
Abstract
Background
The most frequent manifestation in adult hypophosphatasia (HPP) is musculoskeletal pain. The unspecific nature of its clinical presentation may prevent correct diagnosis. The aim of the study was to assess the prevalence of ALPL mutations in adult patients treated in rheumatological outpatient facilities with evident musculoskeletal symptoms typical for HPP.
Methods
Over a period of 10 years 9,522 patients were screened in the rheumatology outpatient clinic of the Hanusch hospital Vienna. Serum ALP levels ≤ 40 U/L were found in 524 patients. After screening for secondary causes, 73 patients were invited for clinical evaluation. Genetic testing was performed in 23 patients with suspected HPP. Logistic regression models with Firth penalisation were used to estimate the unadjusted and BMI-adjusted association of each clinical factor with HPP.
Results
Mutations in the ALPL gene were observed in 57% of genetically screened patients. Arthralgia, fractures, and pain were the leading symptoms in individuals with ALPL mutation. Chondrocalcinosis (OR 29.12; 95% CI 2.02–1593.52) and dental disease (OR 8.33; 95% CI 0.93–143.40) were associated with ALPL mutation, independent of BMI. Onset of symptoms in patients with ALPL mutation was at 35.1 (14.3) years, with a mean duration from symptoms to diagnosis of 14.4 (8.1) years. Bone mineral density (BMD) and trabecular bone score (TBS) as well as bone turnover markers were not indicative for HPP or ALPL mutation.
Conclusion
HPP can mimic rheumatologic diseases. Thus, HPP should be considered as a possible diagnosis in adult patients presenting with musculoskeletal pain of unknown origin in rheumatology outpatient clinics. In patients with persistently low ALP serum levels and unclear musculoskeletal pain, HPP as the underlying cause has to be considered.
Similar content being viewed by others

Introduction
Since the introduction of the specific enzyme replacement therapy asfotase alfa, awareness for hypophosphatasia (HPP) has increased. HPP is a hereditary metabolic bone disease caused by a loss of function mutation in the ALPL gene encoding for the tissue non-specific alkaline phosphatase (TNSALP) [1]. Clinical manifestation of HPP is heterogeneous. Its primary hallmark is hypomineralisation, which can lead to bone deformities, fractures, delayed fracture healing as well as premature tooth loss, especially in children and adolescents. Extra-skeletal manifestations such as pulmonary abnormalities, cerebral seizures, impaired motor skills or nephrocalcinosis occasionally occur [2].
In adults, the leading clinical manifestation of HPP is musculoskeletal pain, often mimicking rheumatologic diseases [3,4,5,6]. In the study of Genest et al. concerning adults with paediatric onset HPP, all patients reported a history of muscular, dental, or skeletal complaints as well as pain at baseline. Rheumatic manifestations were stated by 21% of patients, while no detailed information was given [7]. Anecdotally, chondrocalcinosis, enthesitis and arthralgia/arthritis were described to occur in patients with HPP [8]. In a global registry, pain, orthopaedic procedures, and recurrent fractures were the most common manifestations in adult HPP patients [9]. Notably, most HPP patients suffer from more than one clinical HPP manifestation including dental disease, fractures or pain [10], [11].
Diagnosis of HPP can be challenging. Persistently low alkaline phosphatase (ALP) is the serological hallmark of HPP. However, low ALP is a common and often overlooked phenomenon in clinical routine and low ALP levels are not pathognomonic for HPP. Eating disorders, thyroid dysfunction, Milk-Alkali syndrome, multiple myeloma, Celiac Disease, zinc deficiency, surgery as well as anti-resorptive therapy can cause hypophosphatasemia [12, 13]. In a study by Schmidt et al., the prevalence of low ALP in adults was 8.46% for values < 30 U/L and more than 9% for ALP < 40 (but > 30 U/L) [13].
In the event of low ALP activity, substrates of ALP, such as pyridoxal 5’-phosphate (PLP) accumulate [12]. Elevated PLP levels are indicative of HPP and can be used to differentiate from secondary reasons for low ALP levels [14, 15]. Therefore concomitant PLP measurement in individuals with low ALP and clinical symptoms of HPP has been recommended [13]. Other established bone turnover markers such as osteocalcin, procollagen type 1 N-propeptide (P1NP), tartrate-resistant acid phosphatase 5b (TRAP5b), or N-terminal telopeptide of type 1 collagen (NTx) are usually in normal range and therefore not useful for the diagnosis of HPP [16]. Bone mineral density (BMD) by Dual Energy X-ray Absorptiometry (DXA), which is considered the gold standard for diagnosis and follow up of osteoporosis does not reflect metabolism or severity of the disease in patients with HPP [16].
Misdiagnosis is common and the diagnosis of HPP is often delayed due to the heterogeneity of the disease, unspecific symptoms as well as a lack of awareness for low ALP values [9, 17].
The aim of this study was to assess the prevalence of ALPL mutation in adult patients treated in the rheumatologic outpatient unit with evident musculoskeletal symptoms typical for HPP.
The primary objective was to identify ALPL mutations in patients presenting with musculoskeletal pain but an inconclusive diagnosis.
The secondary objective was to investigate the clinical differences between patients with ALPL gene mutation and those without mutation.
Patients and methods
In this prospective single-centre study, all patients who presented at the rheumatology outpatient unit of the Hanusch Hospital Vienna, a specialized centre for rheumatology and rare bone diseases, between 2008 and 2018 were retrospectively screened for ALP serum levels ≤ 40 U/L.
Patients with at least one low ALP measurement were further evaluated using patient charts. Only adult patients (age ≥ 18 years) were included. Patients who presented with musculoskeletal symptoms (arthralgia, myalgia, arthritis, enthesitis) were included. All patients with secondary causes for low ALP levels (including eating disorders, thyroid dysfunction, surgery, sepsis) or, if available, consecutively normal ALP levels were excluded. Patients who fulfilled these inclusion criteria were invited for clinical evaluation. Clinical features and demographic data were documented with an HPP-specific questionnaire. To exclude secondary causes for low ALP, other metabolic bone diseases and assess HPP comorbidities, detailed laboratory examinations were performed in all participants. The following parameters in venous blood samples were evaluated: blood count, kidney and liver parameters, alkaline phosphatase, calcium, ionised calcium, phosphate, thyroid markers, 25(OH)Vitamin D, albumin, bone turnover markers such as osteocalcin (OC) and serum beta-crosslaps (CTX), as well as intact parathyroid hormone (PTH).
If laboratory results showed consistently low ALP ≤ 40 U/L and patients had clinical features of HPP but (i) no definitive rheumatologic diagnosis or (ii) an inconclusive rheumatologic diagnosis or (iii) were non-responders to their treatment, genetic testing was initiated.
Written informed consent was obtained from all individuals prior to any study related procedures. The study was approved by the Ethics Committee of the City of Vienna (EK 18-239-VK).
Genetic testing was performed on DNA extracted from peripheral blood samples by use of standard methods (QIASymphony, QIAGEN). Coding regions of ALPL-gene were enriched using the TruSight One Expanded panel and sequenced on a NextSeq device (both Illumina). Sequence data was aligned against the reference sequence ENST00000374840.8 and variants detected in coding and flanking intronic regions were classified following the ACMG guidelines [18]. Mutations were classified as Class 5 (pathogenic), Class 4 (likely pathogenic), Class 3 (VUS—variant of uncertain significance), Class 2 (likely benign) and Class 1 (benign).
After receiving genetic results, the study population was divided in two groups (HPP and non-HPP) to compare their clinical characteristics, biochemical profile, and demographic data. In addition, HPP patients underwent dual energy X-ray absorptiometry (DXA, GE Healthcare Lunar Prodigy (GE Healthcare, Madison, WI, USA)) to assess bone mineral density (BMD) and trabecular bone score (TBS). DXA analysis was conducted using the software Encore 16 and TBS was analysed using TBS iNsight 3 software. No information on DXA or TBS were available for the non-HPP group.
Statistical methods
Characteristics of patients were described using frequencies and percentages for categorical variables and means and standard deviation (± SD) or medians and interquartile ranges (IQR) for continuous variables. Normality of parameters distribution in HPP and non-HPP was assessed by the Shapiro–Wilk test. Differences between the two groups were evaluated with T-test/Mann Whitney U test for continuous variables and Chi-squared/Fisher test for categorical variables, as appropriate.
The association between clinical parameters and HPP was assessed with logistic regression using the Firth method [19]. This method provides bias-reduction for small sample size and yields finite and consistent estimates even in case of separation. Crude model and model adjusted for body mass index (BMI) were fitted by penalised maximum likelihood.
All computations were performed in SPSS version 26 [20] and in R version 1.3, package “logistf”[21].
Results
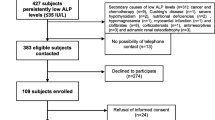
In the initial screening phase, 9,522 patients from the rheumatologic outpatient unit were screened for ALP levels. In total, 524 patients had at least one ALP measurement < 40 U/L. The remaining 8,998 patients presented with a serum ALP level > 40 U/L.
After screening the medical records, 426 patients were excluded due to secondary causes for ALP, transient low ALP, or an absence of typical musculoskeletal symptoms. Nineteen patients were excluded, as no medical records were available. Six patients had died.
The remaining 73 patients were invited to undergo clinical examination. Of these, 29 patients consented to clinical re-evaluation. Six more patients were excluded because serum ALP was > 50 U/L in subsequent blood analysis. In total, 23 patients showed repeatedly low ALP levels, typical musculoskeletal symptoms, no secondary causes, and underwent genetic testing.
A mutation in the ALPL gene was observed in 13 patients (56.5%) (see Fig. 1). One patient was classified as Class 3 variant of uncertain significance but remained in the study due to typical clinical manifestations. Including only patients with Class 4 and 5 mutations, the prevalence of ALPL mutation was 52.2%.

Study design. ALP alkaline phosphatase; ALPL-positive genetically confirmed mutation in the ALPL-gene; ALPL-negative negative genetic testing or ALP > 50 U/L
Characteristics of patients with ALPL mutation and those without are presented in Table 1. Patients with ALPL mutation did not significantly differ in age, sex, or height from the control group without mutation. Mean age of heterozygous mutation positive individuals was 50.0 ± 14.2 (SD) with more women affected (61.5%). However, higher body weight (78.6 ± 19.9 kg vs. 62.0 ± 8.5 kg, p = 0.013) and BMI (27.5 ± 6.6 vs. 21.7 ± 2.1, p = 0.009) were found in ALPL mutation positive individuals. Patients with heterozygous mutation in the ALPL gene had significantly lower levels of ALP (27.7 ± 6.5 vs. 37.7 ± 7.2, p = 0.001) and higher vitamin D levels (89.0 ± 55.0 nmol/l vs. 78.0 ± 38.3 nmol/l, p = 0.045). No differences were found regarding calcium, phosphate, PTH, CTX or OC.
DXA measurements showed normal BMD values at the lumbar spine and hip as well as normal TBS values.
Logistic regression models with Firth penalisation adjusted for BMI showed that periarticular calcification was significantly associated with ALPL mutation (OR 29.12; 95% CI 2.02–1593.52). Similarly, patients with dental disease had a higher likelihood of ALPL mutation, although with borderline statistical significance (OR 8.33; 95% CI 0.93–143.40). A history of fractures, pneumonia, and impaired physical function in childhood were also more likely to be present in patients with ALPL mutation, albeit without statistical significance (see Table 2).
Onset of symptoms was at 35.1 (SD 14.3) years, with a duration from symptoms to diagnosis of 14.4 (SD 8.1) years in heterozygous mutation positive individuals.
In 13 patients, rare genetic variants were identified of which 12 were classified as likely pathogenic or pathogenic (Class 4 or 5), one was classified as variant of unknown significance (Class 3) according to the ACMG guidelines [22]. All variants were in heterozygous state, which is in concordance with the mild clinical presentation of the patients (see Table 3).
Discussion
In the present study we investigated the prevalence of ALPL-mutations in a cohort of adult patients with musculoskeletal symptoms, typical for both HPP and rheumatological diseases. Screened patients were treated in the rheumatologic outpatient unit but had inconclusive diagnoses such as fibromyalgia, chronic back pain, pseudogout, calcific tendinitis, osteoarthritis, spondylarthopathy, or undifferentiated connective tissue diseases. Main symptoms were pain in general, back pain, arthralgia, and myalgia. Notably, several patients were treated with disease-modifying anti-rheumatic drugs and did not show any response to therapy. Genetic screening of our patients revealed a high prevalence of heterozygous mutation in the ALPL gene in those with persistently low ALP levels. Every second genetically tested patient showed a mutation in the ALPL gene, indicating a hereditary origin of the musculoskeletal symptoms. Low ALP was detected in 5% of our patient cohort and 14% of these patients presented with musculoskeletal complaints and repetitive low ALP compatible with a HPP diagnosis. Although only mild symptoms were detected in unselected populations with low ALP levels [23, 24] musculoskeletal pain, dental disease, and fractures were associated to ALPL variations [25]. Since musculoskeletal pain was an inclusion criterion in the present study, no significant differences were found between patients with and without ALPL mutation. However, periarticular calcification discriminated both groups. Moreover, fractures, pneumonia, dental disease, and impaired function in childhood were more common in heterozygous mutation positive individuals than in patients with low ALP, but without mutation in the ALPL gene.
Although the absolute number of patients with mutation in the ALPL gene identified in the entire study population is relatively low, it should be noted that HPP is a rare genetic disease with a low prevalence of 1/6370 for mild HPP [26]. In literature the prevalence of ALPL mutations depends on the investigated population and the geographic background [26, 27]. In an unselected population of adults from Spain with unexplained low ALP, ALPL mutations were found in 50% of patients. Musculoskeletal pain was again the predominant symptom in all patients with no difference between patients with and without genetic mutation [23]. In another Spanish cohort of more than 78,000 subjects, persistently low ALP was found in 0.12% of subjects. Fifty-six patients with low ALP and no secondary causes were identified. A mutation in the ALPL gene was found in seven out of 16 tested individuals. Most of them had the diagnosis of osteoarthritis, ankylosing spondylitis, fibromyalgia, or osteoporosis, respectively [24].
A comparable high prevalence of ALPL mutations were also found by others in more specific cohorts. More than 26,000 adults with endocrinological diagnoses were screened for ALP-levels in Denmark. Fifty-one subjects with persistently low ALP levels were identified after exclusion of risk factors for low ALP. Of these, 24 were genetically tested and more than 50% showed a mutation in the ALPL gene and musculoskeletal pain as leading symptom. PLP levels were higher in HPP than non-HPP subjects [15].
To date, there is only one study reporting on ALP levels in rheumatologic patients. Retrospective screening of medical records including existing genetical testing for ALPL gene mutation in adult patients with rheumatological diseases and repeatedly low ALP levels was done by a study group from Germany [28]. Persistently low ALP levels were detected in 1.31%, which is higher than reported in non-rheumatologic patients by others [24]. In 13 out of 19 previously genetically tested individuals (68.4%) a mutation in ALPL gene was found. Due to the nature of rheumatologic diseases and HPP, all subjects suffered from musculoskeletal pain. The most common rheumatologic diagnoses in ALPL-positive patients were osteoarthritis, rheumatoid arthritis, spondyloarthritis, and collagenosis. Chondrocalcinosis was not linked to HPP in this study [28]. It remains unclear if the patients in this and our study were misdiagnosed or ALPL gene mutation was a concomitant finding.
In contrast to the above-mentioned studies, other authors found a much lower prevalence of low ALP and ALPL mutations, respectively. In a Brazilian cohort of almost 290,000 chemical tests, only 12 patients with repeatedly low ALP and no secondary reason were identified. None of them showed typical symptoms such as musculoskeletal pain, fragility fractures, or chondrocalcinosis. Genetic testing was not performed. The authors hypothesised a lower HPP prevalence in Brazil compared to other countries [29].
In a paediatric setting, 393 out of 6,731 screened children had low ALP. The authors state that most of them had a feasible explanation. From the remaining 30 children only 3 had low ALP in combination with elevated PLP and ultimately an ALPL-mutation. One child was diagnosed with HPP, 2 were considered mutation carriers [30].
HPP is often overlooked in adult patients with typical manifestations. In adulthood, diagnosis is usually made above the age of 40 years. Whereas respiratory and dental symptoms usually occur in childhood, onset of pain and fractures was reported at the median age between 33 and 44 years [11]. In patients with first onset of symptoms in adulthood, symptoms preceded diagnosis by ten years or more [9, 25]. This is in line with our findings. Duration of musculoskeletal symptoms to detection of ALPL gene mutation was more than 14 years in our study. However, 23% of patients had also reported functional impairment in childhood pointing to the fact that HPP can remain undiagnosed for decades.
Although most adults do not qualify for enzyme replacement therapy with asfotase alfa, diagnosis of HPP in adults is still important, because an overlap to osteoporosis may occur. In case of uncertain bone metabolism, anti-resorptive drugs should be avoided. Bisphosphonates and denosumab suppress bone turnover and TNSALP activity and might lead to atypical femoral fractures [31]. More recent data suggest a rather lower risk in HPP patients with heterozygous mutation and lack of histological signs of osteomalacia [32].
Low ALP is the hallmark of HPP. Nevertheless, reference values for ALP depend on age and sex [33] and the lower limit of normal is often not stated in lab reports. Moreover, as low ALP is a common finding, secondary reasons must be ruled out. The measurement of natural substrates such as PLP or PEA is an important diagnostic step in patients with persistently low ALP-levels and typical symptoms. In case of repeatedly low ALP levels in combination with increased values for PLP and typical clinical signs such as musculoskeletal pain, HPP must be considered [13]. Due to the lack of accessibility no PLP plasma concentrations were evaluated in our study. Considering that, predictive models and machine learning algorithms identified HPP patients more accurately based on levels of ALP and PLP than ALP alone [34]. Established bone turnover markers are not indicative for HPP [16] and BMD by means of DXA is not necessarily reduced in HPP, as was shown in our population, and supported by other studies [35]. While not mandatory, genetic testing confirms the diagnosis of HPP because Sanger sequencing and MLPA allow the detection of 95% of all ALPL mutations [15, 36].
It has to be considered, that a mutation in the ALPL gene does not necessarily confirms the diagnosis of HPP. The combination of low ALP, musculoskeletal pain and a mutation in the ALPL gene can be both, mild HPP or a heterozygous carrier state with unspecific symptoms. Moreover, chronic pain, which was used as inclusion criterion in the present study, is a common finding also in the general population. International studies have reported a prevalence of chronic pain between 11% and 51.3% in population-based studies [37,38,39,40]. Especially in the absence of other skeletal and extra-skeletal manifestations such as recurrent fractures, atypical femoral fractures, tooth loss, chondro- or nephro-calcinosis, patients are more likely heterozygous mutation positive individuals, rather than HPP patients. Thus, diagnosis of HPP should be made with caution, in order to not overdiagnoses HPP.
In conclusion, a mutation in the ALPL gene should be considered as a possible explanation in adult patients presenting with musculoskeletal pain of unknown origin in rheumatology outpatient clinics. In patients with persistently low ALP serum levels and unclear musculoskeletal pain, HPP as the underlying cause has to be considered.
Availability of data and materials
The datasets generated and analysed during the current study are not publicly available, but are available from the corresponding author on reasonable request.
References
Whyte MP. Hypophosphatasia—aetiology, nosology, pathogenesis, diagnosis and treatment. Nat Rev Endocrinol. 2016;12(4):233–46.
Salles JP. Hypophosphatasia: biological and clinical aspects, avenues for therapy. Clin Biochem Rev. 2020;41(1):13–27.
Durrough C, Colazo JM, Simmons J, Hu JR, Hudson M, Black M, et al. Characterization of physical, functional, and cognitive performance in 15 adults with hypophosphatasia. Bone. 2021;142: 115695.
Seefried L, Kishnani PS, Moseley S, Denker AE, Watsky E, Whyte MP, et al. Pharmacodynamics of asfotase alfa in adults with pediatric-onset hypophosphatasia. Bone. 2021;142: 115664.
Kuehn K, Hahn A, Seefried L. Mineral intake and clinical symptoms in adult patients with hypophosphatasia. J Clin Endocrinol Metab. 2020;105(8).
Weber TJ, Sawyer EK, Moseley S, Odrljin T, Kishnani PS. Burden of disease in adult patients with hypophosphatasia: results from two patient-reported surveys. Metab Clin Exp. 2016;65(10):1522–30.
Genest F, Rak D, Petryk A, Seefried L. Physical function and health-related quality of life in adults treated with asfotase alfa for pediatric-onset hypophosphatasia. JBMR Plus. 2020;4(9): e10395.
Bangura A, Wright L, Shuler T. Hypophosphatasia: current literature for pathophysiology, clinical manifestations, diagnosis, and treatment. Cureus. 2020;12(6): e8594.
Högler W, Langman C, Gomes da Silva H, Fang S, Linglart A, Ozono K, et al. Diagnostic delay is common among patients with hypophosphatasia: initial findings from a longitudinal, prospective, global registry. BMC Musculoskelet Disord 2019;20(1):80.
Seefried L, Dahir K, Petryk A, Högler W, Linglart A, Martos-Moreno G, et al. Burden of illness in adults with hypophosphatasia: data from the global hypophosphatasia patient registry. J Bone Mineral Res. 2020;35(11):2171–8.
Szabo SM, Tomazos IC, Petryk A, Powell LC, Donato BMK, Zarate YA, et al. Frequency and age at occurrence of clinical manifestations of disease in patients with hypophosphatasia: a systematic literature review. Orphanet J Rare Dis. 2019;14(1):85.
Simon S, Resch H, Klaushofer K, Roschger P, Zwerina J, Kocijan R. Hypophosphatasia: from diagnosis to treatment. Curr Rheumatol Rep. 2018;20(11):69.
Schmidt T, Schmidt C, Amling M, Kramer J, Barvencik F. Prevalence of low alkaline phosphatase activity in laboratory assessment: Is hypophosphatasia an underdiagnosed disease? Orphanet J Rare Dis. 2021;16(1):452.
Whyte MP, Zhang F, Wenkert D, Mack KE, Bijanki VN, Ericson KL, et al. Hypophosphatasia: Vitamin B(6) status of affected children and adults. Bone. 2022;154: 116204.
Hepp N, Frederiksen AL, Duno M, Præst Holm J, Rye Jørgensen N, Beck Jensen JE. Biochemical, clinical and genetic characteristics in adults with persistent hypophosphatasaemia; data from an endocrinological outpatient clinic in Denmark. Bone Rep. 2021;15: 101101.
Seefried L, Rak D, Petryk A, Genest F. Bone turnover and mineral metabolism in adult patients with hypophosphatasia treated with asfotase alfa. Osteopor Int. 2021;32(12):2505–13.
Koga M, Kinoshita Y, Kato H, Kobayashi H, Shinoda Y, Nangaku M, et al. Massive calcification around large joints in a patient subsequently diagnosed with adult-onset hypophosphatasia. Osteopor Int 2021.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24.
Heinze G, Schemper M. A solution to the problem of separation in logistic regression. Stat Med. 2002;21(16):2409–19.
Corp. I. IBM SPSS Statistics for Windows. Armonk, NY: IBM Corp. Released 2019. ;Version 26.0.
Heinze G, Ploner, M., Jiricka L. logistf: Firth's Bias-Reduced Logistic Regression. 2020(R package version 1.24).
Nykamp K, Anderson M, Powers M, Garcia J, Herrera B, Ho YY, et al. Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria. Genet Med. 2017;19(10):1105–17.
Riancho-Zarrabeitia L, García-Unzueta M, Tenorio JA, Gómez-Gerique JA, Ruiz Pérez VL, Heath KE, et al. Clinical, biochemical and genetic spectrum of low alkaline phosphatase levels in adults. Eur J Intern Med. 2016;29:40–5.
García-Fontana C, Villa-Suárez JM, Andújar-Vera F, González-Salvatierra S, Martínez-Navajas G, Real PJ, et al. Epidemiological, clinical and genetic study of hypophosphatasia in a spanish population: identification of two novel mutations in the alpl gene. Sci Rep. 2019;9(1):9569.
Tornero C, Navarro-Compán V, Tenorio JA, García-Carazo S, Buño A, Monjo I, et al. Can we identify individuals with an ALPL variant in adults with persistent hypophosphatasaemia? Orphanet J Rare Dis. 2020;15(1):51.
Mornet E, Yvard A, Taillandier A, Fauvert D, Simon-Bouy B. A molecular-based estimation of the prevalence of hypophosphatasia in the European population. Ann Hum Genet. 2011;75(3):439–45.
Taketani T, Onigata K, Kobayashi H, Mushimoto Y, Fukuda S, Yamaguchi S. Clinical and genetic aspects of hypophosphatasia in Japanese patients. Arch Dis Child. 2014;99(3):211–5.
Karakostas P, Dolscheid-Pommerich R, Hass MD, Weber N, Brossart P, Schäfer VS. [Prevalence of hypophosphatasia in adult patients in rheumatology]. Zeitschrift fur Rheumatologie. 2021.
Vieira LHR, Peixoto KC, Flósi CL, de Farias MLF, Madeira M. Active search of adult patients with persistently low serum alkaline phosphatase levels for the diagnosis of hypophosphatasia. Arch Endocrinol Metab. 2021;65(3):289–94.
Held CM, Guebelin A, Krebs A, Sass JO, Wurm M, Lausch E, et al. Screening for hypophosphatasia: Does biochemistry lead the way? J Pediatr Endocrinol Metab JPEM. 2021.
Sutton RA, Mumm S, Coburn SP, Ericson KL, Whyte MP. “Atypical femoral fractures” during bisphosphonate exposure in adult hypophosphatasia. J Bone Mineral Res. 2012;27(5):987–94.
Rassie K, Dray M, Michigami T, Cundy T. Bisphosphonate use and fractures in adults with hypophosphatasia. JBMR Plus. 2019;3(10): e10223.
Rockman-Greenberg C. Hypophosphatasia. Pediatr Endocrinol Rev PER. 2013;10(Suppl 2):380–8.
Garcia-Carretero R, Olid-Velilla M, Perez-Torrella D, Torres-Pacho N, Darnaude-Ortiz MT, Bustamate-Zuloeta AD, et al. Predictive modeling of hypophosphatasia based on a case series of adult patients with persistent hypophosphatasemia. Osteopor Int 2021.
Genest F, Claußen L, Rak D, Seefried L. Bone mineral density and fracture risk in adult patients with hypophosphatasia. Osteopor Int. 2020
Shapiro JR, Lewiecki EM. Hypophosphatasia in adults: clinical assessment and treatment considerations. J Bone Mineral Res. 2017;32(10):1977–80.
Dahlhamer J, Lucas J, Zelaya C, Nahin R, Mackey S, DeBar L, et al. Prevalence of chronic pain and high-impact chronic pain among adults—United States, 2016. MMWR Morb Mortal Wkly Rep. 2018;67(36):1001–6.
Fayaz A, Croft P, Langford RM, Donaldson LJ, Jones GT. Prevalence of chronic pain in the UK: a systematic review and meta-analysis of population studies. BMJ Open. 2016;6(6): e010364.
Jakobsson U. The epidemiology of chronic pain in a general population: results of a survey in southern Sweden. Scand J Rheumatol. 2010;39(5):421–9.
Wong WS, Fielding R. Prevalence and characteristics of chronic pain in the general population of Hong Kong. J Pain. 2011;12(2):236–45.
Acknowledgements
The authors would like to thank T.A. Vacca for proofreading and the health care professionals of the rheumatology outpatient unit at the Hanusch Hospital Vienna.
Funding
The authors of this study did not receive any funding for this study.
Author information
Authors and Affiliations
Contributions
JF performed clinic evaluations, analyzed patients data regarding clinical and serological features, and contributed to writing the manuscript. MB performed statistical analysis, and helped drafting the manuscript. JH participated in the study design and coordination. KR performed molecular genetic analysis and participated in writing the manuscript. GU and MWB carried out molecular genetic studies. GS and BH helped drafting the manuscript. JZ participated in the study design and coordination. RK conceived and designed the study, interpreted patient’s data, and was a major contributor in writing the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by the Ethics Committee of the City of Vienna (EK 18-239-VK). Written informed consent was obtained from all individuals prior to any study related procedures.
Consent for publication
Not applicable.
Competing interests
There are no competing interests for any authors regarding this project.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Feurstein, J., Behanova, M., Haschka, J. et al. Identifying adult hypophosphatasia in the rheumatology unit. Orphanet J Rare Dis 17, 435 (2022). https://doi.org/10.1186/s13023-022-02572-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-022-02572-7