
Abstract
One of the primary genetic risk factors for Alzheimer’s disease (AD) is the presence of the Ɛ4 allele of apolipoprotein E (APOE). APOE is a polymorphic lipoprotein that is a major cholesterol carrier in the brain. It is also involved in various cellular functions such as neuronal signaling, neuroinflammation and glucose metabolism. Humans predominantly possess three different allelic variants of APOE, termed E2, E3, and E4, with the E3 allele being the most common. The presence of the E4 allele is associated with increased risk of AD whereas E2 reduces the risk. To understand the molecular mechanisms that underlie APOE-related genetic risk, considerable effort has been devoted towards developing cellular and animal models. Data from these models indicate that APOE4 exacerbates amyloid β plaque burden in a dose-dependent manner. and may also enhance tau pathogenesis in an isoform-dependent manner. Other studies have suggested APOE4 increases the risk of AD by mechanisms that are distinct from modulation of Aβ or tau pathology. Further, whether plasma APOE, by influencing systemic metabolic pathways, can also possibly alter CNS function indirectly is not complete;y understood. Collectively, the available studies suggest that APOE may impact multiple signaling pathways and thus investigators have sought therapeutics that would disrupt pathological functions of APOE while preserving or enhancing beneficial functions. This review will highlight some of the therapeutic strategies that are currently being pursued to target APOE4 towards preventing or treating AD and we will discuss additional strategies that holds promise for the future.
Similar content being viewed by others
Background
Apolipoprotein E4 is the major genetic risk factor in Alzheimer’s disease
Alzheimer’s disease (AD), neuropathologically characterized by extracellular amyloid β (Aβ) deposition and intracellular neurofibrillary tangles (NFT) of tau protein, is the most prevalent neurodegenerative dementia affecting millions of people worldwide [1]. One of the primary genetic risk factors for sporadic AD, also referred to as late onset AD (LOAD), is the presence of the E4 isoform of apolipoprotein E (APOE) protein [2].
Humans have three major APOE alleles (E2, E3, and E4) [3]. APOE3 is the reference allele present in the majority of the population; the APOE4 allele increases the risk of AD in a dose- and age-dependent manner whereas the APOE2 allele is associated with reduced risk of AD [4,5,6]. Though there are variations based on sex and ethnicity, it is estimated that APOE2 homozygotes have a 40% reduced risk of developing AD [7]. Presence of APOE2 delays the age of onset in the Paisa kindred of familial AD cases [8], reinforcing the idea that APOE2 isoforms are protective against familial AD. In humans, the APOE2 allele while being protective against AD, is associated with elevated plasma levels of cholesterol and triglycerides and a condition called dysbetalipoproteinemia that is associated with coronary artery disease [9]. On the other hand, APOE4 is associated with increased risk of atherosclerosis and increasing risk of AD by as much as 8-12x in homozygotic humans. There is a general consensus in the literature that AD patients with the APOE4 isoform have accelerated onset of dementia, worse memory performance and higher Aβ burden than APOE4 non-carriers [10], though the isoform-dependent effects on tauopathy remain unclear [11, 12]. APOE4 can also exacerbate functional abnormalities such as neuronal network connectivity independent of gross structural changes or AD type proteinopathy [13]. These topics have been covered previously in excellent reviews and therefore not discussed further [10, 14, 15]. These data suggest that it may be necessary to both restore some critical APOE function in E4 carriers while also inhibiting the activity of APOE4 in promoting AD-related Aβ proteinopathy [14].
Peripheral and CNS pools of APOE are independent
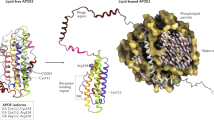
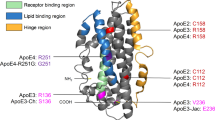
APOE is a 299 amino acid protein, with an apparent molecular mass of ~36kDa whose primary function is that of a cholesterol transporter [14]. The three isoforms differ by one amino acid each at positions 112 and 158 that has profound effects on their functions. Both APOE2 (Cys112, Cys158) and APOE3 (Cys112, Arg158) preferentially interact with small, phospholipid-enriched high-density lipoproteins (HDL), while APOE4 (Arg112, Arg158) has higher propensity to be associated with larger, triglyceride-enriched lipoproteins or VLDL [16]. A further distinction is that among all the isoforms, APOE2 has the lowest binding affinity for low-density lipoprotein (LDL) receptors [17]. Mice have a single allele of Apoe that differs at multiple positions from human APOE, but encodes Arg at the positions cognate to 112 and 158 of human APOE. Most of what is known about APOE has been derived from studies in mice and human cell culture models. Studies in mice have examined both endogenous mouse Apoe and expressed human APOE. For the purposes of this review, we will use the human and mouse nomenclature interchangeably as appropriate for the model systems used, defaulting to APOE when discussing general features of APOE biology.
In the CNS, APOE is primarily synthesized by astrocytes and in certain circumstances, it is also produced by microglia and neurons [18,19,20,21]. APOE has myriad functions in the CNS that include immunomodulation, signal transduction, proteostasis regulation and synaptic plasticity [14, 22]. The peripheral pool of plasma APOE is produced mainly in the liver, and to a lesser extent by the adrenal gland and macrophages. In the periphery, in addition to regulating lipid metabolism, APOE has a key role in controlling cardiovascular function and systemic inflammation [23]. This pool of APOE exists mostly independent of the CNS pool under normal circumstances [24, 25]. An important difference between the CNS and peripheral APOE pools is that only peripheral APOE4 shows faster turnover rate compared to APOE3 and APOE2 in humans and humanized mice [22, 23]. Astrocytic and plasma APOE lipoprotein particles are also structurally different and the former is thought to lack the cholesteryl ester core [26]. Therefore, it is possible that the structure-function relationship of peripheral and CNS pools of APOE to the development of AD and non-AD pathologies might be distinct, suggesting that these two pools of APOE can potentially act independently as risk factors in regulating pathogenesis during normal aging or in neurodegenerative dementias.
Because of the pleiotropic functions of APOE isoforms in the CNS and periphery, mechanistically dissecting the role of APOE in the context of AD and related disorders is fraught with complications. This uncertainty over potential mechanism of action creates a conundrum in that the E4 allele may cause disease by both a loss of function or gain of function, depending on the cellular context [14]. A recent report had serendipitously identified a mouse model with intact peripheral Apoe levels and thus normal plasma lipid profile but with extremely low levels of brain Apoe. These mice have impaired synaptic plasticity but their spatial memory skills are intact [24], suggesting that peripheral and CNS APOE may have distinct effects on CNS function. On the other hand, absence of hepatic APOE does not affect the APOE4-dependent induction of Aβ pathologies in young APP/PS1 female mice, suggesting that plasma APOE4 may have little influence on initiation of Aβ pathologies in the brain [27]. With this knowledge, it is reasonable to explore treatment options that would preferentially modify the CNS pool of APOE without affecting the peripheral sources, thus also avoiding systemic metabolic syndromes.
Rodent models as exemplars of human APOE function
The Apoe deficient mice, Apoe hypomorphic mice and APOE knock-in mice have been key resources in the field of atherosclerosis biology, cardiovascular disease and peripheral inflammation [28]. For the most part, the data are concordant between mouse studies and humans [29]. However, there are some critical differences between mouse and human lipoprotein biology that can impact the interpretation of APOE-related studies in mice. In mice, circulating cholesterol is predominantly associated with HDL whereas it is bound to LDL in humans [30]. In addition, mice lack the cholesteryl ester transfer protein (CETP) gene which transfers cholesteryl esters and triglycerides between lipoproteins [31].
Perhaps the most commonly used models to study human APOE function in the CNS are the human APOE targeted replacement (TR) mice from Nobuyo Maeda’s lab [32,33,34]. The APOE4 TR mice, in which the endogenous Apoe gene has been replaced with human APOE4, display various phenotypes including altered cholesterol trafficking in the brain, blood brain barrier (BBB) leakiness and cognitive deficits [35,36,37,38,39]. However, simply replacing the endogenous mouse Apoe gene with the human APOE4 gene does not produce the primary neuropathologies (Aβ and NFT) found in AD patients. Overall, the lack of spontaneously occurring AD-type pathology in APOE4 TR mice has limited its use as a stand-alone model of AD.
Concurrence of experimental data across different systems
A large number of studies have used rodent models (such as APOE TR), in vitro models including human induced pluripotent stem (iPS) cells and primary rodent cultures as well as data from human biosamples to delineate apoE-related pathologies. Most of the studies show isotype-specific and directionality-specific concordance between these experimental paradigms (Fig. 1). For example, the isoform-dependent effects of APOE on Aβ clearance and Aβ aggregation are in complete agreement in these different systems [42,43,44,45,46,47,48,49,50,51,52,53]. As in humans, presence of APOE4 increases Aβ deposition burden in APP transgenic mice relative to age-matched APOE2 TR mice. This has also been demonstrated in human iPSC-derived glial cultures, where APOE4 impairs glial Aβ uptake and phagocytosis compared to APOE3 [48]. On the other hand, there is a current lack of consensus regarding the relationship between tauopathy and APOE isoform as demonstrated by studies showing a pathogenic interaction of tau to APOE4 [11, 54] or APOE2 [12]. Further, human iPSC derived neurons [55] as well as organoids [49] that express APOE4 accumulate higher levels of phosphorylated tau when compared to neurons expressing APOE3. Importantly, data from humans present no clear association between APOE4 genotype and severity of NFT pathology [56, 57]. Different APOE isoforms have differential pathogenic effect on various metabolic pathways such as cardiovascular function, lipid transport, insulin signaling and glucose metabolism across these model systems [36, 37, 58,59,60,61,62,63,64,65,66,67]. There is a clear consensus regarding APOE4 isoform-dependent pathogenic effect on cardiovascular function in mouse models, in vitro studies and human studies [34, 35, 41, 68]. In the case of lipid transport, several studies have shown that APOE4 carriers have increased hypolipidated APOE compared to APOE3 and APOE2 carriers [69] along with reduced APOE levels in the CSF of Aβ-positive APOE4 carriers [70]. These observations hold true in primary rat neuron cultures and human iPS cell-derived astrocytes [71, 72] as well as APOE TR mice [73, 74]. However, there are some conflicting reports from human studies, which did not observe any isoform-dependent differences in APOE levels in the CSF collected from individuals at different ages [41].

Congruence of the effects of apoE between human studies, mouse models of AD and in vitro cell culture models. apoE influences multiple pathways in the AD cascade in an isoform-dependent manner. We compared the concurrence of available research data in mouse models and in vitro models versus clinical studies with human patients. Pathways indicated in the green color indicate a broad consensus of APOE isoform effect between mice, men and in vitro models where E4 is associated with an increased pathological risk when compared to E3 or E2 isoforms (E4>E3>E2). Data from the pathways indicated in yellow background are not in complete congruence between human studies, mouse model experiments and in vitro data. Interestingly, even within a set of studies in a given experimental system, there is disagreement in between the observations, which is marked by superscripted symbols that refers to the disparate studies. The symbols (< or >) indicate the order of increased pathological effect for the APOE isoforms. The effects listed here are specific to only classical AD pathology and excludes data on α-synuclein and TDP43 which are associated with diseases such as PDD and DLB. *, conflicting reports [see ref 40]; #, conflicting reports [see ref 41]; ¶, studies compared APOE4 TR, Apoe KO, and wild type C57BL6J mice. The references presented are representative and not an exhaustive list
APOE has been implicated in other metabolic and cognitive functions. In the case of insulin signaling and glucose metabolism, data from human brain scans, mice and in vitro cell culture largely agree that APOE4 isoforms cause the most functional impairment [58,59,60,61,62,63]. In terms of regulating brain function, APOE4 is the most pathogenic in terms of brain connectivity and default mode network function in humans whereas the evidence comparing network connectivity in mouse models carrying APOE4 genotype is uncertain as the experimental controls did not include the APOE3 or APOE2 mice cohorts [13, 49, 75,76,77,78,79,80,81,82,83]. In human iPS derived neurons, APOE4 led to elevated number of synapses and increased frequency of synaptic transmission [49]. Regarding inflammation, there is a general agreement across models that APOE4 is the most pathogenic [11, 84,85,86,87,88]. In contrast, there is evidence implicating APOE4 in impaired BBB integrity in humans, mouse models and cell culture models, although a study in APOE4 TR mice did not reveal any APOE-related dysfunction in BBB [36, 37, 40, 67, 89,90,91]. Overall, most of the data from rodent models and human patients show congruence (Fig.1). However, given that there are basic differences between mice and human lipid profiles as well as the structure of apoE itself, it is important to remain cautious of the inherent variations that might affect directly translating APOE targeted therapeutics from mouse models to humans.
Therapies in AD
AD still has no effective treatments or therapies despite years of research. Dozens of drugs have proceeded to clinical trials, ranging from Aβ targeting antibodies to therapies targeting tau or metabolic pathways [92]. Several factors may have influenced these discouraging outcomes - perhaps the drugs are targeting the wrong pathological substrates, or that the treatments are being administered too late, or that a multi-target drug design is needed [93]. With the steady growth of an aging population, the increasing cost for care, and the failure of therapies in clinic, there is a call for more targeted ‘precision therapy’ - treating AD patients stratified based on their APOE genotype.
It is well-known that targeting anti Aβ immunotherapies to patients stratified for APOE genotype can lead to better outcomes. In particular, in MCI patients the APOE4 allele seems to adversely affect the therapy outcomes by modulating the treatment efficacy (disease progression) or safety profile (vulnerability to brain edema) relative to other APOE alleles [94]. Having said that, AD therapies directly targeting specific APOE isoforms are still mostly in developmental phases [95]. It is also possible that such APOE targeted therapies could help with co-morbidities associated with dementia or aging, such as diabetes and cardiovascular disease for E4 carriers, vascular dementia for E4 carriers, neuroinflammation for E4 carriers and type III hyperlipoproteinemia for E2 carriers. Whether APOE by itself is druggable is debatable; however, it is tempting to suggest that targeting CNS APOE specifically early in the disease process could alter the pathologic trajectory of AD either directly by altering CNS pathologies like Aβ and tau and perhaps indirectly by influencing related sequelae, such as inflammation, metabolic impairment and vascular dysfunction. Ultimately, it is possible that a cocktail of drugs targeting APOE function in conjunction with other anti-Aβ approaches that either limit Aβ plaques or inhibit Aβ production can be used at different stages of the disease to achieve significant disease modification.
APOE as a therapeutic target in AD
In the next few sections, we will consider currently available preclinical interventions, therapies that are in early clinical studies in AD as well as some new research on emerging targets that target APOE specifically (Table 1).
Altering levels of APOE4 as a potential disease modifying therapy
APOE, especially APOE4, binds to Aβ, playing a key role in Aβ deposition and clearance. Several studies have shown that simply reducing APOE4 levels (such as cre-mediated excision of APOE4 or creating haploinsufficient APOE4 models) lowers brain Aβ levels in APP transgenic mice [96, 97]. Other approaches such as blocking Aβ-APOE4 interaction can also lead to beneficial effects, prompting the development of strategies to either reduce the availability of APOE4 or prevent its toxic interactions.
Anti-APOE4 immunotherapies
Similar to anti-Aβ antibody-based therapies, the idea behind anti-APOE4 antibodies is that these antibodies will be able to cross the BBB and neutralize the negative effects of APOE4, even if only a small amount of antibodies can effectively enter the brain [98]. APOE4 has already been implicated in Aβ deposition, and along with other amyloid-associated proteins it is found in Aβ deposits. Thus, the idea is that if isoform-specific antibodies can sequester pathogenic forms of APOE, it can prevent Aβ build-up in the brain. Indeed, in mouse models, anti-APOE antibodies can efficiently inhibit the formation of Aβ deposits when introduced before the onset of pathology [98]. More interestingly, these antibodies were also able to attenuate plaque burden when introduced in mice with pre-existing Aβ deposits, suggesting that this antibody could work as a therapeutic agent [98]. In a subsequent study, anti-APOE antibodies also led to improved spatial learning performance and resting-state functional connectivity while having no effect on total plasma cholesterol in APP transgenic mice [99]. In this study, topical application of anti-APOE antibodies directly onto the brain prevented deposition of new Aβ plaques as well as cleared pre-existing plaques. The fact that these anti-APOE antibodies can disrupt the direct binding of apoE to Aβ deposits is very promising, as this might work synergistically with anti Aβ immunotherapy in APOE4 patients to achieve a higher degree of Aβ reduction. More recently, Liao et al. reported that the antibody ‘HAE-4’ that preferentially recognizes the nonlipidated forms of APOE4/APOE3 over the lipidated versions is highly effective in preventing Aβ deposition by a FcγR-dependent mechanism in an APP/APOE4 mouse model [100]. Following direct infusion into the brain or following intraperitoneal administration, HAE-4 reduces total Aβ plaque burden but does not alter the fibrillar plaque load. Surprisingly, this antibody when administered peripherally was more efficient at CNS target engagement than when administered via direct brain infusion. This study is important in two ways – one that it demonstrates that non-lipidated forms of APOE4 may be preferentially pathogenic and second, that since the non-lipidated form of APOE4 is a small fraction of the total CNS APOE burden, this antibody would not be titered out by total APOE and could be efficacious at a lower or less frequent dose [101].
Antisense oligonucleotide therapy
Based on the hypothesis that the reduction of APOE4 expression could reduce Aβ accumulation and thereby alleviate Aβ pathology and cognitive deficits that typically follow, some groups have used antisense knockdown approaches. Antisense oligonucleotides (ASOs) are synthetic polymers that can be used as therapeutic agents by disrupting the synthesis of a particular protein and are considered as first line treatments in several neurodegenerative disorders such as polyneuropathy, muscular dystrophies and spinal muscular atrophy [102]. With regard to AD, there are only a few investigational ASO mediated therapies in clinical testing – a notable one being the anti tau ASO in Phase1/2 (BIIB080 from Ionis/Biogen/Washington University) [103]. In regard to targeting APOE expression with ASOs, the effort is still in the preclinical phases of testing.
ASOs targeting APOE receptors have also been tested in AD mouse models, for example an ASO specific for corrective splicing of ApoER2 resulted in improved synaptic function as well as learning and memory in the CRND8 mouse model of Aβ pathology [104]. In mouse models, ASO treatment targeting specifically APOE have been successful in reducing Aβ pathology in APP/PS1 mice when treated before the onset of Aβ deposition [105] (Table 1). However, it is unclear whether such knock-down strategies will work in the presence of pre-existing Aβ deposits, suggesting that this type of therapy may have a limited window of opportunity. On a positive note, compared to agonists of ApoE receptors that result in systemic adverse effects in lipid metabolism, ASOs do not show such side effects. Combined with the successful ASO based therapies being tested in Huntington’s and Amyotrophic Lateral Sclerosis (ALS) patients and the latest developments in ASO chemistry that can dramatically improve its pharmacokinetic and pharmacodynamic properties, ASO based therapies do have potential as promising future therapeutic for AD patients [106].
Upregulating APOE expression through nuclear receptor agonism
APOE expression is induced by the nuclear receptors, peroxisome proliferator-activated receptor gamma (PPARγ) and liver X receptors (LXR) in coordination with retinoid X receptors (RXRs) [107]. GW3965, an agonist for LXR, increases Abca1 and Apoe protein levels, reduces Aβ levels and improves cognition in the APP/PS1 mouse model [108]. This result was Abca1-dependent, as GW3965 failed to alter Aβ levels in APP transgenic mice lacking Abca1. In another study, the RXR agonist Bexarotene (Targretin®), used to treat T cell lymphoma, was used in the APP/PS1 transgenic mouse model. In a study involving a relatively small cohort, Bexarotene treatment reduced Aβ accumulation in an APOE-dependent manner when orally administered to these mice [109], though several groups were unable to recapitulate the beneficial effect on plaque burden in similar mouse models [110,111,112]. Based on the original study that showed that bexarotene was effective in both preventive and therapeutic modes, it was incorporated in a proof of mechanism Phase IB trial in E3/E3 healthy adults [113] (Table 1) as well as tested in a small cohort of AD patients, called BEAT-AD study [114]. In the BEAT-AD study, bexarotene lowered CNS Aβ levels (by PET imaging) but did not produce any cognitive benefits [114]. Unfortunately, bexarotene treatment increased blood lipid levels in these patients increasing their risk for stroke and heart attack. In a Phase IB proof of mechanism trial in young healthy APOE3 carriers, researchers were able to measure plasma and CSF levels of APOE using the stable isotope leveling kinetics (SILK) method. Though APOE levels increased moderately in the CSF, there was no effect on synthesis or clearance of Aβ in CSF in these individuals [113]. One reason for this may be the poor CNS penetration of bexarotene in human patients (~low nM range) [113]. Notably, in mice the BBB is extremely permeable to bexarotene [115]. This finding raises a general cautionary issue regarding translation of drugs from rodents to humans. Coupled with the hepatotoxicity of bexarotene, the low CNS penetrance of drug resulted in disappointing forecast for translation to AD patients.
Stimulating APOE expression through HDAC inhibition
Histone deacetylase (HDAC) is a class of enzymes that remove acetyl groups from histones in DNA leading to gene silencing [116]. HDACs have been shown to play a central role in regulation of genes involved in the lipid metabolism pathway [117] as well as genes involved in long term memory formation and cognition [118]. A recent study in human astrocytoma cells showed that HDAC inhibition can stimulate APOE expression, independent of LXR and RXR [119]. Through the use of a phenotypic screening strategy utilizing various chemogenomics libraries, pan Class I HDAC inhibitors (MS275 and CI994) were found to increase APOE expression and secretion by astrocytes via an LXR-independent pathway [119]. These recent studies offer a new approach towards modulating APOE function.
Restoring or recalibrating APOE functions can also alleviate CNS and peripheral pathologies
Another option for potentially exploiting APOE functionality for AD treatment is regulating or restoring the normal function of APOE that is typically lost, especially in patients carrying the APOE4 isoform. Investigators have been pursuing strategies to raise overall levels of APOE function by increasing its lipidation as well as using small molecules to modulate APOE4 structure or function to more closely resemble APOE3. Some of these methodologies are primarily geared to recoup the loss of function in APOE in the APOE4 patients whereas others target the toxic gain of function aspects that APOE4 may have on AD-related pathology.
Small molecules that enhance ABCA1-mediated APOE4 lipidation
Among all APOE isoforms, APOE4 is unique in that it has increased propensity of domain-domain interactions that reduces lipid binding to the C terminal domain leading to loss of stability and function [120, 121]. The presence of Arg112 in APOE4 enhances the intramolecular interaction between its N-terminal domain and the C-terminal domain via a salt bridge known as the APOE4 domain interaction. As a result, APOE4 is typically hypolipidated or ‘lipid-depleted’ which has been postulated to correlate with the pathogenicity inherent to APOE4 [69]. In general, APOE lipidation is highly reliant on the ATP-binding cassette transporter A1, or ABCA1, which moves lipids into apolipoproteins and is known to protect against atherosclerosis [122]. Indeed, humans lacking functional ABCA1 have lower APOE levels and increased risk of AD and cardiovascular disease [123]. Consistent with this observation, deficiency of Abca1 exacerbates amyloidogenesis while overexpression of ABCA1 reduced the amyloid load in PDAPP transgenic mice [124]. Supporting the hypothesis that ABCA1-mediated lipidation is crucial Aβ clearance, subsequent studies have upregulated ABCA1 with peptides and various small molecules. An example of a small peptide that activates Abca1 is CS-6253 (Table 1). Intraperitoneal injection of CS-6253 into APOE4 TR mice 1) upregulated Abca1; 2) induced lipidation of APOE4; and 3) reduced cognitive deficits, tau hyperphosphorylation and Aβ accumulation [125]. In a follow-up study using APOE4 TR and APOE3 TR mice, the authors showed that CS-6253 also normalizes plasma APOE4 lipidation and stability to match APOE3 mice and additionally, this peptide was able to partially normalize plasma apoA-I and apoJ levels in APOE4 TR mice [126]. Another strategy to upregulate Abca1 is by using ASOs against microRNA-33. Inhibition of microRNA-33 by ASOs in cultured neurons and APP transgenic mice reduces Aβ levels [127, 128]. With the assumption that these drugs do not disrupt lipidation state and the normal biological function of APOE3, these studies support the notion that activation of ABCA1 to stabilize lipidation profile of APOE4 is a viable therapeutic target. Taken together, these studies demonstrate that correcting the hypolipidation state of APOE4 may be enough to alleviate AD-type pathologies.
Small molecules as APOE4 structure correctors
The domain interaction property of APOE4 reduces its secretion from cells [129] and concurrently makes it protease-labile [130], leading to pathogenic effects [131]. Thus, another potential therapy would be the disruption of this APOE4 domain interaction using ‘structural correctors’ which are expected to negate the pathological consequences of this domain interaction (Table 1). A study using a FRET system coupled with high throughput screening identified several small molecules that could be used as structural correctors [132]. Treatment of Neuro-2a cells expressing APOE4 with such structure correctors caused the protein to become more ‘APOE3-like’ both structurally and functionally. By restoring mitochondrial cytochrome c oxidase levels, this treatment reversed some of the detrimental effects of APOE4 in Neuro-2a cells. In another study using a human cell line, similar effects were observed using the small molecule structure corrector PH002. The compound decreased APOE4 fragmentation, increased GABAergic neuron numbers, reduced phosphorylated tau and Aβ levels in a dose-dependent manner [55]. The studies provide proof of concept that disrupting the APOE4 domain interaction using structure correctors could be druggable target in AD.
APOE mimetic peptides regulate function via competing for receptor binding
Using peptide mimetics that are structurally similar to the lipid binding class A amphipathic helix found in apoE, it is possible to regulate the lipidation and secretion of APOE. These peptides are so designed as to promote cholesterol trafficking, anti-inflammatory signaling and anti-thrombotic effects – properties that have been used in targeting systemic disorders such as atherosclerosis and coronary artery disease [133] or acute brain injury models [134,135,136]. One example is an 18 amino acid peptide with no known natural homologs called 4F (Ac-D-W-F-K-A-F-Y-D-K-V-A-E-K-F-K-E-A-F-NH2) that binds to LDL (particularly oxidized phospholipids and unsaturated fatty acids) and HDL at a site that is recognized by APOE [137]. In primary glial cell cultures derived from humans or mice, 4F increased APOE lipidation and APOE secretion [137] and reversed aggregated Aβ-induced blockage of glial APOE secretion. In a second study using APP overexpressing Drosophila, two novel APOE mimetics, COG 112 and COG 113, prevented neurodegeneration and improved memory, though Aβ deposition was not changed [138]. This suggests that such APOE mimetics can alter AD-type dysfunction through altering lipid metabolism that may be independent of Aβ pathology. These peptides, when used in CVND-AD transgenic mice (SwDI-APP/NOS2(-/-)) improved memory as well as reduced Aβ plaques and phosphorylated tau levels [139]. One study showed that such mimetic peptides are efficacious in APOE3 TR or APOE2 TR mice, but had no effect in APOE4 TR mice [140], suggesting isoform-specificity. Another APOE mimetic peptide derived from the receptor binding region of APOE α helix, CN-105 (Ac-V-S-R-R-R-NH2) has successfully completed Phase I clinical trial in patients with intracerebral hemorrhage (ICH) (Table 1). This peptide is BBB penetrant and reduces neuroinflammation and neuronal injury in mouse models of acute brain injury mouse models [135, 136] but this peptide has not been tested in rodent AD models. Given the beneficial role of APOE mimetics, future studies in AD mouse models and cell culture systems with such mimetics are warranted.
Small molecule inhibitors of APOE-Aβ interactions
As previously stated, APOE, especially APOE4, is normally found within Aβ deposits [141]. Inhibitors of protein–protein interactions (PPI), once considered undruggable, are now emerging as a tour de force because of dramatic improvement in understanding of PPI scaffold chemistry [142]. An advantage of this method is that these are often naturally occurring molecules that can be very selective because of their precise targeting [143]. One such inhibitor that disrupts the binding of APOE to Aβ is a peptide mimetic called Aβ12-28P, which is a non-fibrillogenic and non-toxic Aβ derivative that happens to be BBB permeant [144]. This peptide, by blocking the binding of APOE and Aβ at residues 12 to 28, reduced Aβ-induced neurotoxicity in cell culture. Further studies revealed that Aβ12-28P had a strong pharmacological effect in vivo where systemic administration of the peptide resulted in reduction of Aβ deposits and in general a reduction of CNS Aβ in two different APP transgenic mouse lines [145]. Administration of Aβ12-28P also prevented working memory deficits in mice, bolstering its further translatability [145, 146].
APOE is expressed predominantly from astrocytes in the CNS [147, 148]. However, APOE synthesized by astrocytes can be neurotoxic, to the extent that specifically deleting astrocytic Apoe rescues spatial learning and memory deficits in the APP/PS1 mouse model [149]. This is also supported by a study that used a co-culture system of neurons and astrocytes to investigate the role of APOE on intraneuronal accumulation of Aβ [150]. Intraneuronal Aβ accumulation was higher in neurons co-cultured with wild-type mouse astrocytes compared to the cultures exposed to Apoe KO astrocytes reinforcing the idea that APOE plays a key role in Aβ proteinopathy. Treatment with Aβ12-28P, which disrupts APOE-Aβ interaction, significantly lowered the amount of intraneuronal Aβ as well as inhibited the loss of synaptic proteins in this co-culture system [150].
Another example of an inhibitor of APOE-Aβ interaction is the 6KApoEp peptide that inhibits the binding of APOE to the N-terminus of APP [151]. This peptide contains residues 133-152 of APOE protein conjugated to six lysine residues at the N terminus. When 6KApoEp was injected into the 5XFAD mouse model of amyloid pathology, both Aβ and tau pathologies were reduced concomitant with improved memory and hippocampal-dependent learning. However, 6KAPOEp therapy did not alter the cholesterol or APOE levels in 5xFAD mice. These results demonstrate that apoE-Aβ interaction inhibitors could potentially be used for therapeutic reduction of Aβ and tau burden in the CNS.
HDAC inhibition regulates endolysosomal function
Another group of researchers reported that HDAC regulates endolysosomal function [152, 153]. Initially, using yeast microarray databases, they identified Nhx1 as a major HDAC regulated factor induced during nutrient-limiting conditions [152]. Nhx1 is an endosomal Na+/H+ exchanger (eNHE) whose main function in yeast is vacuolar alkanization. The mammalian homolog of NHx1 was identified as Nhe6 that is regulated by cAMP-response element-binding protein (CREB) and plays a key role in nutrient- and HDAC-dependent regulation of endosomal pH [152]. This research group used three different pharmacological strategies to activate HDAC/CREB-dependent Nhe6 expression in immortalized astrocytes expressing APOE3 or APOE4 and observed that Creb-dependent Nhe6 expression corrected Aβ clearance deficits observed in APOE4 astrocytes. In a second report, this research group could mechanistically relate this finding to dysfunction in LRP1 endocytosis [153]. Using both Nhe6 deficient mice and immortalized APOE4 astrocytes, they showed that Nhe6 deficiency causes endosomes to become hyperacidic, which impedes Aβ clearance by impairing endocytosis of LRP1 [153]. Inhibition of HDAC could normalize Aβ clearance by restoring Nhe6 in the APOE4 astrocytes. Though these HDAC inhibitors are efficacious in other systemic disorders such as heart failure [154] and cancer [155], the widespread clinical applications is limited due to selectivity issues and toxicity issues.
Recalibrating APOE function using gene editing and gene therapy
Several experimental strategies have been tested to alter the prevalent apoE isoform in rodent models and human-derived induced pluripotent stem cells (iPScs) as a means to rectify the neurotoxic functions of APOE4. Various studies have used CRISPR-mediated or adeno-associated virus (AAV)-mediated gene delivery in these model systems. However, these strategies have to contend with ethical and safety hurdles before these can be translated to clinical settings.
CRISPR/Cas9 mediated gene editing
One promising method for gene editing is using the CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) system that has just entered Phase 1 trial for treatment of relapsed refractory multiple myeloma and related cancers (NCT03399448: University of Pennsylvania, Parker Institute for Cancer Immunotherapy, Tmunity Therapeutics). CRISPR/Cas9 basically functions like a pair of molecular scissors where an editable guide RNA leads the Cas9 ‘scissor’ to a specific site of the genome to cut where a different nucleotide sequence can then be inserted to correct a genetic defect [156]. CRISPR/Cas9 has already been proven successful in iPS cells, where cells derived from a healthy E3/E4 individual were converted into E2/E2, E3/E3, E4/E4, or an APOE KO genotype [157]. A second group used iPS cells derived neurons from an APOE4 carrier and found that CRISPR-editing the APOE4 reduced tau phosphorylation and inomycin-induced cell death [158]. Interestingly, though in the CNS APOE is mostly synthesized by astrocytes, this study showed that editing neuronal APOE to the E3 isoform in these iPS-derived neurons is sufficient to protect them from cytotoxic injury [158]. Another study generated different brain cell types and organoids from iPS cells derived from a human subject – on editing the APOE4 allele to APOE3 in these iPS-derived cells increased Aβ clearance and reduced Aβ in the organoid cultures [49]. This study shows that targeting APOE in various CNS cell types can lead to beneficial functional alterations in patient-derived in vitro systems. In animal models, CRISPR/Cas9 is relatively safe and has been successfully used to generate APOE KO in pigs and rats with little to no off-target incidents or mosaicism [159,160,161]. However, there is always the possibility of unexpected edits in the targeted and non-targeted portions of the genome leading to unanticipated side effects as well as triggering cancer risk [162, 163]. Inherent issues of CRISPR/Cas9 include off-target gene editing and mosaicism or where only some of the copies of the target gene are actually edited which could result in harmful side effects or unreliable treatment. Though data from iPS cells is extremely promising, much more research and ethical hurdles need to be cleared before gene editing with CRISPR/Cas9 is ready to be used as a clinical intervention.
AAV-APOE2 biologic therapy
APOE4 has been established as the risk allele for AD, and APOE2 is protective. This set the foundation for the idea that if APOE2 could replace or be overexpressed in APOE4 carriers, there would be a compensatory beneficial therapeutic effect. Indeed, there is a current trial scheduled to start that intends to test the safety of AAV-APOE2 expression in APOE4 carriers (Table 1). Patients will be infused with AAV-APOE2 in the cisterna magna and then followed up for at least 2 years to assess safety of this biologic therapy.
There is a robust rodent literature showing the effects of AAV-mediated APOE expression in primarily mouse models of amyloidosis. For example, intracerebral injection of AAV-APOE4 in APP/PS1 and Tg2576 mice resulted in increased Aβ burden whereas AAV-APOE2 lowers Aβ burden [164]. However, a limiting factor of this study is that this was done in the presence of murine Apoe that may itself influence Aβ deposition. In a subsequent study from a second group, expression of AAV-APOE2 was shown to reduce Aβ plaque burden in a trigenic mouse (APP/PS1/APOE4 TR) [165]. This study also showed that gene delivery of APOE2 was most effective before the onset of amyloid burden, suggesting that in order to be a successful therapy the AAV would need to be injected much before the onset of symptoms in patients which poses its own challenges. Further translational studies on non-human primates revealed that intra-cisternal delivery of AAV-APOE2 led to widespread expression in the CNS which established a safe procedure for CNS delivery of biologics [166]. Because of the inherent risk in any surgical procedure inside the CNS, whether such AAV-APOE2 biologics can be directly delivered into the AD-affected areas of the human CNS needs to be cautiously determined. However as Zhao et al showed in mice, even intrathalamic injections were modestly effective in reducing Aβ burden in neuroanatomically distant areas such as hippocampus [165]. Another confounding variable is that while APOE2 may decrease Aβ plaque formation, it may increase tau phosphorylation [12]. By injecting AAV-P301L tau into APOE TR mice, this research group found that mice expressing APOE2 had higher NFT levels compared to mice expressing APOE3 or APOE4. Along with data showing genetic association between APOE2 with PSP in humans, this brings up the question if overexpression of APOE2 could inadvertently exacerbate tau pathology while alleviating amyloid burden. In addition, questions regarding effective dosage to achieve optimum biodistribution and cell type transduction, pre-existing host immunity and long term CNS consequences still remain safety concerns in AAV therapies. In addition, while AAV-APOE2 gene therapy has promise, more knowledge on the neuropsychological and neuropathologic consequences of APOE2 overexpression is needed.
Lifestyle and diet can also regulate APOE function
Metabolic syndrome (MetS) can be characterized as a cluster of disorders that are associated with atherosclerosis, diabetes, hypertension, and has been linked to dementia in general. A few studies have indicated that the APOE4 allele is associated with increased risk of MetS leading to dementia [167]. Multiple case studies have thus examined the relationship of interventions in lifestyles such as, but not limited to, diet and exercise to reduce the risks associated with the APOE4 isoform. Though there is still no strong precedent for these lifestyle factors to effectively reduce metabolic dysfunction and AD risk via affecting APOE function, these interventions hold promise as future and easily translatable strategies in the personalized medicine niche due to their safety profiles.
Exercise
Based on epidemiological records and rodent studies, an intuitive therapeutic strategy for AD patients is exercise. Exercise increases cerebral blood flow, neurogenesis, and hippocampal volume as well has a positive impact on memory in humans [168, 169]. In wild type mice, exercise resulted in prevention of age-related neurovascular changes, especially in the context of the Apoe gene [170]. This was consistent with the idea that APOE plays a key role in functional impairment of the neurovascular unit during aging and exercise can reverse these effects by modulating neurovascular health.
Physical exercise can have a beneficial effect in AD type dementias by altering neuroplasticity as observed in both human case studies and rodent studies [171, 172]. However, there are still unresolved issues regarding the relative efficacies of different exercise regimens and presence of sex-dependent effects [173]. In a cohort of 201 cognitively normal adults, APOE4 carriers who do not exercise frequently were shown to have an increased risk of Aβ deposition [174]. However, this study did not report how many of these sedentary APOE4 carriers went on to develop actual AD-type dementia. A more recent study with 200 individuals diagnosed with mild AD addressed this issue, examining if exercise held any cognitive or physical improvements for APOE4 carriers [175]. The data did support that exercise intervention improved cognitive function, and was found to be more beneficial to APOE4 carriers. However, out of the five tests for cognition, only one test showed a statistically relevant correlation between exercise and APOE4 status. Along with the small sample number and lack of information on the ethnicities of the cohort, larger studies would be required to validate any of the conclusions and extend its application in the clinical setting.
Statins
Statins, or HMG-CoA reductase inhibitors, are a class of drugs that are typically prescribed to lower cholesterol levels in the blood. Researchers have postulated that increased brain cholesterol levels, or at the very least disruption of lipid homeostasis, influences AD pathology and risk. Epidemiological studies support that higher serum cholesterol levels are linked to increased risk of AD independent of APOE genotype [176,177,178]. A series of epidemiological studies looked at the effect of statins on dementia in general, spurred by observations that usage of statin led to a significantly lower rate of cognitive decline over 6 months [179]. However, more recently the LEADe trial of 2010 and the CLASP study of 2011 that assessed the use of statins in AD patients found no net benefit or harm in terms of cognitive decline relative to the placebo group [180, 181]. Further support for the idea that statins do not generally benefit AD patients comes from another systematic review [182]. These findings, however, contradict another large study of Medicare beneficiaries which established a beneficial association between statin use and reduced AD incidence in specific populations [183]. The data, however, showed wide variations in efficacy of statins based on race and sex; for example, pravastatin was associated with reduced AD risk only among white women whereas atorvastatin was efficacious in white women, black women and Hispanic men. This finding suggests that overall statin use may not be beneficial for all people at risk for AD, but in a future of personalized medicine, physicians should consider whether statins could have higher health impact in specific patient populations based on sex, ethnicity, prevalent health conditions, and APOE genotype.
Ketogenic diet
Modern diets that are high in carbohydrates and low in fats elevate blood glucose levels after ingestion and can alter APOE function through glycation and oxidative damage [184]. These diets are associated with impaired brain glucose metabolism, which is an AD biomarker. Feeding APOE TR rodents a high fat diet affected the plasma levels (E4>E3) and hippocampal levels (E3<E4) of APOE in an isoform-dependent manner [185]. Given that APOE4 carriers have abnormally low rates of glucose metabolism compared to other APOE genotypes, it is possible that these diets may profoundly alter metabolic status in APOE4 patients [186]. A proposed method to supplement brain health could be the use of ketone bodies that are produced by using a ketogenic diet, or a high-fat low-carbohydrate diet (reviewed in [187]) that can alter the microbiome and improve neurovascular functions in young healthy mice [188]. In a small clinical study on AD patients with mild cognitive impairment (NCT02984540), certain gut bacteria showed significant correlation with AD CSF biomarkers (Aβ and phosphorylated tau) and further a modified Mediterranean-ketogenic diet altered gut bacterial profile [189], suggesting that such diets can regulate AD biomarkers through regulating gut microbiome and associated metabolites. Some diet intervention trials have reported that such regulated diets might have an effect on the neuropsychiatric profile of early AD patients [190], and some interventions show an APOE-dependent effect [191, 192]. These studies, however promising, need to be considered as they are – isolated case studies or small trials that require larger placebo controlled investigation for validation and further studies in rodent models are warranted.
Insulin resistance and APOE
As previously stated, diabetes and impaired insulin signaling are factors that increase the risk for MetS and are associated with increased AD risk [193, 194]. Peripheral insulin resistance is associated with lower cerebral glucose metabolism, which is also generally true of APOE4 carriers, and this is further associated with poorer memory performance [195]. However, a clinical trial using insulin nasal sprays showed a complicated sex/APOE interaction. In the APOE4 negative group, male AD patients improved in cognitive function whereas women worsened, whereas in the APOE4 group, both sexes remained equally stable [196, 197].
In mice, the role of Apoe in insulin signaling was established in a study that showed that deletion of Lrp1, a major Apoe receptor, led to impaired brain insulin signaling and glucose metabolism [198]. Studies in APOE4 TR mice showed that age along with peripheral insulin resistance contribute to the insulin signaling impairment in the brain by trapping the insulin receptor inside endosomes and contributing to impaired glycolysis [60]. With the current emerging knowledge on the regulation and function of brain insulin signaling, there is a need for further research into how insulin/glucose metabolism intersects with dementia in APOE isoform-dependent manner.
Neuroinflammation and cerebrovascular integrity in the context of APOE function
Evidence suggests that inflammation as well as cerebrovascular damage play a crucial role in the pathogenesis of AD. APOE has been shown to predispose carriers to different neuroinflammatory profiles depending on the isoform. For example, in the ROS/MAP kindred of LOAD, the protective role of the APOE2 haplotype could be traced to its counteracting a pathologic microglial signature [199] though APOE4 did not show a corresponding pathologic effect on aged microglia [199, 200]. In mouse models, both Apoe KO mice and APOE4 TR mice upregulate pro-inflammatory phenotype when challenged with bacterial lipopolysaccharide [201]. A recent paper suggested that mouse Apoe and human APOE4 act as a direct checkpoint inhibitor by binding to the complement C1q and attenuating the classical complement cascade [202]. This work has spurred interest to not only investigate the function of glia-specific APOE in the CNS but how this impacts the neurovascular unit including the BBB. This line of research has not yet identified any druggable candidates but future research into neuroinflammation and peripheral inflammation may yield potential targets that can be targeted in an APOE genotype-dependent manner.
TREM2
Microglia are resident immune cells in the brain that help maintain CNS homeostasis and can initiate inflammatory reactions when this homeostasis is perturbed. In AD, microglia can become chronically dysfunctional [203]. Recent genome wide association studies have identified several microglial genes that regulate AD risk, foremost among them being TREM2 [204]. The current state of thought is that variants of TREM2 that increase AD risk are loss of function mutations [205,206,207]. Recent studies have implicated a close relationship between TREM2 and APOE. APOE has been found to regulate the function of a subset of microglia, which under the control of TREM2 can adopt a damage associated microglia (DAM) phenotype [208] that is analogous to a toxic molecular signature of disease-associated microglia (MGnD) observed in several animal models including AD model [209]. This APOE-dependent phenotype is induced in phagocytic microglia in the presence of apoptotic neurons and activation of the TREM2-APOE signaling pathway results in functional impairment of the microglia. The authors suggested that the switch from homeostatic to neurodegenerative state in AD-associated microglia is an initial response to neuronal injury compounded by a failure to switch back to a functional state. Several follow-up studies have now shown that Aβ is also a ligand of Trem2 [210, 211], implicating the TREM2-APOE pathway directly in AD pathogenesis. A recent report showed that loss of Trem2 accelerates amyloidogenesis in mice by reducing microglial function but these newly seeded deposits show reduced amounts of Apoe compared to mice carrying Trem2 [212]. Together this data suggests that microglia, through Trem2 mediated signaling, can regulate apoE co-deposition around Aβ deposits, which further has significance in terms of Aβ clearance based on specific APOE isoform [164]. Independently, in a mouse model of tauopathy-mediated neurodegeneration, reducing microglial activity through pharmacological methods increases soluble APOE, reduces tauopathy and rescues neurodegeneration in APOE4 mice [54]. This report did not specifically look at Trem2 though another previous report had ahown that attenuating microglial Trem2 is protective against tau-mediated neurodegeneration [213]. Given that this scenario of tripartite interactions between Aβ, tau and APOE is mediated through microglial homeostasis, it is tempting to suggest that targeting microglial TREM2 functions can result in APOE-isoform dependent therapeutic benefits. Of note, a recent report showed that the ectodomain form of TREM2, soluble TREM2, is protective in an amyloid mouse model by enhancing microglial metabolism of Aβ [214] and triggering microglia to an active state [215]. Given that TREM2 facilitates microglial degradation of Aβ preferentially complexed with LDL [210], this raises the intriguing possibility that soluble TREM2 may have therapeutic promise. However, another cell culture study seemed to indicate that AD-associated TREM2 risk variants do not show altered binding affinity for Aβ or APOE [211], raising the conundrum regarding whether TREM2-Aβ interaction is functionally dependent on specific APOE genotype.
Blood Brain Barrier
The BBB is composed of a layer of tightly packed endothelial cells which keeps out neurotoxins and pathogens from the brain, imparting a sort of unique ‘immune privileged’ milieu during healthy conditions. In AD, BBB dysfunction and leakiness precedes neurodegenerative changes, brain atrophy, and dementia [216]. This finding has encouraged researchers to look into how BBB breakdown relates to neurodegeneration in a series of AD mouse models including APOE models. It has been suggested that APOE is essential for maintaining BBB integrity as the BBB is leaky in Apoe KO models [35, 36, 217]. Further, APOE triggers BBB breakdown in an isoform-dependent manner in an in vitro model (E4>E3) [91], though another group reported that the BBB is largely intact at least in young APOE4 TR mice [40]. This raises the possibility that APOE4 mediated BBB disruptions can be localized to selectively vulnerable brain regions or may depend on other factors, such as aging or presence of amyloid angiopathy. Another group of researchers found that APOE4 mice had higher levels of the cyclophilin A (CypA)-matrix metalloproteinase 9 (MMP-9) in the pericytes. Since pericytes make up the BBB, this can lead to degradation of tight junctions and basement membranes and leakiness of the BBB [36]. Other studies using radioactive tracers in mouse models or using in vitro model of mouse brain microcapillaries showed that APOE3 and APOE2 mediate Aβ clearance through a faster route via LRP1 across the BBB while APOE4 mediates Aβ clearance through VLDR at a much slower rate possibly contributing to CNS accumulation of Aβ [43]. These studies revealed a potential therapeutic target, where researchers genetically and pharmacologically inhibited the CypA-MMP-9 pathway which resulted in repairing the BBB and reversing the neurodegeneration [36]. Curiously, loss in BBB integrity would also imply that drugs (such as antibodies) administered peripherally could gain easier access into the brains of APOE4 individuals, leading to higher bioavailability. On the whole, more studies are still required to establish the relationship between APOE genotype and BBB integrity and how this is altered in the context of neurodegenerative dementia of the elderly.
Critical Challenges for targeting CNS resident APOE
One of the most critical challenges for any AD therapeutic is optimizing the route and mode of administration so as to achieve effective bioavailability by bridging the BBB. A major area of research is now devoted to discovering cutting edge technologies that can safely breach the BBB. One option is to use the so-called Trojan horse strategy utilizing bifunctional molecules, one arm of which can be used to shuttle the APOE therapeutic across the BBB as has been demonstrated for anti-Aβ antibodies [218]. Another new technique is using pulsed ultrasounds that would create transient openings in the BBB allowing the APOE therapeutic to reach the substrate, as has been done to optimize chemotherapy in glioblastoma patients [219]. Perhaps another alternative would be to use gene therapy vectors to deliver a beneficial (E2) or even the neutral form (E3) of APOE, using specific AAV capsid serotypes that are preferentially neurotropic even when administered in the periphery [220, 221]. Each one of these tools have their shortcomings – for example, the bridging molecules used for the Trojan horse strategy are not particularly specific for the BBB, leading to potential dilution or even unwanted peripheral side effects. Likewise, the pulsed ultrasound and AAV based approaches have unknown long-term health implications. Interestingly, a 20 amino acid stretch of the APOE protein itself has been successfully utilized for shuttling therapeutics across the BBB in a mouse model of lipofuscinosis, a pediatric neurodegenerative disorder [222, 223], suggesting the possibility of using endogenous shuttling signals for efficacious delivery through the BBB. Even with these exciting breakthroughs, several challenges remain: if administered peripherally, how do we prevent the APOE therapeutic to be titered by the peripheral pools of APOE, or worse, cause systemic metabolic dysfunction and additionally, how to safely guide the therapeutic to the affected brain regions or cell types once inside the brain.
Targeting APOE in other dementias
Aside from its established role in AD, not much is known about how APOE influences disease pathogenesis in AD related dementias such as Fronto-temporal dementias (FTD), dementia with Lewy bodies (DLB) and vascular dementia. Consequently, very few mechanistic and therapeutic studies in mouse models are available.
The APOE2 allele is associated with an increased risk of ALS-FTD [224]. In another study, APOE2 and APOE4 alleles showed protective and increased disease risk effects, respectively, for FTD subtypes such as behavioral variant FTD and semantic dementia, though potential overlaps between clinical diagnosis of FTD and AD cannot be completely ruled out in this study [225]. Similarly, APOE4 appears to be a risk factor for DLB [226] and vascular dementia [227]. There is no direct association of APOE with other atypical parkinsonism syndromes with dementia such as corticobasal degeneration (CBD), multiple system atrophy (MSA) and progressive supranuclear palsy (PSP) [228]. Knocking out mouse Apoe resulted in delayed neurodegeneration in a mouse model of synucleinopathy [229]. In mouse studies, both APOE4 as well as APOE2 increased tauopathy burden in two different mouse models [11, 12], raising intriguing possibilities of how APOE might interact with tau in the presence of co-morbidities (such as Aβ and α-synuclein).
Conclusions
APOE not only impacts lipid metabolism but various CNS functions and neurodegenerative proteinopathy in AD in an isoform-dependent manner. The current evidence highlights how APOE isoform determines physiological homeostasis in the brain and how several APOE-targeted therapeutic approaches can have corrective or preventive outcome(s) in neurodegenerative proteinopathies, particularly in AD (Fig. 2). Many of these experimental approaches are validated in various cellular or animal models, with the overall perception that current APOE-targeted therapies would be more effective at prevention rather than treatment of those already in the throes of the neurodegenerative cascade. If or when these treatments make it through clinical trials, the potential benefit could be greatest for APOE4 carriers, where early intervention would slow the rate of decline (neuropathologic or neuropsychiatric) though it is unlikely to entirely stop the progression of disease. This is exemplified in some of the rodent amyloid models, where early intervention led to reduction of Aβ deposits but not complete clearance. However, if future research shows that APOE alters other AD related proteinopathies in these patients, such as tau or α-synuclein or inflammation either directly or through altering Aβ levels, then certain APOE directed therapies may have more profound multi-target effects in an APOE isoform-dependent manner. Additionally, APOE4 targeted therapies might also become adjuvants to other multimodal treatments that would target the more age-advanced pathologies, such as neuroinflammation or BBB leakiness [230]. Advancements in biomarkers for earlier diagnosis and prognosis of AD, especially in an APOE-informed population, would be invaluable for targeted therapies in an emerging era of precision medicine. Additionally, how such interventions will alter peripheral lipid homeostasis and vascular function would also need to be determined. The safety profile of any therapeutic will thus need to balance the total amount of APOE, lipidation profile of APOE, vascular risk factors, inflammatory phenotype and systemic effects. With this taken together, APOE-targeted therapeutic strategies remain a propitious area of research for preventing or delaying the onset of AD type dementias.

A schematic to illustrate the various targets for APOE-based AD therapeutics. See the main text for details. Black arrows pointing to the red text boxes indicate a mode of intervention while blue arrows indicate the movement of the drug or protein. Black solid arrows crossing the BBB show permeability while black dashed arrows show BBB semi-permeability. ABCA1: ATP-binding cassette transporter A1; LDLR: low density lipoprotein receptor; APOE: apolipoprotein E; Aβ: amyloid-β; TREM2: triggering receptor expressed on myeloid cells 2
Availability of data and materials
Not applicable.
Abbreviations
- AAV:
-
Adeno-associated virus
- AD:
-
Alzheimer’s disease
- ALS:
-
Amyotrophic Lateral Sclerosis
- APOE:
-
Generic apolipoprotein E including human form
- Apoe:
-
Mouse apolipoprotein E
- APP:
-
Amyloid precursor protein
- ASO:
-
Antisense oligonucleotides
- Aβ:
-
Amyloid β
- BBB:
-
Blood brain barrier
- CBD:
-
Corticobasal degeneration
- CETP:
-
Cholesteryl ester transfer protein
- CNS:
-
Central Nervous System
- CREB:
-
cAMP-response element-binding protein
- CRISPR:
-
Clustered Regularly Interspaced Short Palindromic Repeats
- CypA:
-
Cyclophilin A
- DAM:
-
Damage associated microglia
- DLB:
-
Dementia with Lewy bodies
- EGCG:
-
Epigallocatechin gallate
- eNHE:
-
Endosomal Na+/H+ exchanger
- FTD:
-
Fronto-temporal dementias
- HDAC:
-
Histone deacetylase
- HDL:
-
High Density Lipoprotein
- ICH:
-
Intracerebral hemorrhage
- iPSC:
-
Induced pluripotent stem cell
- LDL:
-
Low-density lipoprotein
- LOAD:
-
Late onset AD
- LXR:
-
Liver X receptors
- MetS:
-
Metabolic syndrome
- MMP-9:
-
Matrix metalloproteinase 9
- MSA:
-
Multiple system atrophy
- NFT:
-
Neurofibrillary tangle
- PNS:
-
Peripheral nervous system
- PPARγ:
-
Peroxisome proliferator-activated receptor γ
- PPI:
-
Protein–protein interactions
- PS1:
-
Presenilin-1
- PS2:
-
Presenilin-2
- PSP:
-
Progressive Supranuclear Palsy
- RXR:
-
Retinoid X receptor
- sAD:
-
Sporadic AD
- SILK:
-
Stable isotope leveling kinetics
- TR:
-
Targeted replacement
- TREM2:
-
Triggering receptor expressed on myeloid cells 2
- VLDL:
-
Very low density lipoprotein
References
Selkoe DJ, Lansbury PJJ. Alzheimer's Disease Is the Most Common Neurodegenerative Disorder. Siegel GJ AB, Albers RW, et al., editor. Philadelphia: Lippincott-Raven1999.
Roses AD. Apolipoprotein E alleles as risk factors in Alzheimer's disease. Annu Rev Med. 1996;47:387-400.
Mahley RW, Rall SC. Apolipoprotein E: far more than a lipid transport protein. Annu Rev Genomics Hum Genet. 2000;1:507–37.
Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 1993;261(5123):921–3.
Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, et al. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90(5):1977–81.
Corder EH, Saunders AM, Risch NJ, Strittmatter WJ, Schmechel DE, Gaskell PC, et al. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat Genet. 1994;7(2):180–4.
Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA. 1997;278(16):1349–56.
Vélez JI, Lopera F, Sepulveda-Falla D, Patel HR, Johar AS, Chuah A, et al. APOE*E2 allele delays age of onset in PSEN1 E280A Alzheimer's disease. Mol Psychiatry. 2016;21(7):916–24.
Marais AD, Solomon GA, Blom DJ. Dysbetalipoproteinaemia: a mixed hyperlipidaemia of remnant lipoproteins due to mutations in apolipoprotein E. Crit Rev Clin Lab Sci. 2014;51(1):46–62.
Yamazaki Y, Zhao N, Caulfield TR, Liu CC, Bu G. Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat Rev Neurol. 2019.
Shi Y, Yamada K, Liddelow SA, Smith ST, Zhao L, Luo W, et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature. 2017;549(7673):523–7.
Zhao N, Liu CC, Van Ingelgom AJ, Linares C, Kurti A, Knight JA, et al. APOE ε2 is associated with increased tau pathology in primary tauopathy. Nat Commun. 2018;9(1):4388.
Machulda MM, Jones DT, Vemuri P, McDade E, Avula R, Przybelski S, et al. Effect of APOE ε4 status on intrinsic network connectivity in cognitively normal elderly subjects. Arch Neurol. 2011;68(9):1131–6.
Liu CC, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nature reviews Neurology. 2013;9(2):106–18.
Kanekiyo T, Xu H, Bu G. ApoE and Aβ in Alzheimer's disease: accidental encounters or partners? Neuron. 2014;81(4):740–54.
Wu L, Zhao L. ApoE2 and Alzheimer's disease: time to take a closer look. Neural Regen Res. 2016;11(3):412–3.
Weisgraber KH, Innerarity TL, Mahley RW. Abnormal lipoprotein receptor-binding activity of the human E apoprotein due to cysteine-arginine interchange at a single site. J Biol Chem. 1982;257(5):2518–21.
Xu Q, Bernardo A, Walker D, Kanegawa T, Mahley RW, Huang Y. Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the ApoE locus. J Neurosci. 2006;26(19):4985–94.
Huang Y, Weisgraber KH, Mucke L, Mahley RW. Apolipoprotein E: diversity of cellular origins, structural and biophysical properties, and effects in Alzheimer's disease. J Mol Neurosci. 2004;23(3):189–204.
Kockx M, Traini M, Kritharides L. Cell-specific production, secretion, and function of apolipoprotein E. J Mol Med (Berl). 2018;96(5):361–71.
Saura J, Petegnief V, Wu X, Liang Y, Paul SM. Microglial apolipoprotein E and astroglial apolipoprotein J expression in vitro: opposite effects of lipopolysaccharide. J Neurochem. 2003;85(6):1455–67.
Holtzman DM, Herz J, Bu G. Apolipoprotein E and apolipoprotein E receptors: normal biology and roles in Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2(3):a006312.
Tudorache IF, Trusca VG, Gafencu AV. Apolipoprotein E - A Multifunctional Protein with Implications in Various Pathologies as a Result of Its Structural Features. Comput Struct Biotechnol J. 2017;15:359–65.
Linton MF, Gish R, Hubl ST, Bütler E, Esquivel C, Bry WI, et al. Phenotypes of apolipoprotein B and apolipoprotein E after liver transplantation. J Clin Invest. 1991;88(1):270–81.
Liu M, Kuhel DG, Shen L, Hui DY, Woods SC. Apolipoprotein E does not cross the blood-cerebrospinal fluid barrier, as revealed by an improved technique for sampling CSF from mice. Am J Physiol Regul Integr Comp Physiol. 2012;303(9):R903–8.
Fagan AM, Holtzman DM, Munson G, Mathur T, Schneider D, Chang LK, et al. Unique lipoproteins secreted by primary astrocytes from wild type, apoE (-/-), and human apoE transgenic mice. J Biol Chem. 1999;274(42):30001–7.
Huynh TV, Wang C, Tran AC, Tabor GT, Mahan TE, Francis CM, et al. Lack of hepatic apoE does not influence early Aβ deposition: observations from a new APOE knock-in model. Mol Neurodegener. 2019;14(1):37.
Getz GS, Reardon CA. Animal models of atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32(5):1104–15.
von Scheidt M, Zhao Y, Kurt Z, Pan C, Zeng L, Yang X, et al. Applications and Limitations of Mouse Models for Understanding Human Atherosclerosis. Cell Metab. 2017;25(2):248–61.
Camus MC, Chapman MJ, Forgez P, Laplaud PM. Distribution and characterization of the serum lipoproteins and apoproteins in the mouse, Mus musculus. J Lipid Res. 1983;24(9):1210–28.
Davidson MH. Update on CETP inhibition. J Clin Lipidol. 2010;4(5):394–8.
Sullivan PM, Mezdour H, Aratani Y, Knouff C, Najib J, Reddick RL, et al. Targeted replacement of the mouse apolipoprotein E gene with the common human APOE3 allele enhances diet-induced hypercholesterolemia and atherosclerosis. J Biol Chem. 1997;272(29):17972–80.
Sullivan PM, Mezdour H, Quarfordt SH, Maeda N. Type III hyperlipoproteinemia and spontaneous atherosclerosis in mice resulting from gene replacement of mouse Apoe with human Apoe*2. J Clin Invest. 1998;102(1):130–5.
Knouff C, Hinsdale ME, Mezdour H, Altenburg MK, Watanabe M, Quarfordt SH, et al. Apo E structure determines VLDL clearance and atherosclerosis risk in mice. J Clin Invest. 1999;103(11):1579–86.
Methia N, André P, Hafezi-Moghadam A, Economopoulos M, Thomas KL, Wagner DD. ApoE deficiency compromises the blood brain barrier especially after injury. Mol Med. 2001;7(12):810–5.
Bell RD, Winkler EA, Singh I, Sagare AP, Deane R, Wu Z, et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature. 2012;485(7399):512–6.
Alata W, Ye Y, St-Amour I, Vandal M, Calon F. Human apolipoprotein E ɛ4 expression impairs cerebral vascularization and blood-brain barrier function in mice. J Cereb Blood Flow Metab. 2015;35(1):86–94.
Bour A, Grootendorst J, Vogel E, Kelche C, Dodart JC, Bales K, et al. Middle-aged human apoE4 targeted-replacement mice show retention deficits on a wide range of spatial memory tasks. Behav Brain Res. 2008;193(2):174–82.
Grootendorst J, Bour A, Vogel E, Kelche C, Sullivan PM, Dodart JC, et al. Human apoE targeted replacement mouse lines: h-apoE4 and h-apoE3 mice differ on spatial memory performance and avoidance behavior. Behav Brain Res. 2005;159(1):1–14.
Bien-Ly N, Boswell CA, Jeet S, Beach TG, Hoyte K, Luk W, et al. Lack of Widespread BBB Disruption in Alzheimer's Disease Models: Focus on Therapeutic Antibodies. Neuron. 2015;88(2):289–97.
Martínez-Morillo E, Hansson O, Atagi Y, Bu G, Minthon L, Diamandis EP, et al. Total apolipoprotein E levels and specific isoform composition in cerebrospinal fluid and plasma from Alzheimer's disease patients and controls. Acta Neuropathol. 2014;127(5):633–43.
Castellano JM, Kim J, Stewart FR, Jiang H, DeMattos RB, Patterson BW, et al. Human apoE isoforms differentially regulate brain amyloid-β peptide clearance. Sci Transl Med. 2011;3(89):89ra57.
Deane R, Sagare A, Hamm K, Parisi M, Lane S, Finn MB, et al. apoE isoform-specific disruption of amyloid beta peptide clearance from mouse brain. J Clin Invest. 2008;118(12):4002–13.
Kok E, Haikonen S, Luoto T, Huhtala H, Goebeler S, Haapasalo H, et al. Apolipoprotein E-dependent accumulation of Alzheimer disease-related lesions begins in middle age. Ann Neurol. 2009;65(6):650–7.
Polvikoski T, Sulkava R, Haltia M, Kainulainen K, Vuorio A, Verkkoniemi A, et al. Apolipoprotein E, dementia, and cortical deposition of beta-amyloid protein. N Engl J Med. 1995;333(19):1242–7.
Schmechel DE, Saunders AM, Strittmatter WJ, Crain BJ, Hulette CM, Joo SH, et al. Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90(20):9649–53.
Tiraboschi P, Hansen LA, Masliah E, Alford M, Thal LJ, Corey-Bloom J. Impact of APOE genotype on neuropathologic and neurochemical markers of Alzheimer disease. Neurology. 2004;62(11):1977–83.
Huang YA, Zhou B, Nabet AM, Wernig M, Südhof TC. Differential Signaling Mediated by ApoE2, ApoE3, and ApoE4 in Human Neurons Parallels Alzheimer's Disease Risk. J Neurosci. 2019;39(37):7408–27
Lin YT, Seo J, Gao F, Feldman HM, Wen HL, Penney J, et al. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer's Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron. 2018;98(6):1294.
Huang YA, Zhou B, Wernig M, Südhof TC. ApoE2, ApoE3, and ApoE4 Differentially Stimulate APP Transcription and Aβ Secretion. Cell. 2017;168(3):427-41.e21.
Fitz NF, Cronican AA, Saleem M, Fauq AH, Chapman R, Lefterov I, et al. Abca1 deficiency affects Alzheimer's disease-like phenotype in human ApoE4 but not in ApoE3-targeted replacement mice. J Neurosci. 2012;32(38):13125–36.
Liu CC, Zhao N, Fu Y, Wang N, Linares C, Tsai CW, et al. ApoE4 Accelerates Early Seeding of Amyloid Pathology. Neuron. 2017;96(5):1024-32.e3.
Christensen DZ, Schneider-Axmann T, Lucassen PJ, Bayer TA, Wirths O. Accumulation of intraneuronal Abeta correlates with ApoE4 genotype. Acta Neuropathol. 2010;119(5):555–66.
Shi Y, Manis M, Long J, Wang K, Sullivan PM, Remolina Serrano J, et al. Microglia drive APOE-dependent neurodegeneration in a tauopathy mouse model. J Exp Med. 2019;216(11):2546–61.
Wang C, Najm R, Xu Q, Jeong DE, Walker D, Balestra ME, et al. Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector. Nat Med. 2018;24(5):647–57.
Ikeda K, Akiyama H, Arai T, Sahara N, Mori H, Usami M, et al. A subset of senile dementia with high incidence of the apolipoprotein E epsilon2 allele. Ann Neurol. 1997;41(5):693–5.
Zhao N, Liu CC, Qiao W, Bu G, Apolipoprotein E. Receptors, and Modulation of Alzheimer's Disease. Biol Psychiatry. 2018;83(4):347–57.
Wu L, Zhang X, Zhao L. Human ApoE Isoforms Differentially Modulate Brain Glucose and Ketone Body Metabolism: Implications for Alzheimer's Disease Risk Reduction and Early Intervention. J Neurosci. 2018;38(30):6665–81.
Johnson LA, Torres ER, Weber Boutros S, Patel E, Akinyeke T, Alkayed NJ, et al. Apolipoprotein E4 mediates insulin resistance-associated cerebrovascular dysfunction and the post-prandial response. J Cereb Blood Flow Metab. 2019;39(5):770–81.
Zhao N, Liu CC, Van Ingelgom AJ, Martens YA, Linares C, Knight JA, et al. Apolipoprotein E4 Impairs Neuronal Insulin Signaling by Trapping Insulin Receptor in the Endosomes. Neuron. 2017;96(1):115-29.e5.
Jagust WJ, Landau SM, Initiative ADN. Apolipoprotein E, not fibrillar β-amyloid, reduces cerebral glucose metabolism in normal aging. J Neurosci. 2012;32(50):18227–33.
Reiman EM, Caselli RJ, Yun LS, Chen K, Bandy D, Minoshima S, et al. Preclinical evidence of Alzheimer's disease in persons homozygous for the epsilon 4 allele for apolipoprotein E. N Engl J Med. 1996;334(12):752–8.
Small GW, Mazziotta JC, Collins MT, Baxter LR, Phelps ME, Mandelkern MA, et al. Apolipoprotein E type 4 allele and cerebral glucose metabolism in relatives at risk for familial Alzheimer disease. JAMA. 1995;273(12):942–7.
Casey CS, Atagi Y, Yamazaki Y, Shinohara M, Tachibana M, Fu Y, et al. Apolipoprotein E Inhibits Cerebrovascular Pericyte Mobility through a RhoA Protein-mediated Pathway. J Biol Chem. 2015;290(22):14208–17.
Davidson Y, Gibbons L, Purandare N, Byrne J, Hardicre J, Wren J, et al. Apolipoprotein E epsilon4 allele frequency in vascular dementia. Dement Geriatr Cogn Disord. 2006;22(1):15–9.
Schilling S, DeStefano AL, Sachdev PS, Choi SH, Mather KA, DeCarli CD, et al. APOE genotype and MRI markers of cerebrovascular disease: systematic review and meta-analysis. Neurology. 2013;81(3):292–300.
Ulrich V, Konaniah ES, Herz J, Gerard RD, Jung E, Yuhanna IS, et al. Genetic variants of ApoE and ApoER2 differentially modulate endothelial function. Proc Natl Acad Sci U S A. 2014;111(37):13493–8.
Wildsmith KR, Basak JM, Patterson BW, Pyatkivskyy Y, Kim J, Yarasheski KE, et al. In vivo human apolipoprotein E isoform fractional turnover rates in the CNS. PLoS One. 2012;7(6):e38013.
Hanson AJ, Bayer-Carter JL, Green PS, Montine TJ, Wilkinson CW, Baker LD, et al. Effect of apolipoprotein E genotype and diet on apolipoprotein E lipidation and amyloid peptides: randomized clinical trial. JAMA Neurol. 2013;70(8):972–80.
Heinsinger NM, Gachechiladze MA, Rebeck GW. Apolipoprotein E Genotype Affects Size of ApoE Complexes in Cerebrospinal Fluid. J Neuropathol Exp Neurol. 2016;75(10):918–24.
Rapp A, Gmeiner B, Hüttinger M. Implication of apoE isoforms in cholesterol metabolism by primary rat hippocampal neurons and astrocytes. Biochimie. 2006;88(5):473–83.
Zhao J, Davis MD, Martens YA, Shinohara M, Graff-Radford NR, Younkin SG, et al. APOE ε4/ε4 diminishes neurotrophic function of human iPSC-derived astrocytes. Hum Mol Genet. 2017;26(14):2690–700.
Riddell DR, Zhou H, Atchison K, Warwick HK, Atkinson PJ, Jefferson J, et al. Impact of apolipoprotein E (ApoE) polymorphism on brain ApoE levels. J Neurosci. 2008;28(45):11445–53.
Ulrich JD, Burchett JM, Restivo JL, Schuler DR, Verghese PB, Mahan TE, et al. In vivo measurement of apolipoprotein E from the brain interstitial fluid using microdialysis. Mol Neurodegener. 2013;8:13.
Wang Z, Dai Z, Shu H, Liao X, Yue C, Liu D, et al. APOE Genotype Effects on Intrinsic Brain Network Connectivity in Patients with Amnestic Mild Cognitive Impairment. Sci Rep. 2017;7(1):397.
Su YY, Liang X, Schoepf UJ, Varga-Szemes A, West HC, Qi R, et al. APOE Polymorphism Affects Brain Default Mode Network in Healthy Young Adults: A STROBE Article. Medicine (Baltimore). 2015;94(52):e1734.
Wiesmann M, Zerbi V, Jansen D, Haast R, Lütjohann D, Broersen LM, et al. A Dietary Treatment Improves Cerebral Blood Flow and Brain Connectivity in Aging apoE4 Mice. Neural Plast. 2016;2016:6846721.
Zerbi V, Wiesmann M, Emmerzaal TL, Jansen D, Van Beek M, Mutsaers MP, et al. Resting-state functional connectivity changes in aging apoE4 and apoE-KO mice. J Neurosci. 2014;34(42):13963–75.
Koffie RM, Hashimoto T, Tai HC, Kay KR, Serrano-Pozo A, Joyner D, et al. Apolipoprotein E4 effects in Alzheimer's disease are mediated by synaptotoxic oligomeric amyloid-β. Brain. 2012;135(Pt 7):2155–68.
Ji Y, Gong Y, Gan W, Beach T, Holtzman DM, Wisniewski T. Apolipoprotein E isoform-specific regulation of dendritic spine morphology in apolipoprotein E transgenic mice and Alzheimer's disease patients. Neuroscience. 2003;122(2):305–15.
Dumanis SB, Tesoriero JA, Babus LW, Nguyen MT, Trotter JH, Ladu MJ, et al. ApoE4 decreases spine density and dendritic complexity in cortical neurons in vivo. J Neurosci. 2009;29(48):15317–22.
Chen Y, Durakoglugil MS, Xian X, Herz J. ApoE4 reduces glutamate receptor function and synaptic plasticity by selectively impairing ApoE receptor recycling. Proc Natl Acad Sci U S A. 2010;107(26):12011–6.
Arendt T, Schindler C, Brückner MK, Eschrich K, Bigl V, Zedlick D, et al. Plastic neuronal remodeling is impaired in patients with Alzheimer's disease carrying apolipoprotein epsilon 4 allele. J Neurosci. 1997;17(2):516–29.
Maezawa I, Nivison M, Montine KS, Maeda N, Montine TJ. Neurotoxicity from innate immune response is greatest with targeted replacement of E4 allele of apolipoprotein E gene and is mediated by microglial p38MAPK. FASEB J. 2006;20(6):797–9.
Vitek MP, Brown CM, Colton CA. APOE genotype-specific differences in the innate immune response. Neurobiol Aging. 2009;30(9):1350–60.
Rodriguez GA, Tai LM, LaDu MJ, Rebeck GW. Human APOE4 increases microglia reactivity at Aβ plaques in a mouse model of Aβ deposition. J Neuroinflammation. 2014;11:111.
Guo L, LaDu MJ, Van Eldik LJ. A dual role for apolipoprotein e in neuroinflammation: anti- and pro-inflammatory activity. J Mol Neurosci. 2004;23(3):205–12.
Keene CD, Cudaback E, Li X, Montine KS, Montine TJ. Apolipoprotein E isoforms and regulation of the innate immune response in brain of patients with Alzheimer's disease. Curr Opin Neurobiol. 2011;21(6):920–8.
Halliday MR, Pomara N, Sagare AP, Mack WJ, Frangione B, Zlokovic BV. Relationship between cyclophilin a levels and matrix metalloproteinase 9 activity in cerebrospinal fluid of cognitively normal apolipoprotein e4 carriers and blood-brain barrier breakdown. JAMA Neurol. 2013;70(9):1198–200.
Halliday MR, Rege SV, Ma Q, Zhao Z, Miller CA, Winkler EA, et al. Accelerated pericyte degeneration and blood-brain barrier breakdown in apolipoprotein E4 carriers with Alzheimer's disease. J Cereb Blood Flow Metab. 2016;36(1):216–27.
Nishitsuji K, Hosono T, Nakamura T, Bu G, Michikawa M. Apolipoprotein E regulates the integrity of tight junctions in an isoform-dependent manner in an in vitro blood-brain barrier model. J Biol Chem. 2011;286(20):17536–42.
Cummings J, Lee G, Ritter A, Sabbagh M, Zhong K. Alzheimer's disease drug development pipeline: 2019. Alzheimers Dement (N Y). 2019;5:272–93.
Mehta D, Jackson R, Paul G, Shi J, Sabbagh M. Why do trials for Alzheimer's disease drugs keep failing? A discontinued drug perspective for 2010-2015. Expert Opin Investig Drugs. 2017;26(6):735–9.
Kennedy RE, Cutter GR, Schneider LS. Effect of APOE genotype status on targeted clinical trials outcomes and efficiency in dementia and mild cognitive impairment resulting from Alzheimer's disease. Alzheimers Dement. 2014;10(3):349–59.
Solomon A, Turunen H, Ngandu T, Peltonen M, Levälahti E, Helisalmi S, et al. Effect of the Apolipoprotein E Genotype on Cognitive Change During a Multidomain Lifestyle Intervention: A Subgroup Analysis of a Randomized Clinical Trial. JAMA Neurol. 2018;75(4):462–70.
Bien-Ly N, Gillespie AK, Walker D, Yoon SY, Huang Y. Reducing human apolipoprotein E levels attenuates age-dependent Aβ accumulation in mutant human amyloid precursor protein transgenic mice. J Neurosci. 2012;32(14):4803–11.
Kim J, Jiang H, Park S, Eltorai AE, Stewart FR, Yoon H, et al. Haploinsufficiency of human APOE reduces amyloid deposition in a mouse model of amyloid-β amyloidosis. J Neurosci. 2011;31(49):18007–12.
Kim J, Eltorai AE, Jiang H, Liao F, Verghese PB, Stewart FR, et al. Anti-apoE immunotherapy inhibits amyloid accumulation in a transgenic mouse model of Aβ amyloidosis. J Exp Med. 2012;209(12):2149–56.
Liao F, Hori Y, Hudry E, Bauer AQ, Jiang H, Mahan TE, et al. Anti-ApoE antibody given after plaque onset decreases Aβ accumulation and improves brain function in a mouse model of Aβ amyloidosis. J Neurosci. 2014;34(21):7281–92.
Liao F, Li A, Xiong M, Bien-Ly N, Jiang H, Zhang Y, et al. Targeting of nonlipidated, aggregated apoE with antibodies inhibits amyloid accumulation. J Clin Invest. 2018;128(5):2144–55.
Borchelt DR. Targeting the accomplice to thwart the culprit: a new target for the prevention of amyloid deposition. J Clin Invest. 2018;128(5):1734–6.
Scoles DR, Minikel EV, Pulst SM. Antisense oligonucleotides: A primer. Neurol Genet. 2019;5(2):e323.
DeVos SL, Miller RL, Schoch KM, Holmes BB, Kebodeaux CS, Wegener AJ, et al. Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Sci Transl Med. 2017;9(374).
Hinrich AJ, Jodelka FM, Chang JL, Brutman D, Bruno AM, Briggs CA, et al. Therapeutic correction of ApoER2 splicing in Alzheimer's disease mice using antisense oligonucleotides. EMBO Mol Med. 2016;8(4):328–45.
Huynh TV, Liao F, Francis CM, Robinson GO, Serrano JR, Jiang H, et al. Age-Dependent Effects of apoE Reduction Using Antisense Oligonucleotides in a Model of β-amyloidosis. Neuron. 2017;96(5):1013-23.e4.
Schoch KM, Miller TM. Antisense Oligonucleotides: Translation from Mouse Models to Human Neurodegenerative Diseases. Neuron. 2017;94(6):1056–70.
Ory DS. Nuclear receptor signaling in the control of cholesterol homeostasis: have the orphans found a home? Circ Res. 2004;95(7):660–70.
Donkin JJ, Stukas S, Hirsch-Reinshagen V, Namjoshi D, Wilkinson A, May S, et al. ATP-binding cassette transporter A1 mediates the beneficial effects of the liver X receptor agonist GW3965 on object recognition memory and amyloid burden in amyloid precursor protein/presenilin 1 mice. J Biol Chem. 2010;285(44):34144–54.
Cramer PE, Cirrito JR, Wesson DW, Lee CY, Karlo JC, Zinn AE, et al. ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models. Science. 2012;335(6075):1503–6.
Tesseur I, Lo AC, Roberfroid A, Dietvorst S, Van Broeck B, Borgers M, et al. Comment on "ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models". Science. 2013;340(6135):924-e.
Veeraraghavalu K, Zhang C, Miller S, Hefendehl JK, Rajapaksha TW, Ulrich J, et al. Comment on "ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models". Science. 2013;340(6135):924-f.
Price AR, Xu G, Siemienski ZB, Smithson LA, Borchelt DR, Golde TE, et al. Comment on "ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models". Science. 2013;340(6135):924-d.
Ghosal K, Haag M, Verghese PB, West T, Veenstra T, Braunstein JB, et al. A randomized controlled study to evaluate the effect of bexarotene on amyloid-β and apolipoprotein E metabolism in healthy subjects. Alzheimers Dement (N Y). 2016;2(2):110–20.
Cummings JL, Zhong K, Kinney JW, Heaney C, Moll-Tudla J, Joshi A, et al. Double-blind, placebo-controlled, proof-of-concept trial of bexarotene in moderate Alzheimer's disease. Alzheimers Res Ther. 2016;8:4.
Kobayashi T, Furusawa Y, Yamada S, Akehi M, Takenaka F, Sasaki T, et al. Positron emission tomography to elucidate pharmacokinetic differences of regioisomeric retinoid x receptor agonists. ACS Med Chem Lett. 2015;6(3):334–8.
Marks PA, Miller T, Richon VM. Histone deacetylases. Curr Opin Pharmacol. 2003;3(4):344–51.
Ferrari A, Fiorino E, Giudici M, Gilardi F, Galmozzi A, Mitro N, et al. Linking epigenetics to lipid metabolism: focus on histone deacetylases. Mol Membr Biol. 2012;29(7):257–66.
Ganai SA, Ramadoss M, Mahadevan V. Histone Deacetylase (HDAC) Inhibitors - emerging roles in neuronal memory, learning, synaptic plasticity and neural regeneration. Curr Neuropharmacol. 2016;14(1):55–71.
Dresselhaus E, Duerr JM, Vincent F, Sylvain EK, Beyna M, Lanyon LF, et al. Class I HDAC inhibition is a novel pathway for regulating astrocytic apoE secretion. PLoS One. 2018;13(3):e0194661.
Mahley RW, Weisgraber KH, Huang Y. Apolipoprotein E: structure determines function, from atherosclerosis to Alzheimer's disease to AIDS. J Lipid Res. 2009;50(Suppl):S183–8.
Rebeck GW. The role of APOE on lipid homeostasis and inflammation in normal brains. J Lipid Res. 2017;58(8):1493–9.
Van Eck M, Pennings M, Hoekstra M, Out R, Van Berkel TJ. Scavenger receptor BI and ATP-binding cassette transporter A1 in reverse cholesterol transport and atherosclerosis. Curr Opin Lipidol. 2005;16(3):307–15.
Nordestgaard LT, Tybjærg-Hansen A, Nordestgaard BG, Frikke-Schmidt R. Loss-of-function mutation in ABCA1 and risk of Alzheimer's disease and cerebrovascular disease. Alzheimers Dement. 2015;11(12):1430–8.
Wahrle SE, Jiang H, Parsadanian M, Kim J, Li A, Knoten A, et al. Overexpression of ABCA1 reduces amyloid deposition in the PDAPP mouse model of Alzheimer disease. J Clin Invest. 2008;118(2):671–82.
Boehm-Cagan A, Bar R, Liraz O, Bielicki JK, Johansson JO, Michaelson DM. ABCA1 Agonist Reverses the ApoE4-Driven Cognitive and Brain Pathologies. J Alzheimers Dis. 2016;54(3):1219–33.
Boehm-Cagan A, Bar R, Harats D, Shaish A, Levkovitz H, Bielicki JK, et al. Differential Effects of apoE4 and Activation of ABCA1 on Brain and Plasma Lipoproteins. PLoS One. 2016;11(11):e0166195.
Jan A, Karasinska JM, Kang MH, de Haan W, Ruddle P, Kaur A, et al. Direct intracerebral delivery of a miR-33 antisense oligonucleotide into mouse brain increases brain ABCA1 expression. [Corrected]. Neurosci Lett. 2015;598:66–72.
Kim J, Yoon H, Horie T, Burchett JM, Restivo JL, Rotllan N, et al. microRNA-33 Regulates ApoE Lipidation and Amyloid-β Metabolism in the Brain. J Neurosci. 2015;35(44):14717–26.
Ramaswamy G, Xu Q, Huang Y, Weisgraber KH. Effect of domain interaction on apolipoprotein E levels in mouse brain. J Neurosci. 2005;25(46):10658–63.
Mahley RW, Weisgraber KH, Huang Y. Apolipoprotein E4: a causative factor and therapeutic target in neuropathology, including Alzheimer's disease. Proc Natl Acad Sci U S A. 2006;103(15):5644–51.
Chen HK, Ji ZS, Dodson SE, Miranda RD, Rosenblum CI, Reynolds IJ, et al. Apolipoprotein E4 domain interaction mediates detrimental effects on mitochondria and is a potential therapeutic target for Alzheimer disease. J Biol Chem. 2011;286(7):5215–21.
Chen HK, Liu Z, Meyer-Franke A, Brodbeck J, Miranda RD, McGuire JG, et al. Small molecule structure correctors abolish detrimental effects of apolipoprotein E4 in cultured neurons. J Biol Chem. 2012;287(8):5253–66.
White CR, Garber DW, Anantharamaiah GM. Anti-inflammatory and cholesterol-reducing properties of apolipoprotein mimetics: a review. J Lipid Res. 2014;55(10):2007–21.
Liu J, Zhou G, Kolls BJ, Tan Y, Fang C, Wang H, et al. Apolipoprotein E mimetic peptide CN-105 improves outcome in a murine model of SAH. Stroke Vasc Neurol. 2018;3(4):222–30.
Lei B, James ML, Liu J, Zhou G, Venkatraman TN, Lascola CD, et al. Neuroprotective pentapeptide CN-105 improves functional and histological outcomes in a murine model of intracerebral hemorrhage. Sci Rep. 2016;6:34834.
Laskowitz DT, Wang H, Chen T, Lubkin DT, Cantillana V, Tu TM, et al. Neuroprotective pentapeptide CN-105 is associated with reduced sterile inflammation and improved functional outcomes in a traumatic brain injury murine model. Sci Rep. 2017;7:46461.
Chernick D, Ortiz-Valle S, Jeong A, Swaminathan SK, Kandimalla KK, Rebeck GW, et al. High-density lipoprotein mimetic peptide 4F mitigates amyloid-β-induced inhibition of apolipoprotein E secretion and lipidation in primary astrocytes and microglia. J Neurochem. 2018;147(5):647–62.
Sarantseva S, Timoshenko S, Bolshakova O, Karaseva E, Rodin D, Schwarzman AL, et al. Apolipoprotein E-mimetics inhibit neurodegeneration and restore cognitive functions in a transgenic Drosophila model of Alzheimer's disease. PLoS One. 2009;4(12):e8191.
Vitek MP, Christensen DJ, Wilcock D, Davis J, Van Nostrand WE, Li FQ, et al. APOE-mimetic peptides reduce behavioral deficits, plaques and tangles in Alzheimer's disease transgenics. Neurodegener Dis. 2012;10(1-4):122–6.
Wang H, Durham L, Dawson H, Song P, Warner DS, Sullivan PM, et al. An apolipoprotein E-based therapeutic improves outcome and reduces Alzheimer's disease pathology following closed head injury: evidence of pharmacogenomic interaction. Neuroscience. 2007;144(4):1324–33.
Namba Y, Tomonaga M, Kawasaki H, Otomo E, Ikeda K. Apolipoprotein E immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in Alzheimer's disease and kuru plaque amyloid in Creutzfeldt-Jakob disease. Brain Res. 1991;541(1):163–6.
Ran X, Gestwicki JE. Inhibitors of protein-protein interactions (PPIs): an analysis of scaffold choices and buried surface area. Curr Opin Chem Biol. 2018;44:75–86.
Cesa LC, Mapp AK, Gestwicki JE. Direct and Propagated Effects of Small Molecules on Protein-Protein Interaction Networks. Front Bioeng Biotechnol. 2015;3:119.
Sadowski M, Pankiewicz J, Scholtzova H, Ripellino JA, Li Y, Schmidt SD, et al. A synthetic peptide blocking the apolipoprotein E/beta-amyloid binding mitigates beta-amyloid toxicity and fibril formation in vitro and reduces beta-amyloid plaques in transgenic mice. Am J Pathol. 2004;165(3):937–48.
Sadowski MJ, Pankiewicz J, Scholtzova H, Mehta PD, Prelli F, Quartermain D, et al. Blocking the apolipoprotein E/amyloid-beta interaction as a potential therapeutic approach for Alzheimer's disease. Proc Natl Acad Sci U S A. 2006;103(49):18787–92.
Pankiewicz JE, Guridi M, Kim J, Asuni AA, Sanchez S, Sullivan PM, et al. Blocking the apoE/Aβ interaction ameliorates Aβ-related pathology in APOE ε2 and ε4 targeted replacement Alzheimer model mice. Acta Neuropathol Commun. 2014;2:75.
Boyles JK, Pitas RE, Wilson E, Mahley RW, Taylor JM. Apolipoprotein E associated with astrocytic glia of the central nervous system and with nonmyelinating glia of the peripheral nervous system. J Clin Invest. 1985;76(4):1501–13.
Pitas RE, Boyles JK, Lee SH, Foss D, Mahley RW. Astrocytes synthesize apolipoprotein E and metabolize apolipoprotein E-containing lipoproteins. Biochim Biophys Acta. 1987;917(1):148–61.
Zheng JY, Sun J, Ji CM, Shen L, Chen ZJ, Xie P, et al. Selective deletion of apolipoprotein E in astrocytes ameliorates the spatial learning and memory deficits in Alzheimer's disease (APP/PS1) mice by inhibiting TGF-β/Smad2/STAT3 signaling. Neurobiol Aging. 2017;54:112–32.
Kuszczyk MA, Sanchez S, Pankiewicz J, Kim J, Duszczyk M, Guridi M, et al. Blocking the interaction between apolipoprotein E and Aβ reduces intraneuronal accumulation of Aβ and inhibits synaptic degeneration. Am J Pathol. 2013;182(5):1750–68.
Sawmiller D, Habib A, Hou H, Mori T, Fan A, Tian J, et al. A Novel Apolipoprotein E Antagonist Functionally Blocks Apolipoprotein E Interaction With N-terminal Amyloid Precursor Protein, Reduces β-Amyloid-Associated Pathology, and Improves Cognition. Biol Psychiatry. 2019;86(3):208–20.
Prasad H, Rao R. Histone deacetylase-mediated regulation of endolysosomal pH. J Biol Chem. 2018;293(18):6721–35.
Prasad H, Rao R. Amyloid clearance defect in ApoE4 astrocytes is reversed by epigenetic correction of endosomal pH. Proc Natl Acad Sci U S A. 2018;115(28):E6640–E9.
McKinsey TA. Therapeutic potential for HDAC inhibitors in the heart. Annu Rev Pharmacol Toxicol. 2012;52:303–19.
Mohammad HP, Barbash O, Creasy CL. Targeting epigenetic modifications in cancer therapy: erasing the roadmap to cancer. Nat Med. 2019;25(3):403–18.
Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, et al. RNA-guided human genome engineering via Cas9. Science. 2013;339(6121):823–6.
Schmid B, Prehn KR, Nimsanor N, Garcia BIA, Poulsen U, Jørring I, et al. Generation of a set of isogenic, gene-edited iPSC lines homozygous for all main APOE variants and an APOE knock-out line. Stem Cell Res. 2019;34:101349.
Wadhwani AR, Affaneh A, Van Gulden S, Kessler JA. Neuronal apolipoprotein E4 increases cell death and phosphorylated tau release in alzheimer disease. Ann Neurol. 2019;85(5):726–39.
Huang L, Hua Z, Xiao H, Cheng Y, Xu K, Gao Q, et al. CRISPR/Cas9-mediated ApoE-/- and LDLR-/- double gene knockout in pigs elevates serum LDL-C and TC levels. Oncotarget. 2017;8(23):37751–60.
Ma Y, Shen B, Zhang X, Lu Y, Chen W, Ma J, et al. Heritable multiplex genetic engineering in rats using CRISPR/Cas9. PLoS One. 2014;9(3):e89413.
Zhao Y, Yang Y, Xing R, Cui X, Xiao Y, Xie L, et al. Hyperlipidemia induces typical atherosclerosis development in Ldlr and Apoe deficient rats. Atherosclerosis. 2018;271:26–35.
Kosicki M, Tomberg K, Bradley A. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat Biotechnol. 2018;36(8):765–71.
Haapaniemi E, Botla S, Persson J, Schmierer B, Taipale J. CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat Med. 2018;24(7):927–30.
Hudry E, Dashkoff J, Roe AD, Takeda S, Koffie RM, Hashimoto T, et al. Gene transfer of human Apoe isoforms results in differential modulation of amyloid deposition and neurotoxicity in mouse brain. Sci Transl Med. 2013;5(212):212ra161.
Zhao L, Gottesdiener AJ, Parmar M, Li M, Kaminsky SM, Chiuchiolo MJ, et al. Intracerebral adeno-associated virus gene delivery of apolipoprotein E2 markedly reduces brain amyloid pathology in Alzheimer's disease mouse models. Neurobiol Aging. 2016;44:159–72.
Rosenberg JB, Kaplitt MG, De BP, Chen A, Flagiello T, Salami C, et al. AAVrh.10-Mediated APOE2 Central Nervous System Gene Therapy for APOE4-Associated Alzheimer's Disease. Hum Gene Ther Clin Dev. 2018;29(1):24–47.
Brown D, Gibas KJ. Metabolic syndrome marks early risk for cognitive decline with APOE4 gene variation: A case study. Diabetes Metab Syndr. 2018;12(5):823–7.
Erickson KI, Voss MW, Prakash RS, Basak C, Szabo A, Chaddock L, et al. Exercise training increases size of hippocampus and improves memory. Proc Natl Acad Sci U S A. 2011;108(7):3017–22.
Pereira AC, Huddleston DE, Brickman AM, Sosunov AA, Hen R, McKhann GM, et al. An in vivo correlate of exercise-induced neurogenesis in the adult dentate gyrus. Proc Natl Acad Sci U S A. 2007;104(13):5638–43.
Soto I, Graham LC, Richter HJ, Simeone SN, Radell JE, Grabowska W, et al. APOE Stabilization by Exercise Prevents Aging Neurovascular Dysfunction and Complement Induction. PLoS Biol. 2015;13(10):e1002279.
Liang KY, Mintun MA, Fagan AM, Goate AM, Bugg JM, Holtzman DM, et al. Exercise and Alzheimer's disease biomarkers in cognitively normal older adults. Ann Neurol. 2010;68(3):311–8.
Choi SH, Bylykbashi E, Chatila ZK, Lee SW, Pulli B, Clemenson GD, et al. Combined adult neurogenesis and BDNF mimic exercise effects on cognition in an Alzheimer's mouse model. Science. 2018;361(6406).
Pietropaolo S, Sun Y, Li R, Brana C, Feldon J, Yee BK. The impact of voluntary exercise on mental health in rodents: a neuroplasticity perspective. Behav Brain Res. 2008;192(1):42–60.
Head D, Bugg JM, Goate AM, Fagan AM, Mintun MA, Benzinger T, et al. Exercise Engagement as a Moderator of the Effects of APOE Genotype on Amyloid Deposition. Arch Neurol. 2012;69(5):636–43.
Jensen CS, Simonsen AH, Siersma V, Beyer N, Frederiksen KS, Gottrup H, et al. Patients with Alzheimer's disease who carry the APOE ε4 allele benefit more from physical exercise. Alzheimers Dement (N Y). 2019;5:99–106.
Jarvik GP, Wijsman EM, Kukull WA, Schellenberg GD, Yu C, Larson EB. Interactions of apolipoprotein E genotype, total cholesterol level, age, and sex in prediction of Alzheimer's disease: a case-control study. Neurology. 1995;45(6):1092–6.
Kivipelto M, Helkala EL, Laakso MP, Hänninen T, Hallikainen M, Alhainen K, et al. Apolipoprotein E epsilon4 allele, elevated midlife total cholesterol level, and high midlife systolic blood pressure are independent risk factors for late-life Alzheimer disease. Ann Intern Med. 2002;137(3):149–55.
Notkola IL, Sulkava R, Pekkanen J, Erkinjuntti T, Ehnholm C, Kivinen P, et al. Serum total cholesterol, apolipoprotein E epsilon 4 allele, and Alzheimer's disease. Neuroepidemiology. 1998;17(1):14–20.
Sparks DL, Sabbagh MN, Connor DJ, Lopez J, Launer LJ, Browne P, et al. Atorvastatin for the treatment of mild to moderate Alzheimer disease: preliminary results. Arch Neurol. 2005;62(5):753–7.
Feldman HH, Doody RS, Kivipelto M, Sparks DL, Waters DD, Jones RW, et al. Randomized controlled trial of atorvastatin in mild to moderate Alzheimer disease: LEADe. Neurology. 2010;74(12):956–64.
Sano M, Bell KL, Galasko D, Galvin JE, Thomas RG, van Dyck CH, et al. A randomized, double-blind, placebo-controlled trial of simvastatin to treat Alzheimer disease. Neurology. 2011;77(6):556–63.
Mejías-Trueba M, Pérez-Moreno MA, Fernández-Arche M. Systematic review of the efficacy of statins for the treatment of Alzheimer's disease. Clin Med (Lond). 2018;18(1):54–61.
Zissimopoulos JM, Barthold D, Brinton RD, Joyce G. Sex and Race Differences in the Association Between Statin Use and the Incidence of Alzheimer Disease. JAMA Neurol. 2017;74(2):225–32.
Seneff S, Wainwright G, Mascitelli L. Nutrition and Alzheimer's disease: the detrimental role of a high carbohydrate diet. Eur J Intern Med. 2011;22(2):134–40.
Lane-Donovan C, Herz J. High-Fat Diet Changes Hippocampal Apolipoprotein E (ApoE) in a Genotype- and Carbohydrate-Dependent Manner in Mice. PLoS One. 2016;11(2):e0148099.
Reiman EM, Chen K, Alexander GE, Caselli RJ, Bandy D, Osborne D, et al. Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer's dementia. Proc Natl Acad Sci U S A. 2004;101(1):284–9.
Newman JC, Verdin E. Ketone bodies as signaling metabolites. Trends Endocrinol Metab. 2014;25(1):42–52.
Ma D, Wang AC, Parikh I, Green SJ, Hoffman JD, Chlipala G, et al. Ketogenic diet enhances neurovascular function with altered gut microbiome in young healthy mice. Sci Rep. 2018;8(1):6670.
Nagpal R, Neth BJ, Wang S, Craft S, Yadav H. Modified Mediterranean-ketogenic diet modulates gut microbiome and short-chain fatty acids in association with Alzheimer's disease markers in subjects with mild cognitive impairment. EBioMedicine. 2019;47:529–42.
Singh B, Parsaik AK, Mielke MM, Erwin PJ, Knopman DS, Petersen RC, et al. Association of mediterranean diet with mild cognitive impairment and Alzheimer's disease: a systematic review and meta-analysis. J Alzheimers Dis. 2014;39(2):271–82.
Martínez-Lapiscina EH, Galbete C, Corella D, Toledo E, Buil-Cosiales P, Salas-Salvado J, et al. Genotype patterns at CLU, CR1, PICALM and APOE, cognition and Mediterranean diet: the PREDIMED-NAVARRA trial. Genes Nutr. 2014;9(3):393.
Yassine HN, Braskie MN, Mack WJ, Castor KJ, Fonteh AN, Schneider LS, et al. Association of Docosahexaenoic Acid Supplementation With Alzheimer Disease Stage in Apolipoprotein E ε4 Carriers: A Review. JAMA Neurol. 2017;74(3):339–47.
Biessels GJ, Staekenborg S, Brunner E, Brayne C, Scheltens P. Risk of dementia in diabetes mellitus: a systematic review. Lancet Neurol. 2006;5(1):64–74.
Craft S, Cholerton B, Baker LD. Insulin and Alzheimer's disease: untangling the web. J Alzheimers Dis. 2013;33(Suppl 1):S263–75.
Willette AA, Bendlin BB, Starks EJ, Birdsill AC, Johnson SC, Christian BT, et al. Association of Insulin Resistance With Cerebral Glucose Uptake in Late Middle-Aged Adults at Risk for Alzheimer Disease. JAMA Neurol. 2015;72(9):1013–20.
Claxton A, Baker LD, Wilkinson CW, Trittschuh EH, Chapman D, Watson GS, et al. Sex and ApoE genotype differences in treatment response to two doses of intranasal insulin in adults with mild cognitive impairment or Alzheimer's disease. J Alzheimers Dis. 2013;35(4):789–97.
Claxton A, Baker LD, Hanson A, Trittschuh EH, Cholerton B, Morgan A, et al. Long Acting Intranasal Insulin Detemir Improves Cognition for Adults with Mild Cognitive Impairment or Early-Stage Alzheimer's Disease Dementia. J Alzheimers Dis. 2015;45(4):1269–70.
Liu CC, Hu J, Tsai CW, Yue M, Melrose HL, Kanekiyo T, et al. Neuronal LRP1 regulates glucose metabolism and insulin signaling in the brain. J Neurosci. 2015;35(14):5851–9.
Olah M, Patrick E, Villani AC, Xu J, White CC, Ryan KJ, et al. A transcriptomic atlas of aged human microglia. Nat Commun. 2018;9(1):539.
Dai J, Johnson ECB, Dammer EB, Duong DM, Gearing M, Lah JJ, et al. Effects of. Front Mol Neurosci. 2018;11:454.
Zhu Y, Nwabuisi-Heath E, Dumanis SB, Tai LM, Yu C, Rebeck GW, et al. APOE genotype alters glial activation and loss of synaptic markers in mice. Glia. 2012;60(4):559–69.
Yin C, Ackermann S, Ma Z, Mohanta SK, Zhang C, Li Y, et al. Publisher Correction: ApoE attenuates unresolvable inflammation by complex formation with activated C1q. Nat Med. 2019;25(3):529.
Golde TE. Harnessing Immunoproteostasis to Treat Neurodegenerative Disorders. Neuron. 2019;101(6):1003–15.
Carmona S, Zahs K, Wu E, Dakin K, Bras J, Guerreiro R. The role of TREM2 in Alzheimer's disease and other neurodegenerative disorders. Lancet Neurol. 2018;17(8):721–30.
Cheng-Hathaway PJ, Reed-Geaghan EG, Jay TR, Casali BT, Bemiller SM, Puntambekar SS, et al. The Trem2 R47H variant confers loss-of-function-like phenotypes in Alzheimer's disease. Mol Neurodegener. 2018;13(1):29.
Sudom A, Talreja S, Danao J, Bragg E, Kegel R, Min X, et al. Molecular basis for the loss-of-function effects of the Alzheimer's disease-associated R47H variant of the immune receptor TREM2. J Biol Chem. 2018;293(32):12634–46.
Kober DL, Alexander-Brett JM, Karch CM, Cruchaga C, Colonna M, Holtzman MJ, et al. Neurodegenerative disease mutations in TREM2 reveal a functional surface and distinct loss-of-function mechanisms. Elife. 2016;5.
Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer's Disease. Cell. 2017;169(7):1276–90 e17.
Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity. 2017;47(3):566–81 e9.
Zhao Y, Wu X, Li X, Jiang LL, Gui X, Liu Y, et al. TREM2 Is a Receptor for β-Amyloid that Mediates Microglial Function. Neuron. 2018;97(5):1023-31.e7.
Lessard CB, Malnik SL, Zhou Y, Ladd TB, Cruz PE, Ran Y, et al. High-affinity interactions and signal transduction between Aβ oligomers and TREM2. EMBO Mol Med. 2018;10(11).
Parhizkar S, Arzberger T, Brendel M, Kleinberger G, Deussing M, Focke C, et al. Loss of TREM2 function increases amyloid seeding but reduces plaque-associated ApoE. Nat Neurosci. 2019;22(2):191–204.
Leyns CEG, Ulrich JD, Finn MB, Stewart FR, Koscal LJ, Remolina Serrano J, et al. TREM2 deficiency attenuates neuroinflammation and protects against neurodegeneration in a mouse model of tauopathy. Proc Natl Acad Sci U S A. 2017;114(43):11524–9.
Zhong L, Xu Y, Zhuo R, Wang T, Wang K, Huang R, et al. Soluble TREM2 ameliorates pathological phenotypes by modulating microglial functions in an Alzheimer's disease model. Nat Commun. 2019;10(1):1365.
Zhong L, Chen XF, Wang T, Wang Z, Liao C, Huang R, et al. Soluble TREM2 induces inflammatory responses and enhances microglial survival. J Exp Med. 2017;214(3):597–607.
Montagne A, Zhao Z, Zlokovic BV. Alzheimer's disease: A matter of blood-brain barrier dysfunction? J Exp Med. 2017;214(11):3151–69.
Mulder M, Blokland A, van den Berg DJ, Schulten H, Bakker AH, Terwel D, et al. Apolipoprotein E protects against neuropathology induced by a high-fat diet and maintains the integrity of the blood-brain barrier during aging. Lab Invest. 2001;81(7):953–60.
Yu YJ, Atwal JK, Zhang Y, Tong RK, Wildsmith KR, Tan C, et al. Therapeutic bispecific antibodies cross the blood-brain barrier in nonhuman primates. Sci Transl Med. 2014;6(261):261ra154.
Carpentier A, Canney M, Vignot A, Reina V, Beccaria K, Horodyckid C, et al. Clinical trial of blood-brain barrier disruption by pulsed ultrasound. Sci Transl Med. 2016;8(343):343re2.
Foust KD, Nurre E, Montgomery CL, Hernandez A, Chan CM, Kaspar BK. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat Biotechnol. 2009;27(1):59–65.
Chan KY, Jang MJ, Yoo BB, Greenbaum A, Ravi N, Wu WL, et al. Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat Neurosci. 2017;20(8):1172–9.
Meng Y, Sohar I, Sleat DE, Richardson JR, Reuhl KR, Jenkins RB, et al. Effective intravenous therapy for neurodegenerative disease with a therapeutic enzyme and a peptide that mediates delivery to the brain. Mol Ther. 2014;22(3):547–53.
Meng Y, Wiseman JA, Nemtsova Y, Moore DF, Guevarra J, Reuhl K, et al. A Basic ApoE-Based Peptide Mediator to Deliver Proteins across the Blood-Brain Barrier: Long-Term Efficacy, Toxicity, and Mechanism. Mol Ther. 2017;25(7):1531–43.
Chiò A, Brunetti M, Barberis M, Iazzolino B, Montuschi A, Ilardi A, et al. The Role of APOE in the Occurrence of Frontotemporal Dementia in Amyotrophic Lateral Sclerosis. JAMA Neurol. 2016;73(4):425–30.
Mishra A, Ferrari R, Heutink P, Hardy J, Pijnenburg Y, Posthuma D, et al. Gene-based association studies report genetic links for clinical subtypes of frontotemporal dementia. Brain. 2017;140(5):1437–46.
Tsuang D, Leverenz JB, Lopez OL, Hamilton RL, Bennett DA, Schneider JA, et al. APOE ε4 increases risk for dementia in pure synucleinopathies. JAMA Neurol. 2013;70(2):223–8.
Sun JH, Tan L, Wang HF, Tan MS, Li JQ, Xu W, et al. Genetics of Vascular Dementia: Systematic Review and Meta-Analysis. J Alzheimers Dis. 2015;46(3):611–29.
Sabir MS, Blauwendraat C, Ahmed S, Serrano GE, Beach TG, Perkins M, et al. Assessment of APOE in atypical parkinsonism syndromes. Neurobiol Dis. 2019;127:142–6.
Gallardo G, Schlüter OM, Südhof TC. A molecular pathway of neurodegeneration linking alpha-synuclein to ApoE and Abeta peptides. Nat Neurosci. 2008;11(3):301–8.
Jack CR, Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS, et al. Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013;12(2):207–16.
Tu TM, Kolls BJ, Soderblom EJ, Cantillana V, Ferrell PD, Moseley MA, Wang H, Dawson HN, Laskowitz DT. Apolipoprotein E Mimetic Peptide, CN-105, Improves Outcomes in Ischemic Stroke. Ann Clin Transl Neurol. 2017;4(4):246–65.
Champagne D, Pearson D, Dea D, Rochford J, Poirier J. The cholesterol-lowering drug probucol increases apolipoprotein E production in the hippocampus of aged rats: implications for Alzheimer's disease. Neuroscience. 2003;121(1):99–110.
Bakker A, Albert MS, Krauss G, Speck CL, Gallagher M. Response of the medial temporal lobe network in amnestic mild cognitive impairment to therapeutic intervention assessed by fMRI and memory task performance. Neuroimage Clin. 2015;7:688–98.
Harrington C, Sawchak S, Chiang C, Davies J, Donovan C, Saunders AM, et al. Rosiglitazone does not improve cognition or global function when used as adjunctive therapy to AChE inhibitors in mild-to-moderate Alzheimer's disease: two phase 3 studies. Curr Alzheimer Res. 2011;8(5):592–606.
de la Torre R, de Sola S, Hernandez G, Farré M, Pujol J, Rodriguez J. et al; TESDAD study group. Safety and efficacy of cognitive training plus epigallocatechin-3-gallate in young adults with Down's syndrome (TESDAD): a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet Neurol. 2016;15(8):801–10.
Morris JK, Vidoni ED, Johnson DK, Van Sciver A, Mahnken JD, Honea RA, Wilkins HM, Brooks WM, Billinger SA, Swerdlow RH, Burns JM. Aerobic exercise for Alzheimer's disease: A randomized controlled pilot trial. PLoS One. 2017;10;12(2):e0170547.
Acknowledgements
Not Applicable
Funding
This work is supported by NIA R01AG055798 (PC) and RF1AG057933 (PC, DRB).
Author information
Authors and Affiliations
Contributions
TW, PC performed literature search. TW, PC, DRB participated in writing and editing of manuscript. All authors read and approved the final manuscript.
Authors’ Information
All information is provided in the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not Applicable.
Consent for publication
Not Applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Williams, T., Borchelt, D.R. & Chakrabarty, P. Therapeutic approaches targeting Apolipoprotein E function in Alzheimer’s disease. Mol Neurodegeneration 15, 8 (2020). https://doi.org/10.1186/s13024-020-0358-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13024-020-0358-9