
Abstract
Background
Tau hyperphosphorylation plays a critical role in neurodegenerative diseases [EMBO Mol Med. 6:1142-60, 2014; Annu Rev Neurosci. 24:1121-59, 2001]. Recent evidence has shown that Akt is down-regulated in AD [J Pathol. 225:54-62, 2011]. However, it remained unknown which pathological process, e.g. tau pathology or neuron death, Akt may contribute to. In this study, Cre-loxP technique was employed to generate a viable Akt three isoforms conditional knockout (Akt cTKO) mouse in which total Akt levels were dramatically reduced in the adult brain.
Results
Significantly increased levels of tau phosphorylated (p-tau) at various sites were observed in Akt cTKO mice as compared to age-matched littermate controls. Increased levels for phosphorylated GSK3α and phosphorylated PKA substrates were detected in Akt cTKO brains. In contrast, no significant changes on p-tau levels were found in Akt1 −/−, Akt2 −/− or Akt3 −/− mice.
Conclusions
Akt may regulate tau phosphorylation in the adult brain by affecting activities for PKA and GSK3α.
Similar content being viewed by others


Findings
Mice with global deletion of Akt three isoforms (Akt TKO or Akt1 −/− ;Akt2 −/− ;Akt3 −/−) were early embryonic lethal [1], precluding the possibility to study its in vivo function. To obtain viable Akt conditional TKO (cTKO) mouse, we generated floxed Akt1 (Additional file 1: Figure S1A). After several steps of crossing, Akt1 f/f ;Akt2 −/− ;Akt3 −/− ;CAG-CreER mice were obtained and became Akt cTKO after the treatment with tamoxifen. To examine Cre-mediated recombination efficiency in the brain, biochemical analyses were conducted. Dramatic reduction on levels for total Akt (t-Akt) was observed in the brain of cTKO mice (Additional file 1: Figure S1B). Levels of p-AktSer473, an activated form of Akt, were markedly decreased in Akt cTKO (Additional file 1: Figure S1B). Immunohistochemistry (IHC) results showed very weak immuno-reactivity of t-Akt in the brain of Akt cTKO mice (Additional file 1: Figure S1C-d-f, a-c for controls).
Increased levels of tau phosphorylated (p-tau) at the Thr205, Thr231 and Ser396 epitopes have been widely reported in Alzheimer’s brain and mouse models [2]. Our Western results showed highly increased levels for p-tauT205, p-tauT231 and p-tauS396 in Akt cTKO mice (Fig. 1a). Moreover, the position of p-tau bands at the 60, 64 and 68 kDa in the SDS gels was consistently higher in Akt cTKOs than that in controls (Fig. 1a), likely due to the slow migrating rate caused by tau hyperphosphorylation. We conducted additional control experiments and used two groups of Akt2/3 double KO mice, Akt1 f/f ;Akt2 −/− ;Akt3 −/− ;CAG-CreER and Akt1 f/f ;Akt2 −/− ;Akt3 −/−, which were age- matched littermates to Akt cTKOs. We found that Akt DKO mice did not show significant changes on p-tau levels (Additional file 1: Figure S2). Strong p-tauThr205 immuno-reactivity was clearly seen in the cytoplasm of neurons in various brain sub-areas of Akt cTKOs but not controls (Fig. 1b). Increased p-tauSer396 immuno-reactivity was largely detected in glial-like cells in the brain of Akt cTKOs (Additional file 1: Figure S3). Immunostaining on p-tauThr231 showed strong signals in neuronal branches of the cTKO mice (Additional file 1: Figure S4), and double-staining with MAP2 suggested dendritic localization for p-tauThr231 (Additional file 1: Figure S4C: a-d).

Tau hyperphosphorylation in the brain of Akt cTKO mice. a Western blotting of p-tau using cortical and cerebellar samples. Antibodies against tau phosphorylated at the Thr205, Thr231 and Ser396 epitopes were used. The migration of three p-tau bands (60 kDa, 64 kDa and 68 kDa) in SDS gels was slower in Akt cTKO (Akt1 f/f ;Akt2 −/− ;Akt3 −/− ;CAG-CreER with tamoxifen treatment) than in control mice (Akt1 f/f ;Akt2 +/+ ;Akt3 +/+ and Akt1 f/+ ;Akt2 +/+ ;Akt3 +/+). Intensities of the p-tau bands were dramatically increased in Akt cTKO mice. Tau5 antibody was used to detect total tau (t-tau). b Immunohistochemistry of p-tauThr205 in the brain. Strong immuno-reactivity of p-tauThr205 was seen in the cortex (e), hippocampal CA3 (f), the hilus of the dentate gyrus (g) and the cerebellum (h) of Akt cTKO mice. Weak immuno-reactivity of p-tauThr205 was observed in different brain sub-areas of control mice (a-d)
TUNEL assay was performed to examine cell death. No difference in the total number of TUNEL+ cells was observed in brains of Akt cTKO and control mice (Additional file 1: Figure S5), suggesting no significant change in apoptosis. Markers for neurons, astrocytes and microglia were used for biochemical and morphological analyses. First, there was no significant reduction on cortical levels for NeuN, GFAP and Iba1 in Akt cTKO mice (Additional file 1: Figure S6A). Second, there was comparable immuno-reactivity for NeuN, GFAP or Iba1 (Additional file 1: Figure S6B-C) in control and Akt cTKO mice. Therefore, deletion of Akt does not significantly affect the survival of cortical cells.
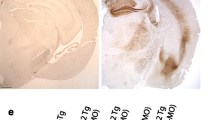
To determine the role of Akt single isoform in tau phosphorylation in the brain, we analyzed p-tau levels using cortical lysates from three lines of Akt single isoform KO mice including Akt1 −/− [3], Akt2 −/− [4] and Akt3 −/− [5]. Nissl staining revealed comparable brain structure between Akt1 −/−, Akt2 −/−, Akt3 −/− and their age-matched WT littermates (Fig. 2). Consistent with previously published observations [5, 6], Akt3 −/− mice displayed smaller brain than WT (Fig. 2e). However, no significant changes in p-tau levels were observed in Akt1 −/−, Akt2 −/− and Akt3 −/− mice (Fig. 2b,d,f: ps > 0.05), suggesting that tau hyperphosphorylation shown in Akt cTKO mice is unlikely to be caused by loss of a single Akt isoform.

Unchanged levels of p-tau in Akt single isoform KO mice. a Nissl staining for the brain of Akt1 −/− mice. Normal brain structure was observed. b Western analyses on p-tau levels using cortical samples of Akt1 −/− mice. There was no significant difference on relative levels of p-tauThr205, p-tauThr231 and p-tauSer396 in Akt1 −/− mice. c Nissl staining for the brain of Akt2 −/− mice. No abnormal brain structure was detected. d Western analyses on p-tau levels using cortical samples of Akt2 −/− mice revealed no significant difference. e Nissl staining for the brain of Akt3 −/− mice. There was a smaller brain in Akt3 −/− mice than in WT animals. f Western analyses on p-tau levels showed no significant difference in Akt3 −/− mice (NS = not significant)
To dissect molecular pathways involved, we conducted Western blotting using an antibody against phosphorylated Akt substrates. We found increased intensities for several bands including those for GSK3α/3β in Akt cTKO mice (Additional file 1: Figure S7A). Since GSK3 is a substrate of Akt, it is expected that deletion of Akt may cause reduced levels for phosphorylated GSK3α/3β [7]. However, we found that relative levels for p-GSK3βSer9 were significantly increased in Akt cTKO mice (Additional file 1: Figure S7B) and those for p-GSK3αTyr279 were also increased (Fig. 3a), suggesting inhibition of GSK3β but activation of GSK3α. Since β-catenin is a well-known substrate of GSK3 [8, 9], p-β-cateninSer33/Ser37/Thr41 was examined and showed significantly increased levels in Akt cTKOs (Additional file 1: Figure S7C), suggesting elevated GSK3α activity. Moreover, we examined several other tau kinases. First, although levels for p-Cdk5Ser159 were increased, those for p25 were decreased (Fig. 3b, p < 0.05). Second, no significant changes on levels for p-Erk1/2 and p-p38 were detected in Akt cTKO mice (Fig. 3c-d, ps > 0.05). Overall, activities for GSK3β, Cdk5, Erk and MAPK p38 were not enhanced.

Increased levels for an activated form of GSK3α in the brain of Akt cTKO mice. a Western blotting on activated forms of GSK3α and GSK3β. Quantitative analysis showed significantly increased levels for p-GSK3αTyr279 (*, p < 0.05) but unchanged levels for p-GSK3βTyr216 (NS, p > 0.05) in Akt cTKO mice. b Western blotting on p-Cdk5, p35 and p25. Relative levels for p-Cdk5Ser159 were significantly increased (*, p < 0.05), but those for p25 were significantly decreased (*, p < 0.05) in the cortex of Akt cTKO mice. GAPDH served as the loading control. c Western blotting on MAPK Erk1/2. Total Erk1/2 levels in Akt cTKO mice were not different from those in control mice. β-actin served as the loading control. Ratio of p-Erk1/2 to the total Erk1/2 in Akt cTKO mice was also not different from that in control mice (p > 0.05). d Western blotting on phosphorylated MAPK p38 (p-p38). Levels of total p38 did not differ between Akt cTKO and control mice (p > 0.05). Ratio of p-p38 to total p38 in Akt cTKO mice was not changed (NS = not significant)
It has been shown that GSK3β is phosphorylated at the Ser9 site not only by Akt but also by PKA [10]. The tyrosine phosphorylation of GSK3 requires the cAMP-PKA signaling [11, 12]. Moreover, it is known that PKA is a kinase to phosphorylate tau at the sites of Thr205, Thr231 and Ser396 [2, 13]. To test the possibility that PKA is involved, we performed the following experiments. First, an antibody against phosphorylated PKA substrates was used to conduct Western blotting. Increased levels for several bands with a wide range of molecular weights were observed (Fig. 4a), suggesting increased PKA activity. Second, we examined VASP, a well-known PKA substrate [14, 15], and observed highly increased p-VASPSer157 levels (Fig. 4b). In contrast, expression levels of PKA regulatory subunits, 1α and 1β, were unchanged (Fig. 4b). Third, since the Ser214 [16] and Ser356 [17, 18] sites of tau are phosphorylated by PKA as well, we analyzed p-tauSer214 and p-tauSer356, which exhibited increased levels in Akt cTKO mice (Fig. 4c).

Increased levels for phosphorylated PKA substrates in Akt cTKO mice. a Western blotting for phosphorylated PKA substrates using cortical samples of Akt cTKO mice. Levels for several phosphorylated PKA substrates with molecular weights at about 100 kDa, 60 kDa and 40 kDa were increased. b Western analyses on VASP and PKA subunits. P-VASPSer157 levels in Akt cTKO mice were highly increased but total VASP levels were not changed. Relative levels for PKA regulatory subunits, 1α and 1β, were not changed. GAPDH served as the loading control. c Western blotting on p-tauSer214 and p-tauSer356. Ratios of p-tau to total tau were significantly increased in Akt cTKO mice, as compared to control animals (*, p < 0.05)
Discussion
Akt levels are decreased in Alzheimer’s brain [19]. However, it remains unknown exactly how Akt is involved in pathophysiological processes of AD, e.g. tau pathology and neurodegeneration. In this study, a novel Akt cTKO mouse model was generated and displayed tau hyperphosphorylation in the brain. We demonstrated that Akt cTKO mice exhibited significantly increased levels for several PKA downstream substrates. We observed increased levels for an activated form of GSK3α in Akt cTKO mice. We reported unchanged levels for activated forms of GSK3β, Cdk5, Erk and MAPK p38 in Akt cTKO mice.
It had been difficult to study normal physiological functions of Akt in the adult brain. First, while there are three Akt isoforms in mammals [20], a recent study did not identify which Akt isoform plays important role in neurodegenerative disease [19]. Second, since global deletion of Akt three isoforms causes early embryonic death in mice [1], a viable Akt cTKO mouse model was generated to overcome developmental problems in this study. Interestingly, Akt cTKO animals but not Akt single isoform KO mice exhibited increased p-tau levels in the brain, suggesting that total Akt but not a single isoform plays a key role in tau phosphorylation. Immunostaining using AT8 or AT100 antibodies revealed no NFTs-like structure in Akt cTKO mice (data not shown). No significant changes on the total number of TUNEL+ cells or NeuN+ cells were detected in Akt cTKO mice. These findings are consistent with the concept that tau hyperphosphorylation is an early event during neurodegenerative process [21].
Tau phosphorylation status is determined by activities of multiple protein kinases [22]. Since there were unchanged levels for activated forms of GSK3β, Cdk5, Erk and p38, it is unlikely that they could account for abnormal tau phosphorylation in Akt cTKO mice. Although it is believed that inactivation of Akt may cause reduced p-GSK3αS21/3βS9 [7] and thus lifts the inhibition on GSK3, levels for p-GSK3βS9 were actually increased in Akt cTKO mice. Since GSK3β is a substrate for both Akt and PKA [10, 12], in the absence of Akt, it can be phosphorylated by PKA. The findings on phosphorylated PKA substrates, p-VASP and p-tau (Fig. 4) suggest enhanced PKA activity, which helps explain why there was tau hyperphosphorylation in Akt cTKO mice. There are two possible mechanisms by which Akt regulates PKA activity. The first possibility is a compensatory mechanism. It is known that the PI3K-Akt [23] and PKA pathways [24] can be activated by extracellular signal molecules such as cytokines. In the absence of the PI3K-Akt pathway, such molecules may over-activate the cAMP-PKA signaling as compensation. The second mechanism is that phosphodiesterases (PDEs) might be involved in the regulation of PAK activity. PDEs are Akt substrates [25] and can degrade cAMP, the second messenger molecule [26]. Therefore, deletion of Akt may inhibit PDEs and cAMP levels may get increased. Upon activation by cAMP, PKA can act on various downstream targets including tau, GSK3 and VASP.
Methods
Generation of Akt1 f/f and Akt cTKO mice
We used a strategy shown in Additional file 1: Figure S1A to generate floxed Akt1 mice. The detailed breeding plan for the generation of Akt cTKO mice was described in the Additional file 1.
Treatment of mice with tamoxifen
Tamoxifen was used to treat Akt1 f/f ;Akt2 −/− ;Akt3 −/− ;CAG-CreER and age-matched littermate controls for 5 consecutive days. Detailed information was described in the Additional file 1.
Brain lysates preparation
Procedures were described previously [27]. Immunoblotting methods and antibody information were described in the Additional file 1.
Statistical analysis
Data were presented as the mean ± SEM. Two-tailed Student’s t-test was performed to examine the difference between control and cKO mice. P < 0.05 (*) was considered statistically significant.
Ethical approval
Mouse breeding was conducted under IACUC approved protocols at the MARC (Model Animal Research Center). All the experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals of the MARC at Nanjing University.
Abbreviations
- AD:
-
Alzheimer’s disease
- Akt:
-
Protein kinase B
- PKA:
-
Protein kinase A
- cAMP:
-
Cyclic adenosine 3′5′ monophosphate
- Akt cTKO:
-
Akt three isoforms conditional knockout
- PI3K:
-
Phosphatidylinositol-3 kinase
- PDE:
-
Phosphodiesterase
- tau:
-
Microtubule associated protein
- p-tau:
-
Phosphorylated tau
- GSK3:
-
Glycogen synthase kinase-3
- Cdk5:
-
Cyclin-dependent kinase 5
- MAPK:
-
Mitogen-activated protein kinase
- Erk:
-
Extracellular signal-regulated kinase
- TUNEL:
-
Terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling
- NFTs:
-
Neurofibrillary tangles
- MAP2:
-
Microtubule-associated protein
- GFAP:
-
Glial fibrillary acidic protein
- Iba1:
-
Ionized calcium-binding adapter molecule 1
- VASP:
-
Vasodilator stimulated phosphoprotein
References
Dummler B, Hemmings BA. Physiological roles of PKB/Akt isoforms in development and disease. Biochem Soc Trans. 2007;35:231–5.
Wang JZ, Xia YY, Grundke-Iqbal I, Iqbal K. Abnormal hyperphosphorylation of tau: sites, regulation, and molecular mechanism of neurofibrillary degeneration. J Alzheimers Dis. 2013;33 Suppl 1:S123–39.
Yang ZZ, Tschopp O, Hemmings-Mieszczak M, Feng J, Brodbeck D, Perentes E, et al. Protein kinase B alpha/Akt1 regulates placental development and fetal growth. J Biol Chem. 2003;278:32124–31.
Cho H, Mu J, Kim JK, Thorvaldsen JL, Chu Q, Crenshaw 3rd EB, et al. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta). Science. 2001;292:1728–31.
Tschopp O, Yang ZZ, Brodbeck D, Dummler BA, Hemmings-Mieszczak M, Watanabe T, et al. Essential role of protein kinase B gamma (PKB gamma/Akt3) in postnatal brain development but not in glucose homeostasis. Development. 2005;132:2943–54.
Easton RM, Cho H, Roovers K, Shineman DW, Mizrahi M, Forman MS, et al. Role for Akt3/protein kinase Bgamma in attainment of normal brain size. Mol Cell Biol. 2005;25:1869–78.
Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–9.
Asuni AA, Hooper C, Reynolds CH, Lovestone S, Anderton BH, Killick R. GSK3α exhibits β-catenin and tau directed kinase activities that are modulated by Wnt. Eur J Neurosci. 2006;24:3387–92.
Liu C, Li Y, Semenov M, Han C, Baeg G-H, Tan Y, et al. Control of β-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell. 2002;108:837–47.
Li M, Wang X, Meintzer MK, Laessig T, Birnbaum MJ, Heidenreich KA. Cyclic AMP promotes neuronal survival by phosphorylation of glycogen synthase kinase 3β. Mol Cell Biol. 2000;20:9356–63.
Plyte SE, O’Donovan E, Woodgett JR, Harwood AJ. Glycogen synthase kinase-3 (GSK-3) is regulated during Dictyostelium development via the serpentine receptor cAR3. Development. 1999;126:325–33.
Kim L, Liu J, Kimmel AR. The novel tyrosine kinase ZAK1 activates GSK3 to direct cell fate specification. Cell. 1999;99:399–408.
Wang JZ, Grundke-Iqbal I, Iqbal K. Kinases and phosphatases and tau sites involved in Alzheimer neurofibrillary degeneration. Eur J Neurosci. 2007;25:59–68.
Li Z, Ajdic J, Eigenthaler M, Du X. A predominant role for cAMP-dependent protein kinase in the cGMP-induced phosphorylation of vasodilator-stimulated phosphoprotein and platelet inhibition in humans. Blood. 2003;101:4423–9.
Butt E, Abel K, Krieger M, Palm D, Hoppe V, Hoppe J, et al. cAMP- and cGMP-dependent protein kinase phosphorylation sites of the focal adhesion vasodilator-stimulated phosphoprotein (VASP) in vitro and in intact human platelets. J Biol Chem. 1994;269:14509–17.
Schneider A, Biernat J, von Bergen M, Mandelkow E, Mandelkow EM. Phosphorylation that detaches tau protein from microtubules (Ser262, Ser214) also protects it against aggregation into Alzheimer paired helical filaments. Biochemistry. 1999;38:3549–58.
Sironi JJ, Yen SH, Gondal JA, Wu Q, Grundke-Iqbal I, Iqbal K. Ser-262 in human recombinant tau protein is a markedly more favorable site for phosphorylation by CaMKII than PKA or PhK. FEBS Lett. 1998;436:471–5.
Litersky JM, Johnson GV, Jakes R, Goedert M, Lee M, Seubert P. Tau protein is phosphorylated by cyclic AMP-dependent protein kinase and calcium/calmodulin-dependent protein kinase II within its microtubule-binding domains at Ser-262 and Ser-356. Biochem J. 1996;316(Pt 2):655–60.
Liu, Y, Liu, F, Grundke-Iqbal, I, Iqbal, K & Gong, CX. Deficient brain insulin signalling pathway in Alzheimer's disease and diabetes. J Pathol. 2011;225:54-62.
Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–74.
Lee, VM, Goedert, M & Trojanowski, JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121-59.
Dolan PJ, Johnson GV. The role of tau kinases in Alzheimer’s disease. Curr Opin Drug Discov Devel. 2010;13:595–603.
Hinton HJ, Welham MJ. Cytokine-induced protein kinase B activation and bad phosphorylation do not correlate with cell survival of hemopoietic cells. J Immunology. 1999;162:7002–9.
Gwosdow AR, O’Connell NA, Abou-Samra AB. Interleukin-1 increases protein kinase A activity by a cAMP-independent mechanism in AtT-20 cells. 1994.
Degerman E, Ahmad F, Chung YW, Guirguis E, Omar B, Stenson L, et al. From PDE3B to the regulation of energy homeostasis. Curr Opin Pharmacol. 2012;11:676–82.
Kitamura T, Kitamura Y, Kuroda S, Hino Y, Ando M, Kotani K, et al. Insulin-induced phosphorylation and activation of cyclic nucleotide phosphodiesterase 3B by the serine-threonine kinase Akt. Mol Cell Biol. 1999;19:6286–96.
Cheng S, Zhang C, Xu C, Wang L, Zou X, Chen G. Age-dependent neuron loss is associated with impaired adult neurogenesis in forebrain neuron-specific Dicer conditional knockout mice. Int J Biochem Cell. 2014;57:186–96.
Acknowledgements
We thank Zenglan Lu, Beibei Lu and Zhengyun Huang for technical assistance. The work was supported by grants from the National Natural Science Foundation of China (31271123),the National Basic Research Program of Ministry of Science and Technology of China (2014CB942804), Jiangsu Provincial Double-Innovation Program and the MARC Initiative Scientific Research Program.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
LW, SC, JH, ZY, CX, SL, XZ, TY, XZ and GC performed experiments. LW and GC analyzed the data. GC, ZY and YX designed the research. GC wrote the manuscript. GC, YP and YX edited the manuscript. All authors read and approved the final manuscript.
Long Wang and Shanshan Cheng contributed equally to this work.
Additional file
Additional file 1:
Conditional inactivation of Akt three isoforms causes tau hyperphosphorylation in the brain. (PDF 3519 kb)
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Wang, L., Cheng, S., Yin, Z. et al. Conditional inactivation of Akt three isoforms causes tau hyperphosphorylation in the brain. Mol Neurodegeneration 10, 33 (2015). https://doi.org/10.1186/s13024-015-0030-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13024-015-0030-y