regulatory region alterations found in human papillomavirus type 16 (HPV-16) isolates from cervical carcinomas increase transcription, ori function, and HPV immortalization capacity in culture. J Virol. 2009;83:7457–66.
Brandsma JL, Sun Y, Lizardi PM, Tuck DP, Zelterman D, Haines GK, et al. Distinct human papillomavirus type 16 methylomes in cervical cells at different stages of premalignancy. Virology. 2009;389(1–2):100–7.
Fernandez AF, Rosales C, Lopez-Nieva P, Grana O, Ballestar E, Ropero S, et al. The dynamic DNA methylomes of double-stranded DNA viruses associated with human cancer. Genome Res. 2009;19(3):438–51.
Wentzensen N, Sun C, Ghosh A, Kinney W, Mirabello L, Wacholder S, et al. Methylation of HPV18, HPV31, and HPV45 genomes and cervical intraepithelial neoplasia grade 3. J Natl Cancer Inst. 2012;104(22):1738-49.
Hong D, Ye F, Lu W, et al. Methylation status of the long control region of HPV 16 in clinical cervical specimens. Mol Med Rep. 2008;1:555–60.
Ding DC, Chiang MH, Lai HC, et al. Methylation of the long control region of HPV16 is related to the severity of cervical neoplasia. Eur J Obstet Gynecol Reprod Biol. 2009;147:215–20.
Kondo Y, Shen L, Cheng AS, Ahmed S, Boumber Y, Charo C, Yamochi T, et al. Gene silencing in cancer by histone H3 lysine 27 trimethylation independent of promoter DNA methylation. Nat Genet. 2008;40:741–50.
Hiragami-Hamada K, Xie SQ, Saveliev A, Uribe-Lewis S, et al. The molecular basis for stability of heterochromatin-mediated silencing in mammals. Epigenetics Chromatin. 2009;2:14.
Gray E, Pett MR, Ward D, et al. In vitro progression of human papillomavirus 16 episome-associated cervical neoplasia displays fundamental similarities to integrant-associated carcinogenesis. Cancer Res. 2010;70:4081–91.
Scarpini CG, Groves IJ, Pett MR, et al. Virus transcript levels and cell growth rates after naturally occurring HPV16 integration events in basal cervical keratinocytes. J Pathol. 2014;2014(233):281–93.
Shivapurkar N, Toyooka S, Toyooka KO, Reddy J, Miyajima K, Suzuki M, Shigematsu H, Takahashi T, et al. Aberrant methylation of trail decoy receptor genes is frequent in multiple tumor types. Int J Cancer. 2004;109:786–92.
Dueñas-González A, Lizano M, Candelaria M, Cetina L, et al. Epigenetics of cervical cancer. An overview and therapeutic Perspectives Molecular Cancer. 2005;4:38.
Widschwendter A, Gattringer C, Ivarsson L, Fiegl H, Schneitter A, Ramoni A, et al. Analysis of aberrant DNA methylation and human papillomavirus DNA in cervicovaginal specimens to detect invasive cervical cancer and its precursors. Clin Cancer Res. 2004;10:3396–400.
Widschwendter A, Muller HM, Hubalek MM, Wiedemair A, Fiegl H, Goebel G, Mueller-Holzner E, Marth C, Widschwendter M. Methylation status and expression of human telomerase reverse transcriptase in ovarian and cervical cancer. Gynecol Oncol. 2004;93:407–16.
Yu MY, Tong JH, Chan PK, Lee TL, Chan MW, Chan AW, Lo KW, To KF. Hypermethylation of the tumor suppressor gene RASSFIA and frequent concomitant loss of heterozygosity at 3p21 in cervical cancers. Int J Cancer. 2003;105:204–9.
Cohen Y, Singer G, Lavie O, Dong SM, Beller U, Sidransky D. The RASSF1A tumor suppressor gene is commonly inactivated in adenocarcinoma of the uterine cervix. Clin Cancer Res. 2003;2003(9):2981–4.
Overmeer RM, Henken FE, Snijders PJ, et al. Association between dense CADM1 promoter methylation and reduced protein expression in high鄄gr ade CIN and cervical SCC. J Pathol. 2008;215:388–97.
Van der Meide WF, Snellenberg S, Meijer CJ, Baalbergen A, Helmerhorst TJ, van der Sluis WB, et al. Promoter methylation analysis of WNT/beta-catenin signaling pathway regulators to detect adenocarcinoma or its precursor lesion of the cervix. Gynecol Oncol. 2011;123:116–22.
Yang N, Eijsink JJ, Lendvai A, Volders HH, Klip H, Buikema HJ, et al. Methylation markers for CCNA1 and C13ORF18 are strongly associated with high-grade cervical intraepithelial neoplasia and cervical cancer in cervical scrapings. Cancer Epidemiol Biomarkers Prev. 2009;18:3000–7.
Yang N, Nijhuis ER, Volders HH, Eijsink JJ, Lendvai A, Zhang B, et al. Gene promoter methylation patterns throughout the process of cervical carcinogenesis. Cell Oncol. 2010;32:131–43.
Kim JH, Choi YD, Lee JS, Lee JH, Nam JH, Choi C. Assessment of DNA methylation for the detection of cervical neoplasia in liquid-based cytology specimens. Gynecol Oncol. 2010;116:99–104.
Milutin Gašperov N, Sabol I, Planinić P, Grubišić G, Fistonić I, Ćorušić A, et al. Methylated Host Cell Gene Promoters and Human Papillomavirus Type 16 and 18 Predicting Cervical Lesions and Cancer. PLoS ONE. 2015;10(6): e0129452.
Clarke Megan A, Nicolas W, Lisa M, Arpita G, Wacholder S, et al. December. Human Papillomavirus DNA Methylation as a Potential Biomarker for Cervical Cancer. Cancer Epidemiol Biomarkers Prev. 2012;21(12):2125–37.
Parfenova M, Pedamallub CS, Gehlenborgc N, Freeman SS, et al. Characterization of HPV and host genome interactions in primary head and neck cancers. PNAS. 2014;111(43):15544–9.
Wilting SM, Snijders PJ, Verlaat W, et al. AlteredmicroRNA expression associated with chromosomal changes contributes to BioMed Research International 11 cervical carcinogenesis. Oncogene. 2013;32(1):106–16.
Wang X, Tang S, Le S-Y, Lu R, Rader JS, et al. Aberrant Expression of Oncogenic and Tumor-Suppressive MicroRNAs in Cervical Cancer Is Required for Cancer Cell Growth. PLoS ONE 2008;3(7):e2557.
van de Putte G, Holm R, Lie AK. Trop´e CG, and Kristensen GB Expression of p27, p21, and p16 protein in early squamous cervical cancer and its relation to prognosis. Gynecol Oncol. 2003;89(1):140–7.
McBee Jr. WC, Gardiner AS, Edwards RP et al. MicroRNA analysis in human papillomavirus (HPV)-associated cervical neoplasia and cancer. J Carcinogene Mutagene. 2011;1:114 doi: 10.4172/2157-2518.1000114.
Melo SA, Esteller M. Dysregulation of microRNAs in cancer: playing with fire. FEBS Lett. 2011;585(13):2087–99.
Long MJ, Wu FX, Li P, Liu M, Li X, Tang H. MicroRNA-10a targets CHL1 and promotes cell growth, migration and invasion in human cervical cancer cells. Cancer Lett. 2012;324(2):186–96.
Budhu A. Ji J, and Wang XW (2010) The clinical potential of microRNAs. J Hematol Oncol. 2010;3:37.
Li JH, Xiao X, Zhang YN, et al. MicroRNA miR-886-5p inhibits apoptosis by down-regulating Bax expression in human cervical carcinoma cells. Gynecol Oncol. 2011;120(1):145–51.
Chang TC, Wentzel EA, Kent OA, et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell. 2007;26(5):745–52.
Wang X, Wang HK, Mccoy JP, et al. Oncogenic HPV infection interrupts the expression of tumor-suppressive miR-34a through viral oncoprotein E6. RNA. 2009;15(4):637–47.
Díaz-González Sacnite del Mar, Deas Jessica, Benítez-Boijseauneau Odelia, Gómez-Cerón Claudia, et al. Utility of MicroRNAs and siRNAs in Cervical Carcinogenesis. BioMed Res Int. 2015;374924:13.
Pereira PM, Marques JP, Soares A, Carreto RL, Santos MAS. Microrna expression variability in human cervical tissues. PLoS ONE. 2010;5(7):e11780.
Yamada T, Manos MM, Peto J, et al. Human papillomavirus type 16 sequence variation in cervical cancers: a worldwide perspective. J Virol. 1997;71(3):2463–72.
Zuna RE, Tuller E, Wentzensen N et al. HPV16 variant lineage, clinical stage, and survival in women with invasive cervical cancer. Infect Agents Cancer. 2011;6:19.
Burk RD, Harari A, Chen Z. Human papillomavirus genome variants. Virology. 2013;445:232–43.
Berumen J, Ordoñez RM, Lazcano E, Salmerón J, Galván SC, Estrada RA, et al. Asian-American variants of human papillomavirus 16 and risk for cervical cancer: a case–control study. J Natl Cancer Inst. 2001;93(17):1325–30.
Tornesello ML, Losito S, Benincasa G, Fulciniti F, Botti G, Greggi S, et al. Human papillomavirus (HPV) genotypes and HPV16 variants and risk of adenocarcinoma and squamous cell carcinoma of the cervix. Gynecol Oncol. 2011;121(1):32–42.
López-Revilla R, Pineda MA, Ortíz-Valdez J, Sánchez-Garza M, Riego L. Human papillomavirus type 16 variants in cervical intraepithelial neoplasia and invasive carcinoma in San Luis Potosi City, Mexico. Infect Agents Cancer. 2009;4:3.
Casas L, Galván SC, Ordoñez RM, López N, Guido M, Berumen J. Asianamerican variants of human papillomavirus type 16 have extensive mutations in the E2 gene and are highly amplified in cervical carcinomas. Int J Cancer. 1999;83(4):449–55.
Niccoli S, Abraham S, Richard C, Zehbe I. The Asian-American E6 variant protein of human papillomavirus 16 alone is sufficient to promote immortalization, transformation, and migration of primary human foreskin keratinocytes. J Virol. 2012;86:12384–96.
Richard C, Lanner C, Naryzhny S, Sherman L, Lee H, et al. The immortalizing and transforming ability of two common human papillomavirus 16 E6 variants with different prevalences in cervical cancer. Oncogene. 2010;29:3435–45.
Jackson R, Togtema M, Lambert PF. Zehbe I (2014) Tumourigenesis Driven by the Human Papillomavirus Type 16 Asian-American E6 Variant in a Three-Dimensional Keratinocyte Model. PLoS One. 2014;9(7):e101540.
Xi LF, Koutsky LA, Hildesheim A, Galloway DA, Wheeler CM, Winer RL, Ho J, Kivia NB. Risk for high-grade cervical intraepithelial neoplasia associated with variants of humanpapilloma virus types16 and18. Cancer Epidemiol Biomarkers Prev. 2007;2007(16):4–10.
Arias-Pulido H, Peyton CL, TorrezMartinez N, Anderson DN, Wheeler CM. Human papilloma virus type18variant lineages inUnitedStatespopula- tions characterized by sequence analysis ofLCR-E6,E2,andL1regions. Virology. 2005;338:22–34.
Schiffman M, Rodriguez AC, Chen Z, et al. A populationbased prospective study of carcinogenic human papillomavirus variant lineages, viral persistence, and cervical neoplasia. Cancer Res. 2010;70(8):3159–69.
Xi LF, Schiffman M, Koutsky LA, He Z, Winer RL, Hulbert A, et al. Persistence of newly detected humanpapillomavirus type 31 infection, stratified by variant lineage. Int J Cancer. 2013;2013(132):549–55.
Xi LF, Schiffman M, Koutsky LA, Hulbert A, Lee SK, DefilippV SZ, Kiviat NB. Association of humanpapillomavirus type31 variants with risk ofcervical intraepithelial neoplasia grades2–3. Int J Cancer. 2012;2012(131):2300–7.
Chang YJ, Chen HC, Lee BH, You SL, et al. Unique variants ofhuman papillomavirus genotypes 52 and 58 and risk of cervical neoplasia. Int J Cancer. 2011;129:965–73.
Acknowledgment
Special thanks to Ms. Itishree Parija, for her help in the preparation of the artwork of the manuscript.
Funding
Author RRS is a recipient of ICMR senior research fellowship, India (Award no-80/780/2012-ECD-I). The funding source was not involved in the preparation of the manuscript, study design or the decision to submit the article for publication.
Competing interest
The authors declare that they have no competing interests.
Authors’ contributions
1. RRS- Conceived and designed the study, collected the literature, performed interpretation and analysis of data, drafted the manuscript. 3. NNS-Participated in interpretations and analysis of data, critically revised the manuscript. 2. BD-Participated in interpretations and analysis of data, critically revised the manuscript. All authors read and approved the final manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Senapati, R., Senapati, N.N. & Dwibedi, B. Molecular mechanisms of HPV mediated neoplastic progression. Infect Agents Cancer 11, 59 (2016). https://doi.org/10.1186/s13027-016-0107-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13027-016-0107-4