
Abstract
Background
Astrocytes play a crucial, yet not fully elucidated role in the selective motor neuron pathology in amyotrophic lateral sclerosis (ALS). Among other responsibilities, astrocytes provide important neuronal homeostatic support, however this function is highly compromised in ALS. The establishment of fully human coculture systems can be used to further study the underlying mechanisms of the dysfunctional intercellular interplay, and has the potential to provide a platform for revealing novel therapeutic entry points.
Methods
In this study, we characterised human induced pluripotent stem cell (hiPSC)-derived astrocytes from FUS-ALS patients, and incorporated these cells into a human motor unit microfluidics model to investigate the astrocytic effect on hiPSC-derived motor neuron network and functional neuromuscular junctions (NMJs) using immunocytochemistry and live-cell recordings. FUS-ALS cocultures were systematically compared to their CRISPR-Cas9 gene-edited isogenic control systems.
Results
We observed a dysregulation of astrocyte homeostasis, which resulted in a FUS-ALS-mediated increase in reactivity and secretion of inflammatory cytokines. Upon coculture with motor neurons and myotubes, we detected a cytotoxic effect on motor neuron-neurite outgrowth, NMJ formation and functionality, which was improved or fully rescued by isogenic control astrocytes. We demonstrate that ALS astrocytes have both a gain-of-toxicity and loss-of-support function involving the WNT/β-catenin pathway, ultimately contributing to the disruption of motor neuron homeostasis, intercellular networks and NMJs.
Conclusions
Our findings shine light on a complex, yet highly important role of astrocytes in ALS, and provides further insight in to their pathological mechanisms.
Graphical Abstract

Similar content being viewed by others


Background
Amyotrophic lateral sclerosis (ALS) is a progressive motor neuron disorder in which the upper motor neurons in the motor cortex as well as the lower motor neurons in the brainstem and the ventral horn of the spinal cord undergo selective and progressive cell death. Symptoms include muscle weakness, spasticity, hyperreflexia, fasciculations, and muscle atrophy. The rapid progression of the disease limits median survival after symptom onset to 2 to 5 years mostly due to respiratory failure [1], and currently no effective treatment is available. In 90% of cases, ALS is a sporadic disease (sALS) clinically indistinguishable from inherited familial forms, but with an unknown aetiology [2]. The remaining 10% covers familial ALS (fALS) cases where mutations in the FUS RNA binding protein (FUS) gene are the fourth most prevalent cause of fALS in the Western world (2.8%), and the second most common form in the Asian ALS population (6.4%) [3]. In addition, FUS mutations cause the majority of juvenile and paediatric cases of ALS [4]. Interestingly, nucleus-to-cytoplasmic mislocalisation of FUS protein, which is a hallmark of FUS mutant pathology, has also been documented in sALS cases [5], and FUS-positive inclusions are present in approximately 10% of frontotemporal lobar degeneration (FTLD) cases despite the lack of genetic mutations in the FUS gene [6]. This suggests that FUS-mediated pathology might have a more general function in neurodegenerative disorders.
Despite the selective degeneration of motor neurons, several studies demonstrate an important role for glial cells in ALS [7,8,9,10]. Astrocyte reactivity is a prominent hallmark in ALS patients [7, 11, 12], and the number of reactive astrocytes has been shown to correlate with disease progression [13]. Normally, astrocytes are the key players in maintaining homeostasis and optimal neuronal function through synaptic activity and plasticity modulation, inter-neuronal crosstalk, nutrition distribution, waste removal and structural support [14]. However, studies in ALS show that astrocytes promote motor neuron toxicity through reduced glutamate uptake [15,16,17], reduced lactate production and shuttling [18, 19] and increased secretion of toxic factors such as tumour necrosis factor α (TNF-α) and reactive oxygen species [20,21,22,23,24]. Despite this insight, many aspects of the astrocytes’ pathologic mechanism in ALS are not fully known.
In this study, we aimed to unravel the role of hiPSC-derived astrocytes in the context of FUS-ALS and include these cells in our previously established human motor unit microfluidics model [25, 26] to evaluate the astrocytic effect in a novel multicellular system. We discovered that FUS-ALS astrocytes showed increased reactivity and secretion of inflammatory cytokines. Once incorporated into the motor unit system, the mutant astrocytes impaired the neurite outgrowth as well as NMJ formation and functionality. Our data establish a mutant FUS-mediated dysregulation of astrocytes, which causes a synergistic gain-of-toxicity and loss-of-support functionality. We propose an auto-regulatory role, in which astrocytes unsuccessfully attempt to counteract this toxicity through secretion of anti-inflammatory cytokines and upregulation of the WNT/β-catenin pathway in motor neurons.
Methods
Cell lines and reagents
Two heterozygous FUS-mutant hiPSC lines from a 17-year-old male ALS patient with a de novo mutation (P525L) and a 71-year-old female ALS patient (R521H) were each compared to their corresponding CRISPR-Cas9 gene-edited isogenic control hiPSC line (P525P and R521R) [25,26,27,28]. The isogenic control hiPSC lines were made by CellSystems (Troisdorf, Germany). hiPSC lines were cultured using Complete Essential 8™ medium (Cat N° A1517001) with 1% penicillin/streptomycin (Cat N° 15070063) on Geltrex®- (Cat N° A1413301) coated plates. Human myoblasts were harvested from biopsies obtained from a healthy 60-year-old male donor via the Human Body Donation program of KU Leuven (KULAK). All cells were routinely tested for mycoplasma contamination with MycoAlert Mycoplasma Detection Kit (Lonza, Rockland, ME, USA, Cat N° LT07-318). Chemicals and reagents used for cell culture were purchased from Thermo Fisher Scientific (Waltham, MA, USA) unless otherwise specified (see Supplementary Table 1, Additional file 1).
Generation of hiPSC-derived astrocytes
hiPSC-derived astrocytes were generated using a slightly modified version of a recently published protocol [29]. In brief, hiPSCs were dissociated with collagenase type IV (Cat N° 10780004), and the cell clusters were collected in Corning® ultra-low attachment flasks (Sigma-Aldrich, St. Louis, MO, USA, Cat N° 734–4140) to support embryoid body (EB) formation. EBs were kept in neuronal induction medium (NIM) consisting of Complete Essential 8™ medium with 1% penicillin/streptomycin supplemented with 0.1 μM LDN-193189 (Stemgent, Beltsville, MA, USA, Cat N° 04–0074-02) and 10 μM SB431542 (Tocris Bioscience, Bristol, UK, Cat N° 1614) for the first week of differentiation. Media changes were performed on days 1, 2 and 4. On day 7, the EBs were plated on Geltrex®-coated plates for neural rosette formation and subsequent neural progenitor cell (NPC) expansion in neuronal maturation medium (NMM) containing 50% DMEM/F12 (Cat N° 11330032) and 50% Neurobasal medium (Cat N° 21103049) with 1% L-glutamine (Cat N° 25030–024), 1% penicillin/streptomycin, 1% N-2 supplement (Cat N° 17502–048) and 2% B-27™ without vitamin A (Cat N° 12587–010) supplemented with 10 ng/ml recombinant murine fibroblast growth factor (FGF)-2 (PeproTech, Rocky Hill, NJ, USA, Cat N° 450–33), 10 ng/ml recombinant human epidermal growth factor (EGF) (ProSpec, Rotherham, UK, Cat N° CYT-217), 0.1 μM LDN-193189 and 10 μM SB431542. The medium was changed daily and NPCs were passaged a few times using accutase (Sigma-Aldrich, Cat N° A6964). On day 16, NPCs were cultured until day 25 in astrocyte differentiation medium (ADM) made of 90% Neurobasal medium, 1% penicillin/streptomycin, 1% N-2 supplement, 1% non-essential amino acids (Cat N° 11140050) and 0.8 μM ascorbic acid (Sigma-Aldrich, Cat N° A4403) supplemented with 10 ng/ml FGF-2, 200 ng/ml recombinant human insulin-like growth factor (IGF)-1 (Peprotech, Cat N° 100–11), 10 ng/ml human Activin A (Cat N° PHC9564) and 10 ng/ml recombinant human Heregulinβ1 (Peprotech, Cat N° 100–03) to convert NPCs to astrocyte progenitor cells (APCs). Medium changes were made every second day during the period. On day 25 (d25/d+0), a glial switch is expected to occur, which commences the astrocyte maturation period. APCs were plated for final experiments or for expansion on new Geltrex®-coated plates and matured for an additional 4 weeks in astrocyte maturation medium (AMM) consisting of 50% DMEM/F12, 50% Neurobasal medium, 1% non-essential amino acids, 1% N-2 supplement, 1% L-glutamine, 1% penicillin/streptomycin, 2% fetal bovine serum (FBS) (Cat N° 10270106), 0.8 μM ascorbic acid, and 1% sodium pyruvate (Cat N° 11360–070) supplemented with 200 ng/ml IGF-1, 10 ng/ml Activin A and 10 ng/ml Heregulinβ1. The medium was changed every second day and maturing astrocytes were cryopreserved in FBS and 10% DMSO (Sigma-Aldrich, Cat N° D2650-100ML) at 2 weeks of maturation for later use. For WB (western blot), qPCR, RNA-sequencing (RNAseq) and secretome experiments, hiPSC-derived APCs were plated at d25/d+0 at 50 000 cells/cm2 in 6-well plates (Cellstar Greiner bio-one, Kremsmünster, Austria, Cat N° 657160) and allowed to mature for 1–4 weeks without passages.
Generation of hiPSC-derived motor neurons
hiPSC-derived motor neurons were generated using a previously published protocol [25,26,27], which is a modified version of the Maury et al. protocol [30]. Briefly, hiPSCs were dissociated with collagenase IV and the cell clusters were transferred to Corning® ultra-low attachment flasks to promote EB formation. EBs were cultured in neuronal medium consisting of 50% DMEM/F12, 50% Neurobasal medium, 1% penicillin/streptomycin, 0.5% L-glutamine, 1% N-2 supplement, 2% B-27™ without vitamin A, 0.5 μM ascorbic acid and 0.1% β-mercaptoethanol (Cat N° 31350010). On day 0 and 1 of differentiation, EBs received neuronal medium with 5 μM Y-27632 (Merck Millipore, Burlington, MA, USA, Cat N° 688001), 0.2 μM LDN-193189, 40 μM SB431542 and 3 μM CHIR99021 (Tocris Bioscience, Cat N° 4423). On days 2 and 4, the EB medium consisted of neuronal medium supplemented with 0.1 μM retinoic acid (Sigma-Aldrich, Cat N° R2625) and 500 nM smoothened agonist (Merck Millipore, Cat N° 566660). On day 7, the EB medium was changed to neuronal medium supplemented with 10 ng/ml brain-derived neurotrophic factor (BDNF) (PeproTech, Cat N° 450–02) and 10 ng/ml glial cell line-derived neurotrophic factor (GDNF) (PeproTech, Cat N° 450–10) in addition to the 0.1 μM retinoic acid and 500 nM smoothened agonist. On day 9, 10 μM DAPT (Tocris Bioscience, Cat N° 2634) was added to the fresh day 7 medium. On day 10, EBs were dissociated into NPCs using 0.05% trypsin (Gibco, Gaithersburg, MA, USA, Cat N° 25300054). NPCs were cryopreserved in knockout serum replacement (Cat N° 10828–028) with 10% DMSO or plated in day 9 medium with 1% RevitaCell™ (Cat N° A2644501) for further experiments. On day 11 of the differentiation, additional day 9 medium was added to the wells. On day 14, cells received neuronal medium supplemented with 10 ng/ml BDNF, 10 ng/ml GDNF and 20 μM DAPT. On day 16, the differentiating motor neurons received 10 ng/ml BDNF, GDNF and ciliary neurotrophic factor (CNTF) (PeproTech, Cat N° 450–13) in addition to 20 μM DAPT. From day 18 on, motor neurons were cultured in neuronal medium with 10 ng/ml BDNF, GDNF and CNTF. The medium was changed every other day until terminal differentiation.
Human myoblasts and myotube differentiation
Human skeletal muscle cells were isolated from a human vastus lateralis muscle biopsy as described before [31]. Briefly, isolated tissue was cut in strips of approximately 2 mm × 10 mm using sterile forceps and scalpel after removing excess connective tissue and fat. Muscle strips were pinned under tension in a Sylgard silicone- (Dow Corning, Midland, MICH, USA, Cat N° 1060040) coated 6-well plate. Two days after pinning the strips, enzymatic digestion was performed by incubating the muscle strips at 37 °C for 1 h in DMEM, high glucose with pyruvate (Cat N° 31966021) supplemented with 0.1% collagenase, type II (Worthington Biochemical Corp, Lakewood, NJ, USA, Cat N° LS004176) and 4 mg/ml dispase II (Roche Diagnostics, Basel, Switzerland, Cat N° 4942078001). After the incubation, cells were collected by filtering through a 100 μm cell strainer (Corning, NY, USA, Cat N° 7340004) and fragments were incubated again to digest the whole tissue. Isolated myoblasts were pooled, centrifuged for 5 min at 200 × g and resuspended in skeletal muscle growth medium consisting of DMEM, high glucose with pyruvate, 10% FBS, 50 µg/ml gentamicin (Cat N° 15750037), and 1% Ultroser solution (Pall Corporation, NY, USA, Cat N° 15950–017). Cells were split at 60–70% confluence and used in experiments before reaching 12 doublings. Myoblasts were differentiated into multinucleated myotubes by changing the medium to DMEM, high glucose with pyruvate, 10 ng/mL recombinant human EGF (Peprotech, Cat N° AF-100–15), 10 µg/ml insulin (Sigma-Aldrich, Cat N° I9278), 50 µg/ml Gentamicin and 50 µg/mL bovine serum albumin (BSA) (Sigma-Aldrich, Cat N° A2153).
Coculturing in microfluidic devices
hiPSC-derived motor neurons and astrocytes were cocultured in microfluidic devices with human primary myoblasts using a slightly modified version of a recently published protocol [26]. On day 10 of the coculture timeline, 250 000 motor neuron-NPCs were plated in a compartment on one side of the microgrooves of the device, while 40 000 myoblasts were plated in the opposite compartment. Motor neuron-NPCs were further differentiated following the standard protocol, while myoblasts were allowed to proliferate until day 14, where the differentiation into myotubes was initiated. On day 18, a volumetric and chemotactic gradient of 10 ng/ml BDNF, GDNF and CNTF in addition to 0.01 μg/ml recombinant human agrin protein (R&D Systems, Cat N° 6624-AG-050) and 20 μg/ml laminin (Sigma-Aldrich, Cat N° L2020-1MG) was implemented to promote motor neuron neurite migration through the microgrooves towards the myotube compartment. The myotube compartment received 200 μl/well motor neuron media with growth factors, while the motor neuron compartment received 100 μl/well without any supplements. The gradient was sustained at every media change until the end of the experiment. On day 21, 250 000 week 3.5 (d + 24) mature astrocytes were plated on top of the maturing motor neurons in the motor neuron compartment in motor neuron medium and the coculture was continued for another 1–2 weeks. Motor unit cultures without astrocytes were kept in parallel as controls. Preassembled microfluidic devices (Xona™ Microfluidics, Temecula, CA, USA, Cat N° XC150 (microgroove length: 150 μm)) were used for immunocytochemistry (ICC) and live-cell calcium recordings of NMJ functionality, while silicone microfluidic devices (Xona™ Microfluidics, Cat N° SND75 (microgroove length: 75 μm)) mounted on Aclar 33C sheets (Electron Microscopy Sciences, Hatfield, PA, USA, Cat N° 50425–25) were used for scanning electron microscopy (SEM).
Bright-field imaging
Bright-field images of astrocyte differentiation were taken with a Nikon Eclipse Ts2 microscope with a DMK 33UX250 camera and NIS-Elements D 5.01.00 software.
Immunocytochemistry (ICC)
ICC analyses of NMJ formation and neurite outgrowth were performed in XC150 microfluidic devices, while remaining stainings were imaged on 13 mm #1.5 coverslips (VWR, Monroeville, PA, USA, Cat N° 631-0150P) in 24-well plates (Greiner bio-one cellstar, Vilvoorde, Belgium, Cat N° 662160) using a previously described method [25, 26]. Briefly, cells were fixed using 4% paraformaldehyde (Cat N° 28908) in DPBS (Cat N° 14190250) for 15–20 min, permeabilized with 0.1% Triton X-100 (Sigma-Aldrich, Cat N° T8787) in DPBS for 20 min, and subsequently incubated with 5% normal donkey serum (NDS) (Sigma-Aldrich, Cat N° D9663) in 0.1% Triton X-100/DPBS for 30 min. Overnight incubation with primary antibodies (see Supplementary Table 1, Additional file 1) diluted in 2% NDS in 0.1% Triton X-100/DPBS was performed at 4 °C. The following day, cells were washed with DPBS and incubated with secondary antibodies in 2% NDS in 0.1% Triton X-100/DPBS for 1 h. Afterwards, nuclei were labelled with DAPI (NucBlue Live Cell Stain ReadyProbes reagent, Cat N° R37605) for 20 min. Coverslips were mounted and devices sealed with Fluorescence Mounting Medium (Dako, Glostrup, Denmark, Cat N° S3023), and images were acquired in a 1024 × 1024 format using an inverted Leica Sp8 DMI8 confocal microscope equipped with a HC PL APO CS2 10x/0.40, 20x/0.75 or HC PL APO CS 40x/0.85 dry objective lens, a Nikon A1R TiE confocal microscope equipped with a Plan Fluor 40X oil DIC objective lens or a Nikon C2 TiE confocal microscope equipped with a Plan Apo 40X oil DIC H objective lens. Analyses were performed utilizing ImageJ 1.52p and NIS-Elements AR 4.30.02 software.
hiPSC differentiation and myotube formation analysis
hiPSC-derived motor neuron-NPCs were plated at 25 000 cells/cm2 and differentiated until day 28, while hiPSC-derived astrocytes were plated at 50 000 cells/cm2 two days before fixation and analysed at maturation weeks 1–4. Myoblasts were plated at 40 000 cells/cm2, differentiated into myotubes and analysed after 8 days. Images were acquired with a Leica Sp8 microscope equipped with a HC PL APO CS2 10x/0.40, 20x/0.75 or HC PL APO CS 40x/0.85 dry objective lens. The presence of cell-type-specific markers was analysed using ImageJ software. For astrocyte quantifications, a minimum of 150 cells per cell line were selected randomly based on positive DAPI staining at 20X magnification. A minimum of 300 cells were quantified per motor neuron cell line. The myotube fusion index was evaluated at 10X magnification from 15 random images from 3 individual differentiations. Myotube nuclei were counted with the ImageJ particle analyser plugin at a size of 100–500 μm2.
FUS mislocalisation analysis
hiPSC-derived astrocytes were analysed at week 4 of maturation. 60 random z-stacks from 3 individual experiments were taken at 40X magnification with a Nikon C2 TiE confocal microscope equipped with a Plan Apo 40X oil DIC H objective lens and analysed using a custom automatic Nikon software script. Images were converted to maximum intensity projections, and the ratio of cytoplasm/nucleus of FUS intensity was quantified in Aquaporin-4 (AQP4)- and glial fibrillary acidic protein (GFAP)-positive astrocytes.
Punctae ICC analysis
For ICC analysis with cleaved caspase 3, hiPSC-derived astrocytes were imaged at week 4 of maturation. 30 random images at 40X magnification from 3 individual experiments were taken per cell line using a Leica Sp8 microscope equipped with a HC PL APO CS 40x/0.85 dry objective lens and analysed using ImageJ 1.28u puncta analyser plugin. Fluorescent punctae areas (μm2) were calculated in each image and normalized to the number of nuclei.
NMJ ICC analysis
NMJ formation was evaluated with ICC at days 28 and 35 of the coculture timeline. Every myosin heavy chain (MyHC)-positive myotube present in the microfluidic device channel was recorded in z-stacks. For very long myotubes, multiple z-stacks were acquired. The number of colocalisations between neurofilament heavy chain (NEFH)/synaptophysin (SYP) and α-bungarotoxin (Btx) was counted manually through each z-stack and normalized to the number of myotubes present in the z-stack. Additionally, the number of innervated myotubes were counted per image.
β-catenin ICC analysis
Monocultured hiPSC-derived astrocytes were analysed at week 4 of maturation. 30 random z-stacks from 3 individual experiments were taken at 40X magnification with a Nikon A1R TiE confocal microscope equipped with a Plan Fluor 40X oil DIC objective lens and analysed using the same custom automatic Nikon software script as for FUS mislocalisation analysis. Images were converted to maximum intensity projections, and the ratio of nucleus/cytoplasm of β-catenin intensity was quantified in AQP4-positive astrocytes.
For cocultures, hiPSC-derived motor neuron-NPCs were plated at day 10 of differentiation at a density of 15 000 cells/cm2 and differentiated until day 21. At day 21 of motor neuron differentiation, astrocytes at week 3.5 of maturation (d + 24) were plated on top of the motor neurons at a density of 7 500 cells/cm2. The coculture was kept for 48 h in motor neuron medium before fixation. 30 random z-stacks at 12-bit and 40X magnification were taken from 3 individual experiments using a Leica Sp8 microscope equipped with a HC PL APO CS 40x/0.85 dry objective lens. For quantification of β-catenin accumulation, images were converted to maximum intensity projections and analysed using ImageJ 1.28u puncta analyser plugin. Fluorescent punctae areas (μm2) were calculated in each image, and the number of accumulations were quantified per βIII-tubulin-positive motor neuron. The cytoplasmic and nuclear content of β-catenin expression were quantified in βIII-tubulin-positive motor neurons using a custom automatic Nikon software script. Cytoplasmic and nuclear β-catenin intensities were normalized to the cytoplasmic and nuclear area (μm2), respectively, within each individual image.
RNA-sequencing
hiPSC-derived astrocytes were collected for RNA-sequencing experiments at week 4 (d + 28) of maturation. RNA was isolated using RNeasy® Mini Kit (Qiagen, Hilden, Germany, Cat N° 74104) according to the manufacturer’s instructions.
RNA quality control
RNA concentration and purity were determined spectrophotometrically using the Nanodrop ND-8000 (Nanodrop Technologies) and RNA integrity was assessed using a Bioanalyzer 2100 (Agilent).
Library preparation
Per sample, an amount of 1 µg of total RNA was used as input. Using the Illumina TruSeq® Stranded mRNA Sample Prep Kit (protocol version: # 1000000040498 v00 October 2017) poly-A containing mRNA molecules were purified from the total RNA input using poly-T oligo-attached magnetic beads. In a reverse transcription reaction using random primers, RNA was converted into first-strand cDNA and subsequently converted into double-stranded cDNA in a second-strand cDNA synthesis reaction using DNA Polymerase I and RNAse H. The cDNA fragments were extended with a single 'A' base to the 3' ends of the blunt-ended cDNA fragments after which multiple indexing adapters were ligated introducing different barcodes for each sample. Finally, enrichment PCR was carried out to enrich those DNA fragments that have adapter molecules on both ends and to amplify the amount of DNA in the library.
Sequencing
Sequence-libraries of each sample were equimolarly pooled and sequenced on Illumina NovaSeq 6000 (S1 flowcell 100 cycles kit v1.5, 100 bp Single Read (100–8-8–0)) at the VIB Nucleomics Core (www.nucleomics.be).
Preprocessing
Low-quality ends and adapter sequences were trimmed off from the Illumina reads with FastX 0.0.14 and Cutadapt 1.15 [32, 33]. Subsequently, small reads (length < 35 bp), polyA-reads (more than 90% of the bases equal A), ambiguous reads (containing N), low-quality reads (more than 50% of the bases < Q25) and artifact reads (all but three bases in the read equal one base type) were filtered using FastX 0.0.14 and ShortRead 1.40.0 [34]. With Bowtie2 2.3.3.1 we identified and removed reads that align to phix_illumina [35].
Mapping
The preprocessed reads were aligned with STAR aligner v2.5.2b to the reference genome of Homo sapiens (GRCh38) [36]. Default STAR aligner parameter settings were used, except for ‘–outSAMprimaryFlag OneBestScore –twopassMode Basic –alignIntronMin 50 –alignIntronMax 500000 –outSAMtype BAM SortedByCoordinate’. Using Samtools 1.5, reads with a mapping quality smaller than 20 were removed from the alignments [37].
Counting
The number of reads in the alignments that overlap with gene features was counted with featureCounts 1.5.3 [38]. Following parameters were chosen: -Q 0 -s 2 -t exon -g gene_id. We removed genes for which all samples had less than 1 count-per-million. Raw counts were further corrected within samples for GC-content and between samples using full quantile normalization, as implemented in the EDASeq package from Bioconductor [39].
Differential gene expression
With the EdgeR 3.24.3 package of Bioconductor, a negative binomial generalized linear model (GLM) was fitted against the normalized counts [40]. We did not use the normalized counts directly but worked with offsets. Differential expression was tested for with a GLM likelihood ratio test, also implemented in the EdgeR package. The resulting p-values were corrected for multiple testing with Benjamini–Hochberg to control the false discovery rate [41]. Differentially expressed genes were selected based on False discovery rate (FDR) < 0.05. log2FC < -1.0 were considered downregulated, and log2FC > 1.0 were considered upregulated. VCP, FUS and SOD1 mutant astrocyte transcriptomic data sets were acquired from Array Express (E-MTAB-10916) [49], while C9orf72 mutant astrocyte transcriptomic data sets were accessed from GSE142730 [42]. The single reads FASTQ files from external experiments were processed in a similar way as the FASTQ files from this study and mapped to the same reference genome. The adapters could not be removed as we did not know the sequence. After counting, the normalization was performed for each independent experiment and control samples were discarded. As low counted genes were discarded for each experiment, only 5249 out of the 6468 initial differentially expressed genes were shared by all experiments. The R package ComplexHeatmap (v2.0.0) [43] was used to visualize the expression profiles for the 5249 genes across all experiments.
Canonical pathway analysis
Canonical pathway analysis was performed with Ingenuity Pathway Analysis (IPA) version 68752261 (Qiagen Inc.) [44]. The data set was compared to the reference set “Whole Human Genome Microarray 4x44 K”. log2FC < -1.0 were considered downregulated, while log2FC > 1.0 were considered upregulated. Expr False Discovery Rate (q-value) < 0.001 was considered significant.
GO analysis
Gene ontology analysis was performed with Panther Gene Ontology-slim molecular function analysis tool (http://www.pantherdb.org/) and Fisher’s exact test [45, 46]. Differentially expressed genes with log2FC < -1.0 were considered downregulated, while genes with log2FC > 1.0 were considered upregulated. FDR < 0.05 was considered significant.
Western blot (WB)
hiPSC-derived astrocytes were collected for WB experiments at week 4 (d + 28) of maturation. Cells were washed in DPBS and harvested in a lysis buffer of 10 ml M-PER (Cat N° 78501), 1 tablet PhosSTOP™ Phosphatase Inhibitor Cocktail (Roche Diagnostics, Cat N° 04906845001) and 1 tablet cOmplete™ Protease Inhibitor (Roche Diagnostics, Cat N° 04693116001). Protein content was measured using a Micro BCA™ Protein Assay kit (Cat N° 23235) per the manufacturer’s instructions. Samples containing 40 μg protein were supplemented with SDS-containing lane marker reducing sample buffer (Cat N° 39000), denatured at 95 °C for 5 min and loaded on NuPAGE® 4–12% Bis–Tris 1.0 mm Mini gel (Cat N° NP0321BOX). The gel was run at a 100 V for 1 h 45 min and transferred to a 0.2 μm nitrocellulose membrane of a Trans-Blot Turbo Mini Transfer Pack (Bio-Rad, Hercules, CA, USA, Cat N° 1704158) using the 7 min, 2.5 A, 25 V program of the Bio-Rad Trans-Blot® Turbo Transfer system. The membrane was blocked in 3% BSA (Sigma-Aldrich, Cat N° A7906) diluted in Tris-buffered saline (Sigma-Aldrich, Cat N° T5912-1L) with Tween (Sigma-Aldrich, Cat N° P1379-500ML) (TBST) for 1 h and subsequently incubated overnight with primary antibodies (see Supplementary Table 1, Additional file 1) in 3% BSA-TBST at 4 °C. The following day, the membrane was washed thrice for 10 min in TBST and incubated with secondary antibodies diluted in TBST for 1 h at RT. Finally, the membrane was washed thrice for 10 min in TBST, treated with Pierce ECL (Cat N° 32106) and imaged with a chemiluminescence ImageQuant LAS4000. Quantifications were made with ImageJ.
Multiplex array
Astrocyte conditioned medium was collected from each condition after 48 h. Cytokine measurements were obtained using the Meso Scale Discovery (MSD) system. The commercially available human-specific V-PLEX Proinflammatory Panel 1 assay (MSD, K15049G-1) was used. Each sample was measured in technical duplicates and the assay was performed according to the manufacturer’s protocol. Cytokine concentrations were measured using the MESO QUICKPLEX SQ 120 machine from MSD with the software DISCOVERY WORKBENCH 4.0. Only values above detection level and with a CV value below 25 were included in the analysis.
qPCR
hiPSC-derived astrocyte pellets were collected with accutase and stored at -80 °C for qPCR experiments at week 1–4 of maturation. For total RNA extraction, each cell pellet was dissolved in 1 ml Tri Reagent® (Sigma-Aldrich, Cat N° T9424). The homogenate was supplemented with 200 μl/sample chloroform (Sigma-Aldrich, Cat N° 319988), inverted for 15 s and incubated at RT for 10 min. Next, the sample was centrifuged for 15 min at 12 000 rpm at 4 °C, and the aqueous phase was transferred to new RNase-free tubes and precipitated with 500 μl/sample isopropanol (Merck Millipore, Cat N° 1.09634.2511). After an incubation of 10 min at RT, the sample was centrifuged for 10 min at 12 000 rpm at 4 °C and the supernatant was discarded. The pellet was washed once with 1 ml 75% ethanol (VWR, Radnor, PA, USA, Cat N° 20.821.296), and dissolved in 11 μl RNase-free water. RNA concentrations were determined with NanoDrop 1000 Spectrophotometer V3.8.1. cDNA was synthesized from 1 μg RNA using SuperScript® III First-Strand Synthesis System for RT-PCR (Cat N° 18080–051) per manufacturer’s instructions. qPCR was performed using Fast SYBR™ Green Master Mix (Cat N° 4385612) and TaqMan™ Fast Universal PCR Master Mix (Cat N° 4352042) (for primers see Supplementary Table 1, Additional file 1) on a StepOnePlus Real-Time PCR machine. Relative gene expression was normalized to β-actin and hiPSCs and calculated with the 2−ΔΔCt method [47].
Live cell calcium recordings of astrocytes
hiPSC-derived astrocytes were plated at 50 000 cells/cm2 in 6-well plates at week 5 of maturation and analysed at week 6. Cells were labelled with 5 μM Fluo-4 AM (Cat N° F14201), and spontaneous Fluo-4 fluorescence was recorded over 90 s with a 10X objective using a Nikon A1R TiE confocal microscope equipped with a Plan Apo 10X objective lens and analyses using a customized Nikon script. The script calculated the difference between min and max fluorescence intensity in individual astrocytes during the 90 s recording. The threshold for active cells was set to an intensity increase > 100.
Scanning electron microscopy (SEM)
Astrocyte, motor neuron and NMJ morphologies were evaluated with SEM at day 28 of the coculture timeline using a previously published method [25, 26]. In brief, cultures were fixed in 2.5% glutaraldehyde (Agar Scientific, Essex, UK, Cat N° R1020) in 0.1 M Na-cacodylate buffer (Sigma-Aldrich, Cat N° C0250) and the SND75 devices were carefully removed from the Aclar sheets. Next, the cultures were incubated in 1% osmium tetroxide (Electron Microscopy Sciences, Cat N° 19150) and dehydrated in a graded ethanol series to 100% ethanol. The sheets were then critical point dried, mounted on SEM support stubs and coated with 5 nm Chromium in a Leica ACE600. Cultures were imaged with a Zeiss Sigma SEM.
LDH analysis
hiPSC-derived motor neuron-NPCs were plated at day 10 of differentiation at a density of 25 000 cells/cm2 and differentiated until day 21. At day 21 of motor neuron differentiation, astrocytes at week 5.5 of maturation (d + 38) were plated on top of the motor neurons in motor neuron medium at a density of 25 000 cells/cm2. The cocultures were kept for 14 days in motor neuron medium, and media samples were collected on days 2, 7 and 14. Monocultured day 21 motor neurons were incubated with 100 μM Arsenite treatment for 48 h as a positive control for cell death. Samples were analysed CyQUANT™ LDH Cytotoxicity Assay Kit (Cat N° C20300) according to the manufacturer’s instructions.
Neurite network quantifications
On days 28 and 35 of coculture (1 and 2 weeks after adding astrocytes, respectively), z-stack tile scan confocal images were taken at 10X magnification of the motor neuron/astrocyte compartment using a Nikon A1R confocal microscope equipped with a Plan Apo 10X objective lens. Images were cropped to display only the channel of the device, and motor neuron neurite (NEFH) volume was quantified in 3D using a customized Nikon software script.
Neurite outgrowth quantifications
Neurite outgrowth was analysed as previously described [25]. On days 28 and 35 of coculture (1 and 2 weeks after adding astrocytes, respectively), tile scan confocal images were taken at 10X magnification of the NEFH fluorescence in the myotube compartment using an inverted Leica SP8 DMI8 confocal microscopy equipped with a HC PL APO CS2 10x/0.40 dry objective lens. Motor neuron neurites were isolated using ilastik 1.3.3post1 Pixel Classification software [48], and the number of pixel intersections was calculated per intersection line using an ImageJ 1.52p software linear Scholl analysis script.
Live-cell calcium recordings of NMJs
Recordings were made using a previously described protocol [25, 26]. On day 28 of the coculture timeline (1 week after adding astrocytes), myotubes were labelled with 5 μM Fluo-4 AM. The motor neuron/astrocyte compartment was stimulated with 50 mM potassium chloride (KCl) and Fluo-4 fluorescence was recorded in the myotubes using a Nikon A1R confocal microscope equipped with a Plan Apo 10X objective lens and analysed with NIS-Elements AR 4.30.02 software’s Time Measurement tool. The myotube compartment was stimulated directly with 50 mM KCl, to calculate the percentage of motor neuron-stimulated myotubes through functional NMJs of the total number of active myotubes.
Quantification and statistical analysis
Statistical analyses were made with GraphPad Prism 9.2.0, where data were tested for normal Gaussian distribution using the D’Agostino-Pearson omnibus normality test, Anderson–Darling test, and Shapiro–Wilk normality test. Statistical details of experiments can be found in the figure legends. For differences of mean between two groups, unpaired t test or Mann–Whitney were used, while one/two-way ANOVA with Tukey’s multiple comparisons test or Kruskal–Wallis test with Dunn’s multiple comparisons test were used for difference of means between multiple groups. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. Each experiment included at least 3 biological replicates, where a biological replicate represents an independent differentiation from hiPSCs to terminal cell type, an independent myoblast differentiation into myotubes or an independent coculture in a one-compartment plate or microfluidic device.
Results
hiPSCs differentiate into functional astrocytes independent of FUS mutations
To evaluate the role of astrocytes in ALS, we differentiated hiPSCs from two ALS-patients with FUS mutations (R521H and P525L) and their corresponding CRISPR-Cas9 gene-edited isogenic controls (R521R and P525P) [25, 27, 28] into astrocytes using a slightly modified version of a recently published protocol [29]. The P525L mutation causes an aggressive juvenile form of ALS, while the more common R521H mutation causes adult-onset ALS [27], and the inclusion of both lines demonstrate a broad disease-onset spectrum. The astrocyte differentiation (25 days) generated APCs, which were further matured into astrocytes during 4 weeks (Fig. 1A). The differentiation efficiency was evaluated with bright-field microscopy (Suppl. Figure 1A, Additional file 2), qPCR (Suppl. Figure 1B, Additional file 2), ICC (Fig. 1B-C, Suppl. Figure 2–3, Additional file 2) and bulk RNAseq (Fig. 1D, Suppl. Figure 1C-D, Additional file 2). During the 4 weeks of maturation, an increase in both gene (Suppl. Figure 1B, Additional file 2) and protein (Suppl. Figure 2, Additional file 2) expression of astrocyte markers S100 calcium-binding protein β (S100β) and AQP4 were observed. In addition, the astrocytes stained increasingly positive for astrocyte-specific markers Aldehyde Dehydrogenase 1 Family Member L1 (ALDH1L1) and SRY-Box Transcription Factor 9 (SOX9), while the neuronal marker Microtubule-Associated Protein 2 (MAP2) remained at a low expression level (Suppl. Figure 2–3, Additional file 2). After 4 weeks of maturation, approximately 95% of astrocytes were positive for all astrocyte markers (Fig. 1B-C) showing no difference in the differentiation potential between patient and gene-repaired hiPSC-lines. Additionally, RNAseq revealed a high expression level of astrocyte-specific genes in comparison to genes specific for other cell type in the central nervous system (Fig. 1D). To assess the functionality, we performed live-cell recordings of spontaneous calcium transients using Fluo-4 (Fig. 1E-G). All four astrocyte lines showed characteristic calcium transients demonstrating clear functionality (Fig. 1E, Additional files 3, 4, 5 and 6). Both mutant astrocyte populations displayed higher peak intensities in comparison to controls (Fig. 1F) suggesting mutant FUS-mediated calcium transient hyperactivity. In addition, P525L also revealed an increase in the number of active astrocytes (Fig. 1G). Next, we looked at the differences in gene expression between FUS-ALS and isogenic control astrocytes. Our principal component analysis demonstrated minor variations between biological replicates (Suppl. Figure 1C, Additional file 2) and a clustering of controls, while R521H and P525L astrocytes showed an almost opposite gene expression profile (Suppl. Figure 1D, Additional file 2). Overall, 635 and 1203 genes were shown upregulated in mutant R521H and P525L astrocytes, respectively, while 919 (R521H) and 1117 (P525L) genes were downregulated in comparison to their respective controls (Fig. 1H). Of all recorded genes, 452 differentially expressed genes overlapped between the two gene-pairs, while 1102 (P525L vs P525P) and 1868 (R521H vs R521R) were unique to the individual mutation (Fig. 1I). In addition, we compared our differential gene expression with previously published transcriptomic data sets from hiPSC-derived astrocytes generated from VCP, FUS, SOD1 and C9orf72 ALS patients (Suppl. Figure 1E, Additional file 2) [42, 49]. Interestingly, we observed some similarity in the gene expression between our P525L astrocytes, the SOD1D90A and the C9orf72 patient astrocytes, while the R521H astrocytes had strikingly opposite gene expression profile. VCPR155C and FUSH517Q were shown to range in between P525L and R521H expression levels. Generally, a large variation in the differential gene expression across lines could be appreciated. Taken together, these results confirm that we are able to generate pure cultures containing functionally active astrocytes from each hiPSC-line independent of the presence of a FUS mutation.

Generation and characterisation of functional hiPSC-derived astrocytes. A Overview of the astrocyte differentiation protocol. hiPSCs were dissociated at day 0 (d0) and cultured for the induction phase as embryoid bodies (EBs) before they were plated for neural rosette formation at day 7 (d7). The neural rosettes underwent expansion to generate neural progenitor cells (NPCs) until day 16 (d16). NPCs were then differentiated into astrocyte progenitor cells (APCs, d25/d+0) after which the APCs were matured into astrocytes for an additional 4 weeks (d + 28). B Representative confocal images of astrocytes at week 4 (d + 28) of maturation stained with astrocytic markers S100β, AQP4, ALDH1L1 and SOX9 in addition to neuronal marker MAP2. Nuclei stained with DAPI. Scale bar: 75 μm. C Quantification of the number of cells positive for astrocyte and neuronal markers. Mean ± s.e.m. of 3 biological replicates (n = 15 images). D Heatmap of the expression of cell type-specific genes in 4 weeks mature astrocytes from RNAseq experiment performed in 3 biological replicates. Dark blue cells: FPKM > 100. E Functional assessment of spontaneous calcium transients in 6 weeks mature astrocytes loaded with the Fluo-4 dye. Y-axis range: ΔF/F0 = 50%. F Quantification of peak Fluo-4 intensity. Mann–Whitney test. ****p < 0.0001. G Percentage of active astrocytes. Inactive astrocyte cut-off: Fluo-4 intensity increase < 100. Unpaired t test. *p < 0.05. H Volcano-plots of up- (red) and down- (blue) regulated genes in week 4 mature astrocytes. Green lines indicate thresholds: log2FC < -1.0 with –log10(FDR) > 2.0 were considered downregulated, and log2FC > 1.0 with –log10(FDR) > 2.0 were considered upregulated. I Comparison of differentially expressed genes between mutation sets. Graphs in panels (F and G) show mean ± s.e.m. of 3 biological replicates with each 2–6 technical replicates. Cell illustrations are modified from Smart Servier Medical Art licensed under a Creative Commons Attribution 3.0 Unported License (https://creativecommons.org/licenses/by/3.0/). See also Suppl. Figure 1–3, Additional file 2, as well as Additional files 3, 4, 5 and 6
FUS-ALS astrocytes display increased reactivity
Glial fibrillary acidic protein (GFAP) is a widely used astrocyte marker but also indicative of astrocyte reactivity [50]. Since an increased GFAP expression has been reported in post-mortem samples from ALS patients [12], we investigated the expression of GFAP within our astrocyte populations using ICC. Our analysis showed a progressive increase in GFAP during astrocyte maturation in all hiPSC-lines (Suppl. Figure 3A-B, Additional file 2), however, especially P525L astrocytes demonstrated a significantly higher percentage of GFAP-positive cells in the population compared to the isogenic control (Fig. 2A-B). These findings were confirmed with Western blot (WB) (Suppl. Figure 3D, Additional file 2), and also our qPCR analysis presented a maturation-dependent increase in GFAP gene expression (Suppl. Figure 3E, Additional file 2).

FUS-ALS astrocytes display increased reactivity. A Confocal images of GFAP expression in 4 weeks mature astrocytes. Nuclei stained with DAPI. Scale bar: 75 μm. B Quantification of GFAP expression. One-way ANOVA with Tukey’s multiple comparisons test. Mean ± s.e.m. of 3 biological replicates (n = 15 images). ****p < 0.0001. C Representative confocal images of FUS protein localisation in 4-week mature P525L and P525P astrocytes. Nuclei stained with DAPI. Scale bar: 50 μm. Inset scale bar: 10 μm. D Violin plot of FUS cytoplasmic mislocalisation in GFAP+ and AQP4+ astrocytes. Data from 3 biological replicates (n = 60 images). Unpaired t test and Mann–Whitney test, respectively. *p < 0.05 and ****p < 0.0001. E Apoptotic assessment in week 4 mature astrocytes stained with cleaved caspase 3. Violin plot from 3 biological replicates (n = 30 images) with outliers (Q = 0.1%) removed. F Secretome analysis of immature (d25/d + 0) and more mature (week 4) astrocytes. Mean ± s.e.m. of 3 biological replicates. G RNAseq reactive astrocyte gene expression profiling of week 4 mature astrocytes. See also Suppl. Figure 3–4, Additional file 2
As we previously demonstrated cytoplasmic mislocalisation of FUS protein in FUS-ALS patient fibroblasts, hiPSCs and hiPSC-derived motor neurons [27], we also evaluated the cellular localisation of FUS protein within our astrocytes. In line with these findings, both FUS-mutant lines showed a cytoplasmic mislocalisation of FUS in the GFAP-positive astrocyte population, while only P525L astrocytes displayed mislocalisation within the more general AQP4-positive population (Fig. 2C-D). No difference in apoptosis as measured by the size of cleaved caspase 3 ICC punctae was observed (Fig. 2E).
ALS astrocytes secrete toxic factors [20, 21, 23], so to investigate this further, we performed a secretome analysis by measuring the presence of 10 inflammatory cytokines (IFN-γ, TNF-α, IL-1β, IL-2, IL-4, IL-6, IL-8, IL-10, IL-12p70 and IL-13) in the media from immature (APCs d25/d+0) and more mature astrocytes (maturation week 4) (Fig. 2F). Remarkably, immature R521H astrocytes showed a high secretion of especially IL-6 and IL-8, while P525L astrocytes showed an increased secretion of the majority of cytokines at this early differentiation state. In more mature mutant astrocytes, the cytokine secretion increased several tenfolds, which signifies that the inflammatory response of astrocytes is differentiation-dependent (Suppl. Figure 4A, Additional file 2). Both IL-4 and IL-10 are involved in suppressing/regulating the immune response [51], and their presence in the FUS-ALS secretome might therefore also indicate an astrocytic attempt to counteract the other pro-inflammatory cytokines. To explore this further we performed a gene-ontology (GO) analysis on our RNAseq data to examine both up- and downregulated molecular functions (Suppl. Figure 4B, Additional file 2). In R521H astrocytes, no upregulated pathways were found, however, several biological pathways correlating with cytokine functions (cytokine activity, cytokine receptor activity and cytokine binding) were downregulated, which supports our hypothesis of an auto-regulatory attempt to counteract the inflammatory response. Interestingly, the P525L astrocytes showed an upregulation of both cytokine receptor binding and cytokine activity, which argues for a mutation- and/or age of disease onset-dependent reactive effect.
Since our data so far favoured a toxic reactive phenotype in our FUS-ALS astrocytes, we turned our attention to a previously suggested binary categorisation of reactive astrocytes; “A1” astrocytes being neurotoxic and “A2” astrocytes being neuroprotective [52]. Our results disclosed neither a clear A1 nor A2 gene expression profile in our FUS mutant astrocytes in comparison to controls but showed both an up- and downregulation of genes within each group as well as for genes present in pan-reactive astrocytes (Fig. 2G). Taken together, these results demonstrate a FUS-mutant-mediated astrocyte reactivity, which affects the transcriptome and secretome in a toxic although heterogeneous mutation-dependent manner.
hiPSC-derived astrocytes are successfully integrated in a microfluidic model of the human motor unit
To evaluate the effect of astrocytes in a novel multicellular model, we optimised our previously established protocol for the generation of a human motor unit in microfluidic devices containing functional NMJs [26]. hiPSC-derived motor neurons were differentiated from each hiPSC-line (Fig. 3A) using our formerly established protocol [27], and the differentiation potential was evaluated with ICC (Fig. 3B, Suppl. Figure 5A-B, Additional file 2). At day 28 of motor neuron differentiation, approximately 85–95% of cells stained positive for motor neuron-specific markers choline acetyltransferase (ChAT) and Islet-1 in addition to the pan-neuronal markers neurofilament heavy chain (NEFH) and βIII-tubulin (Tubulin) with no difference between hiPSC-lines (Fig. 3C). A limitation of our previous motor unit model was the restricted viability of the mesoangioblast-derived myotubes [26], so to prolong the coculture, we instead made use of primary human myoblasts (Fig. 3D). The myoblasts ability to differentiate and fuse into multinucleated myotubes was evaluated with ICC (Fig. 3E, Suppl. Figure 5C-D, Additional file 2), and showed 70–90% expression of myotube-markers myogenin (MyoG), myosin heavy chain (MyHC), desmin, titin and α-actinin (ACTN2) (Fig. 3F). For the coculture in compartmentalized microfluidic devices, day 10 motor neuron-neural progenitor cells (MN-NPCs) were plated in the compartment at one side of the microgrooves, while myoblasts were plated in the opposite compartment (coculture timeline shown in Fig. 3G). Myoblasts were differentiated into myotubes, and a chemotactic and volumetric gradient was established to promote the motor neuron neurite across-microgroove migration in order for motor neurons and myotubes to interact and form NMJs [26]. Week 3.5 mature astrocytes were subsequently plated on top of the maturing motor neurons in the motor neuron compartment of the microfluidic device and the cocultures were evaluated 1 week (day 28) and 2 weeks (day 35) later.

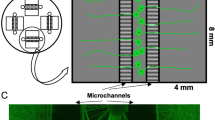
Motor neuron and myotube verification for the establishment of microfluidic coculture model. A Overview of motor neuron differentiation from hiPSCs through an EB state towards MN-NPCs and finally post-mitotic spinal motor neurons. B Representative confocal images of mature motor neurons at day 28 of differentiation stained with motor neuron markers ChAT and Islet-1 in addition to pan-neuronal markers NEFH and βIII-tubulin (Tubulin). Nuclei stained with DAPI. Scale bar: 25 μm. C Quantification of the number of cells positive for neuronal markers. Mean ± s.e.m. of 3 biological replicates (n = 15 images). Kruskal–Wallis test with Dunn’s multiple comparisons test. D Overview of myotube differentiation. Primary human myoblasts were isolated from vastus lateralis muscle, expanded and differentiated into multinucleated elongated myotubes. E Representative confocal images of myotubes stained with markers (AB+) MyoG, MyHC, desmin, titin and ACTN2. Nuclei stained with DAPI. Scale bar: 200 μm. F Quantification of myogenic markers in multinucleated myotubes. Mean ± s.e.m. of 3 biological replicates (n = 15 images). G Schematic overview of coculture between hiPSC-derived astrocytes and motor neurons with human primary myoblast-derived myotubes in a compartmentalized microfluidic device. A chemotactic and volumetric gradient is established to promote motor neuron neurite polarization [25, 26]. Experiments were conducted 1 week (d28) and 2 weeks (d35) after plating the mature astrocytes. Cell illustrations are modified from Smart Servier Medical Art licensed under a Creative Commons Attribution 3.0 Unported License (https://creativecommons.org/licenses/by/3.0/). See also Suppl. Figure 5, Additional file 2
FUS-ALS astrocytes fail to integrate in the motor neuron network
Four coculture conditions were established to assess the effect of astrocytes on the motor neurons; fully-isogenic control (IC) (IC motor neurons and IC astrocytes), fully-mutant (mutant motor neurons and mutant astrocytes) and combined setups (Fig. 4A-B). Interestingly, we observed an increase in GFAP expression in the astrocytes in all conditions, which suggests that the presence of motor neurons further activates the astrocytes independently of FUS mutations (Suppl. Figure 6A, Additional file 2). Overall, the astrocytes displayed a more elongated morphology and appeared well-integrated in the motor neuron networks in the fully-IC conditions as well as for the combined setup of mutant motor neurons and IC astrocytes (Suppl. Figure 6A, Additional file 2). In contrast, the FUS-ALS astrocytes appeared hypertrophic and non-integrated in the fully-mutant conditions as well as in the combined setup of IC motor neurons and mutant astrocytes. These results were confirmed using scanning electron microscopy (SEM), which showed that the mutant R521H and P525L astrocytes generally appeared large, flat and non-engaging with the motor neurons (Fig. 4C). The presence of the FUS-mutant astrocytes had no apparent influence on the motor neuron network, as we did not measure any difference in motor neuron neurite volume in the motor neuron/astrocyte compartment across conditions at either time point (Suppl. Figure 6B-G, Additional file 2). To investigate whether the astrocyte presence caused motor neuron death, we performed a Lactate Dehydrogenase (LDH) activity assay (Suppl. Figure 6H-I, Additional file 2). The R521H astrocytes had an initial mild cytotoxic effect on both R521R and R521H motor neurons after 2 days of coculture. However, this small effect disappeared over the course of the 14 days coculture, and was neither comparable to the 100 μM arsenite-treated motor neurons, where 100% cell death was observed (data not shown). No significant differences in cell death were measured in the P525L and P525P conditions.

FUS-ALS astrocytes fail to integrate in the motor neuron network. A Overview of coculture setup between hiPSC-derived motor neurons (MN) and astrocytes (AC). B Experimental assessment was performed in the highlighted motor neuron/astrocyte compartment of the microfluidic device after 1 and 2 weeks of coculture. C Scanning electron microscopy images of motor neuron and astrocyte cocultures in microfluidic devices after 1 week. Mutant astrocytes are circled with a white dashed line. Scale bar: 10 μm. D Top 25 RNAseq canonical pathways in mutant astrocytes compared to controls at week 4 of maturation. Analysis was performed with a cut-off Log Ratio of -1.0 to 1.0 and an FDR of 0.001. Dotted line marks p-value = 0.05. Cell illustrations are modified from Smart Servier Medical Art licensed under a Creative Commons Attribution 3.0 Unported License (https://creativecommons.org/licenses/by/3.0/). See also Suppl. Figure 6, Additional file 2
To further assess the mechanisms involved in FUS-ALS astrocyte function, we performed an IPA canonical pathway analysis on our RNAseq data. The majority of pathways were downregulated in FUS-ALS astrocytes representing a loss-of-support mechanism since these pathways are largely involved in the maintenance of neuronal homeostasis (Fig. 4D). Notably, finding the synaptogenesis signaling pathway and the axonal guidance signaling pathway in the top 3 of P525L and R521H respectively further supported the dysregulation of astrocyte-neuronal interaction. Collectively, our data demonstrate that FUS-ALS astrocytes fail to integrate and support the motor neuron network.
P525L FUS-ALS astrocytes impair motor neuron neurite outgrowth
We previously showed that P525L hiPSC-derived motor neurons have a reduced neurite outgrowth in microfluidic devices in comparison to controls [25]. As we found a predominant downregulation of neuronal support mechanisms with the axonal guidance signaling pathway ranking high in the transcriptomic data from our FUS-ALS astrocytes, we sought to evaluate whether the astrocytes could have an effect on the motor neuron neurite outgrowth. Therefore, we performed the same linear Scholl analysis on the motor neuron neurites in the myotube compartment on each coculture condition (Fig. 5A-B). For R521H and R521R setups, we only observed an increase in outgrowth in R521R motor neurons cocultured with mutant R521H astrocytes compared to fully IC conditions after 2 weeks (Fig. 5C-D). These findings confirm that the initial cell death observed in the cocultures has no influence on the long term neurite outgrowth. Interestingly, R521H and R521R cultures without astrocytes collectively showed a larger neurite outgrowth than cocultures including astrocytes, however, no difference was found between the mutant and isogenic motor units, which confirms our previous findings [25]. For the P525L and P525P conditions, we observed a reduced neurite outgrowth in fully-mutant cocultures, combined IC motor neuron and mutant astrocyte conditions as well as in the control motor neuron conditions without astrocytes after 1 week (Fig. 5E). These results demonstrate that P525L astrocytes do not exacerbate the reduced outgrowth of P525L motor neurons, which is already seen without the presence of astrocytes, but that they do limit the outgrowth of control P525P motor neuron neurites. Interestingly, P525P astrocytes rescued this effect on the P525L motor neurons after 1-week coculture but were unable to sustain this influence after 2 weeks. P525P astrocytes were likewise able to increase the outgrowth of P525P motor neurons above the level of P525P motor neurons cultured without astrocytes (Fig. 5F). After 2 weeks of coculture, the differences became less pronounced due to the overall increase in neurite outgrowth across weeks (Fig. 5F), which indicates that the motor neuron neurites are able to recover despite the toxic effects of the mutant astrocytes.

P525L FUS-ALS astrocytes impair motor neuron neurite outgrowth. A Example of a tile scan confocal image of motor neuron (MN) neurite (NEFH/SYP) outgrowth in the myotube compartment after 1 week (d28) of coculture with astrocytes (AC). Arrows (right) depict the neurite growth direction upon exit of the microgrooves. Scale bar: 300 μm. B Mask of tile scans with intersection lines at every 50 μm starting from the exit of the microgrooves. C-F Neurite outgrowth quantifications of the number of pixel intersections after 1 week (panel C + E) and 2 weeks (panel D + F) of coculture between motor neurons, myotubes and astrocytes. Graphs in (panel C-F) show mean ± s.e.m. of 3 biological replicates. Overall comparisons between genotypes were performed with two-way ANOVA with Tukey’s multiple comparisons test. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001
FUS-ALS astrocytes impair the formation and functionality of NMJs
Next, we assessed the effect of astrocytes on the NMJ formation in the myotube compartment of the microfluidic device (Fig. 6A). NMJ formation was evaluated with ICC and SEM (Fig. 6B, Suppl. Figure 7A, Additional file 2), and the co-localisation between neuritic (NEFH) and presynaptic (synaptophysin (SYP)) markers with acetylcholine receptor (AChR) marker α-bungarotoxin (Btx) was counted per MyHC-labelled myotube (Fig. 6B, Suppl. Figure 7B-D, Additional file 2). Our analysis revealed that FUS-mutant astrocytes reduced the formation of NMJs in both fully-mutant and combined IC motor neuron with mutant astrocyte cocultures after 1 week (Fig. 6C-D), and that this negative effect was sustained after 2 weeks of coculture (Fig. 6E-F). In contrast, IC astrocytes had a beneficial effect on the NMJ numbers, which increased in both fully-IC cocultures above the control condition of IC motor units without astrocytes, as well as completely or partially rescued the NMJ formation in combined mutant motor neuron and IC astrocyte conditions. From week 1 to week 2 of the coculture timeline, NMJ numbers increased in fully-IC cocultures, which confirms the continued development of connections between motor neurons and myotubes as the system matures (Fig. 6G-H). This effect was also seen in the R521R motor unit culture without astrocytes (Fig. 6G), as well as in the combined P525L motor neurons and P525P astrocytes (Fig. 6H). Remarkably, we saw a time-dependent decrease in NMJ formation in the P525L motor units without astrocytes, which demonstrates a direct loss of NMJs. Additionally, we quantified the percentage of innervated myotubes and found a reduction in both fully mutant systems after 2 weeks of coculture (Suppl. Figure 8, Additional file 2).

FUS-ALS astrocytes impair NMJ formation and functionality. A Experimental assessment was performed in the highlighted myotube compartment of the microfluidic device after 1 and 2 weeks of coculture between motor neurons (MN) and astrocytes (AC). B Confocal image example of an NMJ. NMJs were identified through colocalisation between motor neuron (NEFH) and presynaptic (SYP) markers with postsynaptic AChR marker (Btx) on MyHC-stained myotubes. Scale bar: 20 μm. C-F Quantification of the number of NMJs per myotube after 1 week (panel C-D) and 2 weeks (panel E–F) of cocultures between motor neuron/myotubes and astrocytes. G-H Number of NMJ formations over time. Graphs in panel C-H show mean ± s.e.m. of 3 biological replicates. One-way ANOVA with Tukey’s multiple comparisons test (panel C-F) and unpaired t test (panel G-H). *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001. I Schematic overview of live-cell calcium recordings to assess NMJ functionality. The motor neuron/astrocyte compartment was stimulated with 50 mM KCl to evoke an intracellular response, after which an influx in calcium was recorded in myotubes labelled with calcium-sensitive Fluo-4 dye. J Representative calcium influx curves in myotubes after KCl stimulation (arrow). K Quantifications of peak Fluo-4 intensity. Outliers (Q = 1%) removed. L Percentage of NMJ-excitable myotubes of total active myotubes. Data in (K and L) represent mean ± s.e.m. of 4–5 biological replicates with 2 technical replicates in each experiment. One-way ANOVA with Tukey’s multiple comparisons test. *p < 0.05 and **p < 0.01. Cell illustrations are modified from Smart Servier Medical Art licensed under a Creative Commons Attribution 3.0 Unported License (https://creativecommons.org/licenses/by/3.0/). See also Suppl. Figure 7–8, Additional file 2, as well as Additional files 7, 8, 9, 10, 11, 12 and 13
To assess NMJ functionality, we performed live-cell calcium recordings on P525L and P525P conditions as described before [26]. By chemically stimulating the motor neuron/astrocyte compartment with potassium chloride (KCl), we evoked an influx of calcium in Fluo-4 labelled myotubes, confirming transmission through a functional NMJ connection (Fig. 6I, Additional files 7, 8, 9, 10, 11, 12 and 13). Influx curves could be recorded in all conditions (Fig. 6J), and a reduction in peak size in the fully mutant system compared to fully isogenic cocultures could be appreciated (Fig. 6K). Similarly, a reduction in the percentage of NMJ-excitable myotubes were found in the fully mutant systems (Fig. 6L), while a rescuing trend could be observed in P525L motor units cocultured with P525P astrocytes. No obvious difference was observed between the P525P and P525L cocultures without astrocytes, which indicates that the difference in NMJ formation (Fig. 6D) could be explained by a larger presence of immature non-functional NMJs in P525P motor unit systems. In addition, this could also explain the lack of change in NMJ numbers in P525P motor units between week 1 and week 2 cocultures, as the system might have reached its maximum potential for NMJ formation (Fig. 6H).
Taken together, these results demonstrated that FUS-ALS and especially P525L astrocytes impair motor neuron neurite outgrowth, NMJ formation and functionality, while IC astrocytes are able to rescue many of these aberrations.
FUS-ALS astrocytes activate the WNT/β-catenin pathway in FUS-ALS motor neurons
Based on our RNAseq canonical pathway analysis (Fig. 4D), we identified one pathway, the WNT/β-catenin pathway, which was upregulated in both R521H and P525L astrocytes. Previous studies suggest the involvement of the WNT/β-catenin pathway in ALS [53, 54] as well as other neurodegenerative disorders [55], so to further investigate this, we looked into the gene expression of an array of WNT/β-catenin pathway components. Our data revealed a complex relationship of genes being both up- and downregulated (Fig. 7A). Target genes such as CCND1, FZD4, FZD7, FZD8 and MYC were upregulated in P525L astrocytes, which correlated with an upregulation described in the final disease stages in SOD1-ALS mice [56]. Similarly, FOSL1, another important gene in late ALS disease stages [56], was upregulated in both R521H and P525L astrocytes. The activation of the WNT/β-catenin pathway causes translocation of β-catenin from the cytoplasm to the nucleus [57]. To investigate this further, we measured the nucleus/cytoplasmic ratio of β-catenin intensity in astrocytes, and found an increase in nuclear β-catenin in P525L astrocytes confirming an activation of the WNT/β-catenin pathway (Fig. 7B-C). To evaluate the influence of astrocytes on motor neurons, we cocultured P525L and P525P motor neurons and astrocytes for 48 h in 24-well plates, and evaluated the cellular localisation of β-catenin within Tubulin-positive motor neurons with ICC (Fig. 7D). At first, we observed a predominant accumulation of cytoplasmic β-catenin in P525L motor neurons cocultured with P525L astrocytes, which was successfully lowered by the presence of P525P astrocytes (Fig. 7E). Larger accumulations were found in P525P and P525L motor neurons cocultured with P525L astrocytes, while a smaller β-catenin cluster size was found in P525L motor neurons cultured with P525P astrocytes (Fig. 7F-G). Overall, the cytoplasmic β-catenin expression was less pronounced in the motor neurons in the fully isogenic cocultures and in the combined cocultures of P525P motor neurons and P525L astrocytes (Fig. 7H). Remarkably, we found a higher level of nuclear-translocated β-catenin in the fully mutant cocultures, which could indicate that P525L astrocytes are able to activate the WNT/β-catenin pathway in the mutant motor neurons after as little as two days coculture (Fig. 7I). A similar trend was observed in the P525P motor neurons cocultured with P525L astrocytes, while fully isogenic cocultures as well as combined P525L motor neurons and P525P astrocytes showed low nuclear β-catenin expression. In conclusion, these results establish an important role of WNT/β-catenin pathway activation in FUS-ALS motor neuron-astrocyte crosstalk.

FUS-ALS astrocytes influence motor neurons through the WNT/β-catenin pathway. A RNAseq differential gene expression of WNT/β-catenin pathway components in 4-week mature astrocytes. B Representative confocal images of β-catenin expression in 4-week mature P525L and P525P astrocytes (AQP4). Inset shows a magnification of astrocytes with β-catenin localisation. Nuclei stained with DAPI. Scale bar: 50 μm. Inset scale bar: 5 μm. C Quantification of β-catenin localisation presented as the nuclei/cytoplasmic ratio. Data from 3 biological replicates (n = 30 images). Unpaired t test. **p < 0.01. D Representative confocal images of β-catenin expression in motor neurons (Tubulin), which have been cocultured with astrocytes (GFAP) for 48 h. Inset shows a magnification of motor neurons with β-catenin localisation. Arrowheads show examples of β-catenin accumulation. Nuclei (DAPI) are circled with a white dashed line. Scale bar: 50 μm. Inset scale bar: 5 μm. E Quantification of number of β-catenin accumulations per Tubulin+ motor neuron. F Quantification of individual β-catenin accumulation size in Tubulin+ motor neuron. G Percentage distribution of different size ranges of β-catenin accumulation in Tubulin+ motor neuron. H Quantification of cytoplasmic β-catenin expression per Tubulin+ motor neuron cytoplasmic area (μm2). I Quantification of nuclear β-catenin expression per Tubulin+ motor neuron nuclear area (μm.2). Panel (E-I) show mean ± s.e.m. of 3 biological replicates (n = 30 images). Panel (E and I): One-way ANOVA with Tukey’s multiple comparisons test. Panel (F and H): Kruskal–Wallis test with Dunn’s multiple comparison test. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001
Discussion
In this study, we investigated the influence of hiPSC-derived astrocytes on human motor units cultured in microfluidic devices in the context of FUS-ALS. We discovered that FUS-ALS astrocytes displayed increased reactivity through calcium transient hyperactivity, increased GFAP expression, cytoplasmic FUS mislocalisation and spontaneous secretion of inflammatory cytokines in comparison to isogenic control astrocytes. Once cocultured with hiPSC-derived motor neurons, we observed a FUS-ALS astrocyte-mediated lack of neurite network integration and a cytotoxic effect on motor neuron neurite outgrowth in addition to NMJ formation and functionality. IC astrocytes were able to either improve or fully rescue all these aberrations. Finally, our data argue for a synergistic loss-of-support/gain-of-toxicity astrocyte functionality and an important role of WNT/β-catenin pathway activation as molecular mechanisms in FUS-ALS.
Although astrocyte reactivity has been proposed to be induced by microglia [52], the lack of microglia in our study suggests otherwise. Even though microglia are the primary immune-reactive cell type in the brain, our data demonstrate that astrocytes are equally relevant to produce pro-inflammatory cytokines and thereby play a central role in the abnormal immune response documented in ALS [58]. The early secretion of immune-regulatory cytokines IL-4 and especially IL-10 by immature astrocytes correlates with an initial anti-inflammatory and neuroprotective phase in ALS, where astrocytes and microglia try to modulate the abnormal response [59]. Subsequently, this astrocyte secretion of IL-4 and IL-10 in addition to the downregulation of associated molecular functions in R521H astrocytes suggest an auto-regulatory attempt to counteract the pro-inflammatory reaction and cytotoxic consequences. In ALS, the neuroprotective phase is later taken over by a cytotoxic phase, which is mediated by neurotoxic microglia [59], but likewise correlates with our astrocyte secretome. Notably, the secretion of IL-6 and IL-8 appeared prominent, and especially secreted IL-8 has been shown to be increased in cerebrospinal fluid of sALS patients emphasizing a common nominator in ALS [60, 61]. Interestingly, we observed a differentiation-dependent increase in the secretion of cytokines. This could be related to the clinical severity of the different mutations, as the pro-inflammatory cytokine response was more pronounced early in immature P525L astrocytes and late in more mature R521H astrocytes. Similarly, we saw an increase in active astrocytes and GFAP expression, a more pronounced cytoplasmic mislocalisation of FUS protein, and a more prominent reactive gene expression in P525L astrocytes in line with a more aggressive astrocytic phenotype. Furthermore, we observed a larger cytotoxic effect of P525L astrocytes on motor neuron neurite outgrowth and NMJ formation. Collectively these data demonstrate how the juvenile-onset P525L mutation causes more aggressive phenotypes in-vitro. However, to fully establish whether our observations are truly due to a mutation-dependent effect, further studies incorporating additional hiPSC lines are needed.
Jointly, our data argue for a complex role of astrocytes in FUS-ALS. The reactivity, secretion of cytokines, impairment of motor neuron neurites and NMJs all support an astrocytic involvement through a gain-of-toxicity function. Although we do not observe any cell death in our cocultures, these findings complement previous studies in which ALS astrocytic cytotoxic phenotypes were linked to excitotoxicity and/or motor neuron death [9, 21, 22, 62,63,64,65,66,67]. The lack of cell death in our system could be due to technical differences between protocols and the use of favourable culture conditions rather than a specific ALS-related mutation effect. Interestingly, the IPA canonical pathway analysis revealed a dominant downregulation of pathways, which overall can be divided into their involvement in either an immune response or homeostatic maintenance of neuronal networks. For the former group, a downregulation of HOTAIR (HOX transcript antisense RNA), STAT3 (signal transducer and activator of transcription 3) and Interferon signalling among others suggests a collective attempt to inhibit an immune response. Especially, HOTAIR regulates the NFκB-pathway [68], which is a major activator of the immune system response [69] and is involved in several neurodegenerative disorders [29, 70] including ALS [71, 72]. Notably, PI3K/AKT and BEX2 signalling, which were upregulated in R521H and P525L astrocytes, respectively, are also involved in NFκB-activation [73,74,75,76,77], and this dysregulation supports the heterogeneous reactive response observed in our astrocytes. In addition, modulations of the NFκB-pathway in astrocytes have proven unsuccessful in slowing down disease progression, suggesting that the astrocytic involvement in ALS extends beyond this single pathway [72, 78]. For the latter group, axonal guidance, synaptogenesis, endocannabinoid-, CREB- and CDK5 signalling are all important for neurodevelopment, network modulation and homeostatic maintenance [79,80,81]. Downregulation of these pathways, in addition to downregulation of several important receptors (neurotransmitter, glutamate, acetylcholine, etc.) from our GO molecular function analysis, indicates a loss-of-support function in FUS-ALS astrocytes, and complements previous astrocyte-mediated loss-of-support mechanisms in ALS [15,16,17,18,19]. In favor of this conclusion, a recent meta-analysis on sequencing data from both hiPSC and mouse ALS astrocytes with various ALS mutations confirms this synergistic interplay between loss-of-support and gain-of-toxicity mechanisms within the gene-expression, and further establishes it as a general mechanism in ALS [82]. In this context, our comparative differential gene expression analysis also revealed some overlap in the transcriptomic profile between FUS-P525L, SOD1 and C9orf72 hiPSC-derived astrocytes [42, 49]. This suggest a potential commonality across multiple fALS mutations, and further studies might reveal similar cytotoxic phenotypes.
Finally, our data argue for the involvement of the WNT/β-catenin pathway as an important player in the molecular mechanism of FUS-ALS. The WNT/β-catenin pathway is widely involved in cell survival, axonal guidance and NMJ formation [57]. For example, WNT ligands WNT3, WNT4 and WNT11 are involved in NMJ formation specifically enhancing the clustering of AChR on muscle cells as well as motor neuron axon outgrowth during NMJ innervation [83, 84]. Similarly, dysregulation of WNT ligands causes reduced AChR clustering and consequently disassembly of NMJs [85,86,87,88], which emphasizes the importance of WNT/β-catenin homeostasis. As such, the abnormal WNT/β-catenin pathway activation in our FUS-ALS astrocytes could be a central underlying mechanism of NMJ pathology in our multicellular system. Similarly, abnormal astrocyte activation of WNT ligands can activate a pro-inflammatory response within the astrocytes themselves [89], which could explain our secretory profile. In line with our observations, increased cytoplasmic β-catenin in astrocytes, neuronal β-catenin accumulation and activation [90, 91], β-catenin nuclear translocation [92], and dysregulation of WNT/β-catenin pathway components have been reported in the spinal cord, NMJs and muscles of ALS patients and in transgenic animal and in vitro models [53, 93,94,95,96,97,98], which discloses WNT/β-catenin pathway activation as a general underlying mechanism of ALS. Interestingly, it was demonstrated that WNT/β-catenin pathway activation in astrocytes increased neuroprotection in response to oxidative stress and inflammation [99], subsequently sparking the hypothesis that the activation of the WNT/β-catenin pathway in astrocytes could be a neuroprotective attempt to counteract motor neuron pathology in ALS through activation of the pathway within motor neurons themselves [57]. In support of this neuroprotective hypothesis, our P525L astrocytes successfully activated the WNT/β-catenin pathway within P525L motor neurons. As such, the combined effect of anti-inflammatory cytokine secretion and WNT/β-catenin pathway activation might thereby be an attempt to counteract the motor neuron cytotoxicity in the system, raising the question if mutant astrocytes are “all bad”. The inability to rescue or minimize the cytotoxicity is likely due to the overwhelming opposing toxic effects of the mutant astrocyte reactivity equally fuelled by the astrocytic WNT/β-catenin pathway dysregulation. Since WNT/β-catenin pathway balance is important to maintain optimal cellular homeostasis and NMJ integrity, the trend in activation of the WNT/β-catenin pathway within P525P motor neurons likely contributed to the impairment in neurite outgrowth and NMJ formation inflicted by the P525L astrocytes.
Until recently, astrocyte heterogeneity was a collectively accepted term used for their morphological differences and spatial location within the central nervous system [50]. The dynamic interplay between loss-of-support and gain-of-toxicity functions within our astrocyte populations specifies a heterogeneous functionality and favours a larger yet-to-be explored role of astrocytes in ALS. Transplantation of healthy rat astrocytes into transgenic rats expressing mutant human SOD1 protein has been shown to reduce microgliosis, attenuate motor neuron loss and extend disease duration [100]. In line with this, our IC astrocytes were able to improve or rescue all aberrations, which suggests a beneficial effect of gene therapy targeting the dynamic and migratory astrocytes rather than the static and post-mitotic motor neurons. Moreover, our data on the involvement of astrocytes on NMJ pathology in an all-human system endorse previous findings, where hiPSC-derived astrocytes from sALS patients were injected into mice and caused NMJ denervation and subsequent motor deficits [11]. This similarity between FUS-ALS and sALS demonstrates how astrocyte-induced NMJ toxicity is a general mechanism in ALS. Further integration of additional cell types such as upper motor neurons, interneurons and other glial cells would advance our understanding of the disease process in addition to enhancing the value of our NMJ model in drug testing and therapy development.
Conclusion
Our study demonstrates that astrocytes are important players in ALS pathogenesis causing impairment of motor neuronal network and NMJ formation and functionality through multiple gain-of-toxicity and loss-of-support mechanisms. Furthermore, we propose an astrocytic attempt to counteract the toxicity through WNT/β-catenin pathway upregulation in motor neurons, albeit with limited success. In addition, our fully human multicellular microfluidics model provides a platform for further studies and can be used for drug development and testing.
Availability of data and materials
• RNAseq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the Supplementary Table 1, Additional file 1. Microscopy data reported in this paper will be shared by the corresponding author upon reasonable request.
• ImageJ and Nikon software scripts are available from the corresponding author upon reasonable request.
• Any additional information required to reanalyse the data reported in this paper is available from the corresponding author upon reasonable request.
Abbreviations
- AC:
-
Astrocyte
- AChR:
-
Acetylcholine receptor
- ACTN2:
-
α-Actinin
- ADM:
-
Astrocyte differentiation medium
- ALDH1L1:
-
Aldehyde Dehydrogenase 1 Family Member L1
- ALS:
-
Amyotrophic lateral sclerosis
- AMM:
-
Astrocyte maturation medium
- APC:
-
Astrocyte progenitor cell
- AQP4:
-
Aquaporin-4
- BDNF:
-
Brain-derived neurotrophic factor
- BSA:
-
Bovine serum albumin
- Btx:
-
α-Bungarotoxin
- ChAT:
-
Choline acetyltransferase
- CNTF:
-
Ciliary neurotrophic factor
- EB:
-
Embryoid body
- EGF:
-
Epidermal growth factor
- fALS:
-
Familial ALS
- FBS:
-
Fetal bovine serum
- FDR:
-
False discovery rate
- FGF-2:
-
Fibroblast growth factor 2
- FTLD:
-
Frontotemporal lobar degeneration
- FUS:
-
FUS RNA binding protein
- GDNF:
-
Glial cell line-derived neurotrophic factor
- GFAP:
-
Glial fibrillary acidic protein
- GLM:
-
Generalized linear model
- GO:
-
Gene ontology
- hiPSC:
-
Human induced pluripotent stem cell
- HOTAIR:
-
HOX transcript antisense RNA
- IC:
-
Isogenic control
- ICC:
-
Immunocytochemistry
- IGF-1:
-
Insulin-like growth factor
- IPA:
-
Ingenuity Pathway Analysis
- KCl:
-
Potassium chloride
- LDH:
-
Lactate dehydrogenase
- MAP2:
-
Microtubule-Associated Protein 2
- MN:
-
Motor neuron
- MN-NPC:
-
Motor neuron neural progenitor cell
- MSD:
-
Meso Scale Discovery
- MyHC:
-
Myosin heavy chain
- MyoG:
-
Myogenin
- NDS:
-
Normal donkey serum
- NEFH:
-
Neurofilament heavy chain
- NIM:
-
Neuronal induction medium
- NMJ:
-
Neuromuscular junction
- NMM:
-
Neuronal maturation medium
- NPC:
-
Neural progenitor cell
- S100β:
-
S100 calcium-binding protein β
- sALS:
-
Sporadic ALS
- SEM:
-
Scanning electron microscopy
- SOX9:
-
SRY-Box Transcription Factor 9
- STAT3:
-
Signal transducer and activator of transcription 3
- SYP:
-
Synaptophysin
- TBST:
-
Tris-buffered saline with Tween
- TNF-α:
-
Tumour necrosis factor α
- Tubulin:
-
βIII-tubulin
- WB:
-
Western blot
References
Masrori P, Van Damme P. Amyotrophic lateral sclerosis: a clinical review. Eur J Neurol. 2020;27(10):1918–29.
Shatunov A, Al-Chalabi A. The genetic architecture of ALS. Neurobiol Dis. 2021;147:105156.
Zou ZY, Zhou ZR, Che CH, Liu CY, He RL, Huang HP. Genetic epidemiology of amyotrophic lateral sclerosis: A systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. 2017;88(7):540–9.
Picher-Martel V, Brunet F, Dupré N, Chrestian N. The Occurrence of FUS Mutations in Pediatric Amyotrophic Lateral Sclerosis: A Case Report and Review of the Literature. J Child Neurol. 2020;35(8):556–62.
Tyzack GE, Luisier R, Taha DM, Neeves J, Modic M, Mitchell JS, et al. Widespread FUS mislocalization is a molecular hallmark of amyotrophic lateral sclerosis. Brain. 2019;142(9):2572–80.
Nolan M, Talbot K, Ansorge O. Pathogenesis of FUS-associated ALS and FTD: insights from rodent models. Acta Neuropathol Commun. 2016;4(1):99.
Haidet-Phillips AM, Hester ME, Miranda CJ, Meyer K, Braun L, Frakes A, et al. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat Biotechnol. 2011;29(9):824–8.
Yamanaka K, Chun SJ, Boillee S, Fujimori-Tonou N, Yamashita H, Gutmann DH, et al. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci. 2008;11(3):251–3.
Di Giorgio FP, Boulting GL, Bobrowicz S, Eggan KC. Human Embryonic Stem Cell-Derived Motor Neurons Are Sensitive to the Toxic Effect of Glial Cells Carrying an ALS-Causing Mutation. Cell Stem Cell. 2008;3(6):637–48.
Ajmone-Cat MA, Onori A, Toselli C, Stronati E, Morlando M, Bozzoni I, et al. Increased FUS levels in astrocytes leads to astrocyte and microglia activation and neuronal death. Sci Rep. 2019;9(1):4572.
Qian K, Huang H, Peterson A, Hu B, Maragakis NJ, Ming G li, et al. Sporadic ALS Astrocytes Induce Neuronal Degeneration In Vivo. Stem Cell Reports. 2017;8(4):843–55.
Guttenplan KA, Weigel MK, Adler DI, Couthouis J, Liddelow SA, Gitler AD, et al. Knockout of reactive astrocyte activating factors slows disease progression in an ALS mouse model. Nat Commun. 2020;11(1):3753.
Hall ED, Oostveen JA, Gurney ME. Relationship of microglial and astrocytic activation to disease onset and progression in a transgenic model of familial ALS. Glia. 1998;23(3):249–56.
Haim LB, Rowitch DH. Functional diversity of astrocytes in neural circuit regulation. Nat Rev Neurosci. 2017;18(1):31–41.
Staats KA, Van Den Bosch L. Astrocytes in amyotrophic lateral sclerosis: Direct effects on motor neuron survival. J Biol Phys. 2009;35(4):337–46.
Rothstein JD, Van Kammen M, Levey AI, Martin LJ, Kuncl RW. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerisis. Ann Neurol. 1995;38(1):73–84.
Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, et al. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16(3):675–86.
Madji Hounoum B, Mavel S, Coque E, Patin F, Vourc’h P, Marouillat S, et al. Wildtype motoneurons, ALS-Linked SOD1 mutation and glutamate profoundly modify astrocyte metabolism and lactate shuttling. Glia. 2017;65(4):592–605.
Ferraiuolo L, Higginbottom A, Heath PR, Barber S, Greenald D, Kirby J, et al. Dysregulation of astrocyte-motoneuron cross-talk in mutant superoxide dismutase 1-related amyotrophic lateral sclerosis. Brain. 2011;134(9):2627–41.
Halpern M, Brennand KJ, Gregory J. Examining the relationship between astrocyte dysfunction and neurodegeneration in ALS using hiPSCs. Neurobiol Dis. 2019;132:104562.
Nagai M, Re DB, Nagata T, Chalazonitis A, Jessell TM, Wichterle H, et al. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci. 2007;10(5):615–22.
Tripathi P, Rodriguez-Muela N, Klim JR, de Boer AS, Agrawal S, Sandoe J, et al. Reactive Astrocytes Promote ALS-like Degeneration and Intracellular Protein Aggregation in Human Motor Neurons by Disrupting Autophagy through TGF-β1. Stem Cell Reports. 2017;9(2):667–80.
Kia A, McAvoy K, Krishnamurthy K, Trotti D, Pasinelli P. Astrocytes expressing ALS-linked mutant FUS induce motor neuron death through release of tumor necrosis factor-alpha. Glia. 2018;66(5):1016–33.
Marchetto MCN, Muotri AR, Mu Y, Smith AM, Cezar GG, Gage FH. Non-Cell-Autonomous Effect of Human SOD1G37R Astrocytes on Motor Neurons Derived from Human Embryonic Stem Cells. Cell Stem Cell. 2008;3(6):649–57.
Stoklund Dittlau K, Krasnow EN, Fumagalli L, Vandoorne T, Baatsen P, Kerstens A, et al. Human motor units in microfluidic devices are impaired by FUS mutations and improved by HDAC6 inhibition. Stem Cell Reports. 2021;16(9):2213–27.
Stoklund Dittlau K, Krasnow EN, Fumagalli L, Vandoorne T, Baatsen P, Kerstens A, et al. Generation of Human Motor Units with Functional Neuromuscular Junctions in Microfluidic Devices. J Vis Exp. 2021;175:e62959.
Guo W, Naujock M, Fumagalli L, Vandoorne T, Baatsen P, Boon R, et al. HDAC6 inhibition reverses axonal transport defects in motor neurons derived from FUS-ALS patients. Nat Commun. 2017;8(1):861.
Wang H, Guo W, Mitra J, Hegde PM, Vandoorne T, Eckelmann BJ, et al. Mutant FUS causes DNA ligation defects to inhibit oxidative damage repair in Amyotrophic Lateral Sclerosis. Nat Commun. 2018;9:3683.
Chandrasekaran A, Stoklund Dittlau K, Corsi GI, Haukedal H, Doncheva NT, Ramakrishna S, et al. Astrocytic reactivity triggered by defective autophagy and metabolic failure causes neurotoxicity in frontotemporal dementia type 3. Stem Cell Reports. 2021;16(11):2736–51.
Maury Y, Côme J, Piskorowski RA, Salah-Mohellibi N, Chevaleyre V, Peschanski M, et al. Combinatorial analysis of developmental cues efficiently converts human pluripotent stem cells into multiple neuronal subtypes. Nat Biotechnol. 2015;33(1):89–96.
Gholobova D, Gerard M, Terrie L, Desender L, Shansky J, Vandenburgh H, et al. Co-culture method to obtain endothelial networks within human tissue-engineered skeletal muscle. In: Rønning S, editor., et al., Myogenesis Methods in Molecular Biology, vol 1889 Humana Press. New York: NY; 2019. p. 169–83.
HannonLab. Fastx-toolkit. 2010.
Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17(1):10–12.
Morgan M, Anders S, Lawrence M, Aboyoun P, Pages H, Gentleman R. ShortRead: a bioconductor package for input, quality assessment and exploration of high-throughput sequence data. Bioinformatics. 2009;25(19):2607–8.
Langmead B, Salzberg S. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–9.
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–9.
Liao Y, Smyth GK, Shi W. FeatureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30(7):923–30.
Risso D, Schwartz K, Sherlock G, Dudoit S. GC-Content Normalization for RNA-Seq Data. BMC Bioinformatics. 2011;12:480.
Robinson MD, Smyth GK. Moderated statistical tests for assessing differences in tag abundance. Bioinformatics. 2007;23(21):2881–7.
Benjamini Y, Hochberg Y. Controlling the False Discovery Rate : A Practical and Powerful Approach to Multiple Testing. J R Stat Soc. 1995;57(1):289–300.
Birger A, Ben-Dor I, Ottolenghi M, Turetsky T, Gil Y, Sweetat S, et al. Human iPSC-derived astrocytes from ALS patients with mutated C9ORF72 show increased oxidative stress and neurotoxicity. EBioMedicine. 2019;50:274–89.
Gu Z, Eils R, Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32(18):2847–9.
Krämer A, Green J, Pollard J, Tugendreich S. Causal analysis approaches in ingenuity pathway analysis. Bioinformatics. 2014;30(4):523–30.
Mi H, Muruganujan A, Thomas PD. PANTHER in 2013: Modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 2013;41(D1):D377–86.
Thomas PD, Campbell MJ, Kejariwal A, Mi H, Karlak B, Daverman R, et al. PANTHER: A library of protein families and subfamilies indexed by function. Genome Res. 2003;13(9):2129–41.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods. 2001;25(4):402–8.
Berg S, Kutra D, Kroeger T, Straehle CN, Kausler BX, Haubold C, et al. Ilastik: Interactive Machine Learning for (Bio)Image Analysis. Nat Methods. 2019;16(12):1226–32.
Taha DM, Clarke BE, Hall CE, Tyzack GE, Ziff OJ, Greensmith L, et al. Astrocytes display cell autonomous and diverse early reactive states in familial amyotrophic lateral sclerosis. Brain. 2022;145(2):481–9.
Escartin C, Galea E, Lakatos A, O’Callaghan JP, Petzold GC, Serrano-Pozo A, et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat Neurosci. 2021;24(3):312–25.
Zhang J-M, An J. Cytokines. Inflammation and Pain Int Anesth Clin. 2007;45(2):27–37.
Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541(7638):481–7.
González-Fernández C, Gonzalez P, Andres-Benito P, Ferrer I, Rodríguez FJ. Wnt Signaling Alterations in the Human Spinal Cord of Amyotrophic Lateral Sclerosis Cases: Spotlight on Fz2 and Wnt5a. Mol Neurobiol. 2019;56(10):6777–91.
Jiang X, Guan Y, Zhao Z, Meng F, Wang X, Gao X, et al. Potential roles of the WNT signaling pathway in amyotrophic lateral sclerosis. Cells. 2021;10(4):839.
Jridi I, Canté-Barrett K, Pike-Overzet K, Staal FJT. Inflammation and Wnt Signaling: Target for Immunomodulatory Therapy? Front Cell Dev Biol. 2021;8:615131.
Yu L, Guan Y, Wu X, Chen Y, Liu Z, Du H, et al. Wnt signaling is altered by spinal cord neuronal dysfunction in amyotrophic lateral sclerosis transgenic mice. Neurochem Res. 2013;38(9):1904–13.
Gonzalez-Fernandez C, González P, Rodríguez F. New insights into Wnt signaling alterations in amyotrophic lateral sclerosis: A potential therapeutic target? Neural Regen Res. 2020;15(9):1580–9.
McCauley ME, Baloh RH. Inflammation in ALS/FTD pathogenesis. Acta Neuropathol. 2019;137(5):715–30.
Moreno-Martinez L, Calvo AC, Muñoz MJ, Osta R. Are circulating cytokines reliable biomarkers for amyotrophic lateral sclerosis? Int J Mol Sci. 2019;20(11):2759.
Mennini T, Giordano L, Mengozzi M, Ghezzi P, Tonelli R, Mantegazza R, et al. Increased IL-8 levels in the cerebrospinal fluid of patients with amyotrophic lateral sclerosis. Eur J Inflamm. 2009;7(1):39–44.
Kuhle J, Lindberg RLP, Regeniter A, Mehling M, Steck AJ, Kappos L, et al. Increased levels of inflammatory chemokines in amyotrophic lateral sclerosis. Eur J Neurol. 2009;16(6):771–4.
Rojas F, Cortes N, Abarzua S, Dyrda A, van Zundert B. Astrocytes expressing mutant SOD1 and TDP43 trigger motoneuron death that is mediated via sodium channels and nitroxidative stress. Front Cell Neurosci. 2014;8:24.
Arredondo C, Cefaliello C, Dyrda A, Jury N, Martinez P, Díaz I, et al. Excessive release of inorganic polyphosphate by ALS/FTD astrocytes causes non-cell-autonomous toxicity to motoneurons. Neuron. 2022;110(10):1656-1670.e12.
Madill M, McDonagh K, Ma J, Vajda A, McLoughlin P, O’Brien T, et al. Amyotrophic lateral sclerosis patient iPSC-derived astrocytes impair autophagy via non-cell autonomous mechanisms. Mol Brain. 2017;10(1):22.
Almad AA, Doreswamy A, Gross SK, Richard JP, Huo Y, Haughey N, et al. Connexin 43 in astrocytes contgributes to motor neuron toxicity in amyotrophic lateral scleoris. Glia. 2016;64(7):1154–69.
Almad AA, Taga A, Joseph J, Gross SK, Welsh C, Patankar A, et al. Cx43 hemichannels contribute to astrocyte-mediated toxicity in sporadic and familial ALS. PNAS. 2022;119(13):e2107391119.
Zhao C, Devlin AC, Chouhan AK, Selvaraj BT, Stavrou M, Burr K, et al. Mutant C9orf72 human iPSC-derived astrocytes cause non-cell autonomous motor neuron pathophysiology. Glia. 2019;68(5):1046–64.
Obaid M, Udden SMN, Deb P, Shihabeddin N, Zaki MH, Mandal SS. LncRNA HOTAIR regulates lipopolysaccharide-induced cytokine expression and inflammatory response in macrophages. Sci Rep. 2018;8:15670.
Dresselhaus EC, Meffert MK. Cellular specificity of NF-κB function in the nervous system. Front Immunol. 2019;10:1043.
Snow WM, Albensi BC. Neuronal gene targets of NF-κB and their dysregulation in alzheimer’s disease. Front Mol Neurosci. 2016;9:118.
Swarup V, Phaneuf D, Dupré N, Petri S, Strong M, Kriz J, et al. Deregulation of TDP-43 in amyotrophic lateral sclerosis triggers nuclear factor κB-mediated pathogenic pathways. J Exp Med. 2011;208(12):2429–47.
Frakes AE, Ferraiuolo L, Haidet-Phillips AM, Schmelzer L, Braun L, Miranda CJ, et al. Microglia induce motor neuron death via the classical NF-κB pathway in amyotrophic lateral sclerosis. Neuron. 2014;81(5):1009–23.
Naderi A. Molecular functions of brain expressed X-linked 2 (BEX2) in malignancies. Exp Cell Res. 2019;376(2):221–6.
Downward J. Mechanisms and consequences of activation of protein kinase B/Akt. Curr Opin Cell Biol. 1998;10(2):262–7.
Bai D, Ueno L, Vogt P. Akt-mediated regulation of NFkappaB and the essentialness of NFkappaB for the oncogenicity of PI3K and Akt. Int J Cancer. 2009;125(12):2863–70.
Naderi A, Liu J, Hughes-Davies L. BEX2 has a functional interplay with c-Jun/JNK and p65/RelA in breast cancer. Mol Cancer. 2010;9:111.
Naderi A, Teschendorff AE, Beigel J, Cariati M, Ellis IO, Brenton JD, et al. BEX2 is overexpressed in a subset of primary breast cancers and mediates nerve growth factor/nuclear factor-κB inhibition of apoptosis in breast cancer cell lines. Cancer Res. 2007;67(14):6725–36.
Crosio C, Valle C, Casciati A, Iaccarino C, Carrì MT. Astroglial inhibition of NF-κb does not ameliorate disease onset and progression in a mouse model for amyotrophic lateral sclerosis (ALS). PLoS ONE. 2011;6(3):e17187.
Xu J-Y, Chen C. Endocannabinoids in synaptic plasticity and neuroprotection. Neuroscientist. 2015;21(2):152–68.
Sakamoto K, Karelina K, Obrietan K. CREB: A multifaceted regulator of neuronal plasticity and protection. J Neurochem. 2010;116(1):1–9.
Kim SH, Ryan TA. CDK5 Serves as a Major Control Point in Neurotransmitter Release. Neuron. 2010;67(5):797–809.
Ziff OJ, Clarke BE, Taha DM, Crerar H, Luscombe NM, Patani R. Meta-analysis of human and mouse ALS astrocytes reveals multi-omic signatures of inflammatory reactive states. Genome Res. 2022;32(1):71–84.
Henriquez JP, Webb A, Bence M, Bildsoe H, Sahores M, Hughes SM, et al. Wnt signaling promotes AChR aggregation at the neuromuscular synapse in collaboration with agrin. PNAS. 2008;105(48):18812–7.
Gordon LR, Gribble KD, Syrett CM, Granato M. Initiation of synapse formation by Wnt-induced MuSK endocytosis. Development. 2012;139(5):1023–33.
Messéant J, Ezan J, Delers P, Glebov K, Marchiol C, Lager F, et al. Wnt proteins contribute to neuromuscular junction formation through distinct signaling pathways. Development. 2017;144(9):1712–24.
Wang J, Luo Z-G. The role of Wnt/β-catenin signaling in postsynaptic differentiation. Commun Integr Biol. 2008;1(2):158–60.
Wang J, Ruan NJ, Qian L, Lei WL, Chen F, Luo ZG. Wnt/β-catenin signaling suppresses Rapsyn expression and inhibits acetylcholine receptor clustering at the neuromuscular junction. J Biol Chem. 2008;283(31):21668–75.
Shen C, Li L, Zhao K, Bai L, Wang A, Shu X, et al. Motoneuron Wnts regulate neuromuscular junction development. Elife. 2018;7:e34625.
Gonzalez P, Rodríguez FJ. Analysis of the expression of the Wnt family of proteins and its modulatory role on cytokine expression in non activated and activated astroglial cells. Neurosci Res. 2017;114:16–29.
Pinto C, Medinas DB, Fuentes-Villalobos F, Maripillán J, Castro AF, Martínez AD, et al. β-catenin aggregation in models of ALS motor neurons: GSK3β inhibition effect and neuronal differentiation. Neurobiol Dis. 2019;130:104497.
Hawkins S, Namboori SC, Tariq A, Blaker C, Flaxman C, Dey NS, et al. Upregulation of β-catenin due to loss of miR-139 contributes to motor neuron death in amyotrophic lateral sclerosis. Stem Cell Reports. 2022;17:1650–65.
Bhinge A, Namboori SC, Zhang X, VanDongen AMJ, Stanton LW. Genetic Correction of SOD1 Mutant iPSCs Reveals ERK and JNK Activated AP1 as a Driver of Neurodegeneration in Amyotrophic Lateral Sclerosis. Stem Cell Reports. 2017;8(4):856–69.
McLoon LK, Harandi VM, Brännström T, Andersen PM, Liu JX. Wnt and extraocular muscle sparing in amyotrophic lateral sclerosis. Investig Ophthalmol Vis Sci. 2014;55(9):5482–96.
Wang S, Guan Y, Chen Y, Li X, Zhang C, Yu L, et al. Role of Wnt1 and Fzd1 in the spinal cord pathogenesis of amyotrophic lateral sclerosis-transgenic mice. Biotechnol Lett. 2013;35(8):1199–207.
Li X, Guan Y, Chen Y, Zhang C, Shi C, Zhou F, et al. Expression of Wnt5a and its receptor Fzd2 is changed in the spinal cord of adult amyotrophic lateral sclerosis transgenic mice. Int J Clin Exp Pathol. 2013;6(7):1245–60.
Chen Y, Guan Y, Zhang Z, Liu H, Wang S, Yu L, et al. Wnt signaling pathway is involved in the pathogenesis of amyotrophic lateral sclerosis in adult transgenic mice. Neurol Res. 2012;34(4):390–9.
Chen Y, Guan Y, Liu H, Wu X, Yu L, Wang S, et al. Activation of the Wnt/β-catenin signaling pathway is associated with glial proliferation in the adult spinal cord of ALS transgenic mice. Biochem Biophys Res Commun. 2012;420(2):397–403.
Kwan T, Kazamel M, Thoenes K, Si Y, Jiang N, King PH. Wnt antagonist FRZB is a muscle biomarker of denervation atrophy in amyotrophic lateral sclerosis. Sci Rep. 2020;10(1):16679.
Halleskog C, Dijksterhuis JP, Kilander MBC, Becerril-Ortega J, Villaescusa JC, Lindgren E, et al. Heterotrimeric G protein-dependent WNT-5A signaling to ERK1/2 mediates distinct aspects of microglia proinflammatory transformation. J Neuroinflammation. 2012;9:111.
Lepore AC, Rauck B, Dejea C, Pardo AC, Rao MS, Rothstein JD, et al. Focal transplantation – based astrocyte replacement is neuroprotective in a model of motor neuron disease. Nat Neurosci. 2008;11(11):1294–301.
Acknowledgements
The authors would like to thank VIB Nucleomics core (www.nucleomics.be) for their contributions, Nadine Becker-von Buch for technical assistance and Dr. Jérôme Wahis from Laboratory of Glia Biology (VIB-KU Leuven) for his valuable advice regarding live-cell calcium recordings of astrocytes.
Funding
This research was supported by the VIB, KU Leuven (C1 and “Opening the Future” Fund), the Agency for Innovation by Science and Technology (IWT; SBO-iPSCAF), the “Fund for Scientific Research Flanders” (FWO-Vlaanderen), Target ALS, the ALS Liga België (A Cure for ALS), the Belgian Government (Interuniversity Attraction Poles Program P7/16 initiated by the Belgian Federal Science Policy Office), the Thierry Latran Foundation and the “Association Belge contre les Maladies neuro-Musculaires” (ABMM). P.V.D holds a senior clinical investigatorship of FWO-Vlaanderen and is supported by the E. von Behring Chair for Neuromuscular and Neurodegenerative Disorders, the ALS Liga België and the KU Leuven funds “Een Hart voor ALS”, “Laeversfonds voor ALS Onderzoek” and the “Valéry Perrier Race against ALS Fund”. L.Te. is funded by an FWO mandate (1133520N) and L.Th. receives support via FWO (G0D3620N) and KU Leuven (C24E/20/067).
Author information
Authors and Affiliations
Contributions
K.S.D. designed and performed most of the experiments and data analysis and wrote the paper. L.Te. optimized the culturing of myoblasts. P.B. performed SEM. A.K. and N.C. wrote the Nikon scripts and assisted in the analysis. L.D.S. and R.J. performed RNA-sequencing experiment and data analysis. M.M. performed secretome analysis. L.Te. and L.Th. provided myoblasts. P.M. and P.V.D. provided ideas for the project. P.H. and K.F. provided the astrocyte differentiation protocol and ideas for the project. L.V.D.B. supervised the project and edited the manuscript. All authors read and approved the final version of the paper.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Written informed consent was obtained from all subjects who provided tissue samples. The use of patient fibroblasts for the generation of hiPSCs was approved by the ethics committee of University Hospital Leuven (n° S50354 and S63792), while the use of myoblasts was approved by the ethical commission (n° NH019-2020–04-02).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplementary Table 1.
List of material and resources.
Additional file 2: Supplemental Figure 1.
Astrocyte differentiation verification with bright-field microscopy, qPCR and RNAseq. Related to Fig. 1. Supplemental Figure 2. Astrocyte differentiation verification with ICC. Related to Fig. 1. Supplemental Figure 3. Astrocyte reactivity analysis. Related to Figs. 1 and 2. Supplemental Figure 4. Astrocyte secretome analysis. Related to Fig. 2. Supplemental Figure 5. Motor neuron and myotube differentiation verification with ICC. Related to Fig. 3. Supplemental Figure 6. Astrocyte and motor neuron coculture and LDH analysis. Related to Fig. 4. Supplemental Figure 7. NMJ morphology. Related to Fig. 6. Supplemental Figure 8. Myotube innervation. Related to Fig. 6.
Additional file 3. Astrocyte calcium activity_P525P.
Additional file 4. Astrocyte calcium activity_P525L.
Additional file 5. Astrocyte calcium activity_R521R.
Additional file 6. Astrocyte calcium activity_R521H.
Additional file 7. NMJ functionality_MN_P525P_AC_P525P.
Additional file 8. NMJ functionality_MN_P525P_AC_P525L.
Additional file 9. NMJ functionality_MN_P525L_AC_P525L.
Additional file 10. NMJ functionality_MN_P525L_AC_P525P.
Additional file 11. NMJ functionality_MN_P525P.
Additional file 12. NMJ functionality_MN_P525L.
Additional file 13. NMJ functionality_Myotube positive control
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Stoklund Dittlau, K., Terrie, L., Baatsen, P. et al. FUS-ALS hiPSC-derived astrocytes impair human motor units through both gain-of-toxicity and loss-of-support mechanisms. Mol Neurodegeneration 18, 5 (2023). https://doi.org/10.1186/s13024-022-00591-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13024-022-00591-3