
Abstract
Background
Genetic investigations of inherited neuromuscular disorders in Africans, have been neglected. We aimed to summarise the published data and comment on the genetic evidence related to inherited neuropathies (Charcot-Marie-Tooth disease (CMT)), hereditary spastic paraplegias (HSP) and spinal muscular atrophy (SMA) in Africans.
Methods
PubMed was searched for relevant articles and manual checking of references and review publications were performed for African-ancestry participants with relevant phenotypes and identified genetic variants. For each case report we extracted phenotype information, inheritance pattern, variant segregation and variant frequency in population controls (including up to date frequencies from the gnomAD database).
Results
For HSP, 23 reports were found spanning the years 2000–2019 of which 19 related to North Africans, with high consanguinity, and six included sub-Saharan Africans. For CMT, 19 reports spanning years 2002–2021, of which 16 related to North Africans and 3 to sub-Saharan Africans. Most genetic variants had not been previously reported. There were 12 reports spanning years 1999–2020 related to SMN1-SMA caused by homozygous exon 7 ± 8 deletion. Interestingly, the population frequency of heterozygous SMN1-exon 7 deletion mutations appeared 2 × lower in Africans compared to Europeans, in addition to differences in the architecture of the SMN2 locus which may impact SMN1-SMA prognosis.
Conclusions
Overall, genetic data on inherited neuromuscular diseases in sub-Saharan Africa, are sparse. If African patients with rare neuromuscular diseases are to benefit from the expansion in genomics capabilities and therapeutic advancements, then it is critical to document the mutational spectrum of inherited neuromuscular disease in Africa.
Highlights
-
Review of genetic variants reported in hereditary spastic paraplegia in Africans
-
Review of genetic variants reported in genetic neuropathies in Africans
-
Review of genetic underpinnings of spinal muscular atrophies in Africans
-
Assessment of pathogenic evidence for candidate variants
Similar content being viewed by others

Introduction
Inherited neurological diseases in African populations have been largely neglected. Africans will be left behind in the global quest for targeted genetic therapies without an African perspective on disease-associated mutations. While modern genomic approaches have led to new gene discoveries in complex inherited neuromuscular disorders [1], the genetic landscape of neuromuscular disorders in Africans are barely known.
Inherited neuromuscular disorders, such as hereditary spastic paraplegia (HSP) and Charcot-Marie-Tooth (CMT) disease are not rare in North America, Europe, and Asia with a global prevalence ranging between 4.3/100,000 for HSP and 82.3/100,000 for CMT [1, 2]. There are no epidemiological data for Africa. Akinyemi et al. reported that of the 58 African states, scattered reports related to the genetics of neurological disorders emanated from only 17 countries and these were heavily concentrated in four North African countries [3]. Presently in South Africa, and with relevance to this review, the National Health Laboratory Service offers one genetic screening test for CMT (the common PMP22 gene duplication/deletion) and none for HSP. Although the screening test to detect the most common cause of Spinal Muscular Atrophy (SMA) (homozygous deletion/disruption of SMN1) has been available in South Africa for more than 2 decades, only isolated cases are able to access gene therapies for SMA which are available in resource-rich countries. Therefore, there is an urgent need to address the disparate healthcare in inherited neuromuscular diseases which exist between the developed world and Africa. However, there are presently a few initiatives such as the International Centre for Genomic Medicine in Neuromuscular Diseases (ucl.ac.uk/genomic-medicine-neuromuscular- diseases/) to prioritise the advancement of genetic research in neuromuscular diseases, and the broader H3Africa Initiative to expand population reference data in sub-Saharan Africans [4], which will facilitate the analysis of pathogenic genetic variants in Africans with rare inherited diseases. This review will synthesize genetic reports from HSP, CMT and SMA in Africans, to give an overview of the genetic variants and their associated phenotypes, which have been reported and can be used as a reference resource for African researchers and clinicians. A separate review of inherited myopathies and muscle dystrophies in Africans, is in progress.
Methodology
PubMed was searched for journal articles related to the molecular genetic causes of HSP, CMT, and SMA in Africa. The following MeSH terms were used (hereditary spastic paraplegia) or (Charcot-Marie-Tooth disease) or (genetic neuropathies) or (inherited neuropathies) or (familial amyloid neuropathies) AND (Africa), and (spinal muscular atrophy) AND (Africa) for searching PubMed. We performed a google search using search terms: “genetic neuropathies Africa”, “neuromuscular inherited Africa”, “hereditary spastic paraplegia Africa”, “Charcot-Marie-Tooth disease Africa”, “spinal muscular atrophy or SMA and Africa”, “Kennedy’s syndrome Africa”. We also manually searched the reference lists of reports and review publications to look for additional references and searched for “Africa” within articles. We confined this review to studies with genetic descriptive components. Studies involving linkage analysis of a large genomic region or single genes where a genetic diagnosis was not reached were excluded as the focus of this paper was on the identified genetic causes of inherited neuromuscular disorders in Africans (European or Indian ancestries were excluded) (Fig. 1). Reports related to infectious disease-associated neuropathies were excluded. Only English articles were reviewed which resulted in the exclusion of two reports from 2002 and 2008 which were published in French.

Flow chart describing the methodology used for the literature curation
The data collected from the reports included: the genetic results of probands with African-genetic ancestry, phenotypic features including age at onset, inheritance pattern and consanguinity, and electrophysiological features. We also noted genetic variants found in Africans but which had been previously reported in non-African families, whether there were attempts to determine segregation of the putative disease-causing variant within the family, and whether population controls were assessed for the variant. Segregation of genetic variation was scored positive if the putative disease-causing variant was (a) excluded in at least one unaffected individual of the same age or older than the affected individual for autosomal dominant inheritance, or (b) confirmed in the heterozygous state in at least one unaffected parent for autosomal recessive inheritance. Variants in which functional studies had been performed were noted. In addition, as many of these publications were published prior to the establishment of large scale public genetic databases, we also interrogated the gnomAD database (last accessed 6 Sept. 2021) to determine the frequency of putative disease-causing variants [5]. For variant nomenclature we followed the Human Genetic Variation Sequence (HGVS)(version 20.05) guidelines [6].
Results
Most reports used the following genetic methodologies: Targeted PCR sequencing and/or Sanger sequencing; multiplex ligation-dependent probe amplification; and HSP or CMT gene panels. Some studies used appropriate microsatellite markers to construct segregating haplotypes to establish linkage in families followed by targeted Sanger sequencing of coding exons. More recent reports (from 2013) used whole exome sequencing (WES) to screen protein coding variants or performed comprehensive whole genome sequence (WGS) analysis.
Hereditary spastic paraplegia
Hereditary spastic paraplegias (HSP) are clinically characterized by a progressive gait disturbance due to increasing spasticity of the legs. Clinicians have recognized two forms of HSP; patients who only have features of HSP (or pure HSP), or those with additional neurological system dysfunction such as ataxia, cognitive/intellectual disability, extrapyramidal signs, and features of sensory ± motor neuropathy. The latter are called complex HSP.
Although the clinical manifestations of HSP usually manifest over years rather than months, it remains important to exclude other non-degenerative conditions by performing imaging studies of the brain and spinal cord. Magnetic resonance imaging (MRI) of the brain may be normal or show atrophy, and/or may show thinning of the corpus callosum and/or increased white matter signal intensities (Table 1). In Africa, infectious causes such as HTLV1-associated tropical spastic paraparesis is a concern in adults, which can be excluded with cerebrospinal fluid examination and/or serology [7]. Lathyrism caused by excessive consumption of the chickpeas of the lathyrism family, is endemic in Ethiopia, and can result in a slowly progressive paraparesis [7].
More than 88 genes have thus far been reported to cause HSP, which are designated as SPastic Gait/Gene or SPG genes [2, 8]. Inheritance patterns in HSP are predominant autosomal dominant (AD), except in areas with high consanguinity, such as in North Africa, where autosomal recessive inheritance (AR) patterns are prevalent [2, 8, 9] (See Table 1). X-linked and mitochondrial maternal inheritance patterns of HSP are rare [8]. World-wide SPG4 is reported to account for up to 79% of HSP cases with AD inheritance, albeit mainly in those with Caucasian ancestry [8]. Other frequent causes of AD HSP include the monoallelic pathogenic variants in KIF1A, as well as SPG3A and SPG31 [8]. Genes accounting for HSP cases with AR inheritance patterns include SPG11 and SPG7, followed by SPG15 and SPG5 in overall frequencies [8]. Interestingly, three genes (KIF1C, SPG7, KIF1A) have been reported to associate with mixed inheritance patterns related to allele-dose-dependent clinical phenotypes i.e. milder phenotypes with heterozygous variants, and more severe phenotypes with homozygous states [8].
HSP in North Africa
Most of the genetic reports on HSP in Africa are from North Africa and are based on targeted linkage analysis in families to identify a candidate gene locus that segregated with the phenotype, followed by direct gene sequencing (Fig. 2). The commonest gene harbouring a pathogenic variant identified in HSP cases, was SPG11 (KIAA1840) associated with thin corpus callosum on MRI [9] (Table 1; Additional file 1: Table A). Only four of the reports used WES for a more comprehensive gene screen in North African cases with HSP conditions, and one used WGS data [10,11,12,13]. The commonly encountered AR-HSP causal genes (SPG11, SPG15) in North African populations were also amongst the top seven genes in a large European cohort [9, 10, 14,15,16,17,18,19]. Other AR-HSP genes amongst North African families included SPG5, SPG7, SPG35, SPG46, SPG48, SPG51, and SPG57, as well as mutations in the ALS2 and SACS genes [9, 13, 14, 20, 21] However, private mutations in novel genes (RNF170, CAPN1, KLC2, B4GALNT1, DDHD1, CCT5) were also reported to be disease-causing in isolated cases or families [10,11,12, 21,22,23,24,25].

Bubble map depicting the number of genetic reports in HSP- and CMT-related disorders in African countries
The most frequent gene variants accounting for autosomal dominant inheritance patterns, were found in SPG4 (SPAST) [26,27,28].
HSP in sub-Saharan Africa
Six reports were found from sub-Saharan Africa of which two screened a targeted panel of 58 HSP genes [29, 30] and two used WES [17, 31] (Table 1; Additional file 1: Table A; Fig. 2). The cases from consanguineous families from Kenya and Mali with homozygous pathogenic alleles were most frequent with SPG11 variants, followed by SPG7, SPG35 and SPG43 [32].
There were two reports on autosomal dominant HSP; one black South African family with a novel SPG3A variant [33] and a family from Mali with SPG10 [30]. Therefore, the common SPG genes present in Europeans [19], viz. SPG3, SPG4 and SPG10, have been found in isolated African cases.
Genetic neuropathies
The largest group of genetic neuropathies are referred to as the Hereditary Sensory Motor Neuropathies or Charcot Marie Tooth (CMT) disease. CMT affects predominantly the motor and sensory nerves, although the CMT-spectrum includes rare forms with autonomic and motor only involvement [34]. The clinical features of CMT disease are progressive and symmetrical weakness and wasting of distal muscles of the foot and ankle which may result in clumsy feet, foot deformities such as pes cavus, and loss of deep tendon jerks. Later, there may be involvement of the distal arms with wasting and weakness although clawing of the hands is less common. Some genetic neuropathies may have early and predominant upper limb involvement. Sensory involvement ranges from mild distal numbness to severe loss of sensation with ulcers, and/or sensory ataxia. The insidious clinical progression of CMT distinguishes it from subacute acquired inflammatory neuropathies in most cases, although rare forms of CMT can give a patchy electrophysiological picture with conduction blocks that may resemble treatment-resistant chronic inflammatory demyelinating polyradiculoneuropathy [34]. In Southern African populations, where HIV-infection is prevalent, small fibre painful neuropathies may be considered in cases with more advanced HIV-infection, and/or with concomitant tuberculosis and isoniazid therapies, but weakness is extremely rare [35]. This contrasts with CMT where the absence of motor involvement is unlikely [34].
In the pre-molecular era, CMT was categorized by the electrophysiological involvement of the sensory and motor nerves, whereas the CMT neuropathies are further categorized according to their electrophysiological findings into three types; the demyelinating forms (nerve conduction velocities (NCVs) < 38 m/s in the upper limbs), axonal forms (NCV > 45 m/s), or the intermediate types of CMT (NCV in the upper limbs between 25 and 45 m/s) [1]. All neuropathies categorized as HSMN or CMT, would show evidence of motor and sensory nerve abnormalities on electrophysiological testing, whereas hereditary motor neuropathy (HMN) by definition would have normal sensory nerve action potential responses. However, there appears to be genetic overlap between CMT2 and HMN subtypes [36].
In North America and European populations, most CMT neuropathies show AD inheritance compared to AR inheritance which comprises < 10% of cases. In contrast, in North Africa, where consanguinity is high [37], most of the cases published showed AR inheritance (Table 2). Similar to what is observed in HSP, CMT shows substantial genetic heterogeneity with > 100 genes identified which can cause genetic neuropathies [1]. The most common autosomal dominantly inherited CMT in North America and Europe, the demyelinating CMT1A caused by a duplication in the PMP22 gene, accounts for ~ 40% of genetic neuropathies [38], yet remains unreported in those with African genetic ancestry.
CMT in North Africa
Due to high levels of consanguinity in Algeria, Morocco, and Tunisia, AR-CMTB1 (LMNA) was by far the commonest, followed by CMT4A (GDAP1), CMT4C (SH3TC2), and CMT4B2 (MTMR13) [24, 39,40,41,42,43,44,45,46,47,48,49,50](Table 2). These are present in < 1% of AR-CMT cases in non-Africans [38]. Two Algerian families had compound heterozygous pathogenic variants with the common GDAP1 S194* variant [51], which has a population frequency of 2.3 × 10–5 (Additional file 1: Table B). Isolated cases were reported with CMT4B1 and CMT4F [37].
Four Algerian families with distal HMN (dHMN5A) and AD inheritance patterns were reported with the rare [38] GARS pathogenic variants characterised by predominant upper limb weakness and hand wasting [52, 53].
CMT in Sub-Saharan Africa
Three reports were found (Fig. 2). One CMT1B (MPZ) Nigerian AD pedigree with late-onset demyelinating neuropathy [54]; and an intermediate CMT phenotype with conduction blocks and a novel PLEKHG5 variant which segregated in the family [55]. A consanguineous pedigree from Mali was reported with a heterozygous GARS variant, but without evidence of segregation or population screening [56]. Caution must be used in interpreting variants with “incomplete penetrance” to explain incomplete segregation of variants particularly in Africans where the population data are sparse and genetic variation is increased [57].
Familial amyloid neuropathies
There are three types of familial amyloid neuropathies (FAP) which are categorised according to the abnormal precursor protein which will result in downstream deposition of amyloid fibrils viz. transthyretin (TTR), apolipoprotein A-1 and gelsolin [58]. Although some TTR mutations can cause FAP, which characteristically manifests with sensory and autonomic nerve dysfunction alone, a rare manifestation is oculoleptomeningeal amyloidosis (OLMA) which may present with additional features such as subarachnoid haemorrhage, epilepsy, hearing and visual loss, and headaches [59]. OLMA was described in a Nigerian adult heterozygous for TTR L21P, a variant which was previously reported in several European-ancestry cases [59]. Another common variant, at least among African-Americans (and found amongst West Africans), is the TTR V122I variant which was detected in the heterozygous state in 4% of African-Americans [60] and is associated with hypertrophic restrictive cardiomyopathy in older individuals, but without neuropathy. A man from Benin was reported with cognitive changes, a sensori-motor neuropathy with autonomic involvement and sensory ataxia, as well as hypertrophic cardiomyopathy, and a TTR I107V variant, which has been found in several Europeans with inherited amyloidosis [61].
Spinal muscular atrophies
Classical Spinal Muscular Atrophy (SMA) due to the homozygous loss of exon 7 (± exon 8) of SMN1 results in a critical loss of protein production and progressive degeneration of the lower motor neurons of the spinal cord [62]. We will refer to this as SMN1-SMA. Clinically, SMN1-SMA is characterized by proximal muscle atrophy and weakness, and eventually distal paresis as well. The clinical subtypes of SMN1-SMA (types I–IV) were categorized based on the disease severity and age at onset, which also informed the prognosis and survival; Type I is most severe and manifests in early infancy, SMA II manifests in late infancy to early childhood (< 18 months), SMA III in childhood (> 18 months)[62] and SMA IV has adult-onset [63].
There is increasing recognition of SMN1-negative SMA, although this groups accounts for < 5% of SMA and is often associated with overlapping central nervous system/brainstem signs, and even cardiomyopathy [63]. However, in reports from Africa there are between 25 and 65% of the clinical cohorts categorised as either congenital hypotonia or SMA phenotypes, which can be categorized as SMN1-negative SMA (absence of homozygous exon 7 deletion). In addition, there are several types of distal SMA (DSMA) which overlap with classifications of distal HMN/dHMN [63] (see Table 2).
The SMN2 gene is a highly homologous centromeric copy of SMN1 in which a C > T variant in exon 7 splicing enhancer distinguishes SMN2 from SMN1 [64]. Although genetic variation in SMN2 does not cause disease, SMN2 copy numbers may modify disease severity and age at onset [65].
SMN1-SMA in North Africa
SMN1-SMA in North African populations have been reported in families with and without high consanguinity rates [66,67,68,69,70,71,72,73,74,75] (Additional file 2: Table C). Similar to European cohorts, 57/60 (95%) Tunisian cases with presumed SMN1-SMA showed homozygous deletion of SMN1 exon 7 [70], although the other samples showed lower proportions of SMN1-SMA particularly in older individuals [69].
SMN1-SMA in sub-Saharan Africa
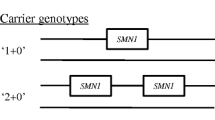
Five reports on SMA in sub-Saharan Africans were found, mostly involving South Africans and one each from Congo and Mali (Additional file 2: Table C) [73,74,75]. Several cases from two regions in South Africa reported SMN1-SMA with homozygous loss of exon 7 (± exons 8) ranging between 35 and 100% of their clinical samples, indicating a substantial number of cases with an alternative molecular diagnosis [76,77,78]. An SMN1 gene dosage assay in 300 random black SA samples showed the heterozygote exon 7 deletion in 6 individuals (1/50 population controls; 2%) which was similar to the frequency of SMN1 copy numbers in Kenyans and Nigerians [74], but roughly half of the heterozygote frequency found in European ancestry controls (3–4%)[77]. In comparison, the heterozygote frequency amongst 628 Malians was found to be 0.5% [74].
Humans have variable copies of an SMN2 gene, between 0 and 8 copies, and transcripts of this gene can modify the expression of SMN1-SMA [63]. Interestingly, the architecture of the SMN region differs substantially between Europeans and Africans, although African-Americans roughly followed the same trends in terms of SMN2 copy numbers as Europeans and Asians [79]. Amongst 75 black South African SMN1-SMA patients, 11% had > 2 SMN2 copies compared with 37% (of 30) SMN1-SMA patients with European ancestry [78]. Taken together, these results underscore the fact that the genetic architecture and disease pathogenic mechanisms in African ancestry individuals may vary from Europeans, and requires further study.
Complex inherited conditions with neuromuscular features
Although there are numerous complex multi-system conditions in which the presence of neuropathy may be present but not prominent [34], we mention two reports in Africans in which the recognition and initiation of appropriate treatment underscores their importance. Two families/probands with Allgrove or Triple A syndrome was described from North Africa/Algeria with the homozygous pathogenic variant in the AAAS gene (IVS14 + 1G > A); 1 family was consanguineous [80] and in the other both parents were heterozygous for the variant [81]. The main features were ACTH-resistant adrenal deficiency, achalasia and dry eyes, as well as features of distal motor neuropathy with/without spasticity, with the clinical onset during childhood. The importance is to recognise the treatable metabolic disturbances. A case of acute intermittent porphyria in a black South African man due to the HMBS R149* variant, was reported to mimic severe subacute motor neuropathy [82].
Conclusion
Although the high rate of consanguinity and occurrence of large families from North Africa have resulted in several molecularly confirmed cases of HSP and CMT, the genetic studies related to identifying the pathogenic variants in these conditions in sub-Saharan Africans, are sparse (Fig. 2). The high proportion of SMN1-negative SMA cases in particularly sub-Saharan Africa, identifies another group of patients with an as yet molecularly undiagnosed condition. Although the low rates of genetic reports in these complex disorders are likely due to the lack of resources and limited access to genetic screening, the clinical and genetic characteristics of these disorders need to be described and identified so that the burden of genetic variants and disorders are curated as the first steps to address accessibility to potential therapeutic trials. Collaborations among African researchers are slowly gaining momentum and will strengthen future funding applications to extend specialist clinical training of clinicians and genetic councellors, as well as increasing the number of cases and genomics capabilities in Africa. Increasing neurogenomics capacity and the development of appropriate genetic screening panels for Africans with inherited neuromuscular diseases, would help improve diagnostic capabilities.
Availability of data and materials
Not applicable.
References
Reilly MM, Rossor AM. Humans: the ultimate animal models. J Neurol Neurosurg Psychiatry. 2020;91:1132–6. https://doi.org/10.1136/jnnp-2020-323016.
Boutry M, Morais S, Stevanin G. Update on the genetics of spastic paraplegias. Curr Neurol Neurosci Rep. 2019;19:18. https://doi.org/10.1007/S11910-019-0930-2.
Akinyemi RO, Owolabi MO, Oyeniyi T, Ovbiagele B, Arnett DK, Tiwari HK, et al. Neurogenomics in Africa: perspectives, progress, possibilities and priorities. J Neurol Sci. 2016;366:213–23. https://doi.org/10.1016/j.jns.2016.05.006.
Matovu E, Bucheton B, Chisi J, Enyaru J, Hertz-Fowler C, Koffi M, et al. Enabling the genomic revolution in Africa. Science. 2014;344:1346–8. https://doi.org/10.1126/science.1251546.
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint quantified from variation in 141,456 humans. Nature. 2020;581:434–44. https://doi.org/10.1038/s41585-020-2308-7.
Den Dunnen JT, Dalgleish R, Maglott DR, Hart RK, Greenblatt MS, McGowan-Jordan J, et al. HGVS recommendations for the description of sequence variants: 2016 update. Hum Mutat 2016;37:564–69. doi:https://doi.org/10.1002/humu.22981
Heckmann JM, Bhigjee A. Tropcial Neurology. In: Farrar J, Hotez P, Junhanss, Kang G, Lalloo D, White N, editors. Manson’s Tropical Infectious Diseases, 23ed. Saunders Ltd 2013, p. 1047–1060.
Elsayed LEO, Eitazi IZ, Ahmed AE, Stevanin G. Insights into clinical, genetic, and pathological aspects of hereditary spastic paraplegias: a comprehensive overview. Front Mol Biosci. 2021;8: 680899. https://doi.org/10.3389/fmolb.2021.698099.
Elsayed LEO, Mohammed IN, Hamed AAA, Elseed MA, Johnson A, Mairey M, et al. Hereditary spastic paraplegias: identification of a novel SPG57 variant affecting TFG oligomerization and description of HSP subtypes in Sudan. Eur J Hum Genet. 2016;25:100–10. https://doi.org/10.1038/ejhg.2016.108.
Gan-Or Z, Bouslam N, Birouk N, Lissouba A, Chambers DB, Veriepe J, et al. Mutations in CAPN1 cause autosomal-recessive hereditary spastic paraplegia. Am J Hum Genet. 2016;98:1038–46. https://doi.org/10.1016/j.ajhg.2016.04.002.
Melo US, Macedo-Souza LI, Figueiredo T, Muotri AR, Gleeson JG, Coux G, et al. Overexpression of KLC2 due to a homozygous deletion in the non-coding region causes SPOAN syndrome. Hum Mol Genet. 2015;24:6877–85. https://doi.org/10.1093/hmg/ddv388.
Wagner M, Osborn DPS, Gehweiler I, Nagel M, Ulmer U, Bakhtiari S, et al. Bi-allelic variants in RNF170 are associated with hereditary spastic paraplegia. Nat Commun. 2019;10:4790. https://doi.org/10.1038/s41467-019-12620-9.
Kong XF, Bousfiha A, Rouissi A, Itan Y, Abhyankar A, Bryant V, et al. A novel homozygous p.R1105X mutation of the AP4E1 gene in twins with hereditary spastic paraplegia and mycobacterial disease. PLoS One 2013;8:e58286. doi: https://doi.org/10.1371/journal.pone.0058286 .
Elleuch N, Depienne C, Benomar A, Hernandez AM, Ferrer X, Fontaine B, et al. Mutation analysis of the paraplegin gene (SPG7) in patients with hereditary spastic paraplegia. Neurology. 2006;66:654–9. https://doi.org/10.1212/01.wnl.0000201185.91110.15.
Stevanin G, Azzedine H, Denora P, Boukhris A, Tazir M, Lossos A, et al. Mutations in SPG11 are frequent in autosomal recessive spastic paraplegia with thin corpus callosum, cognitive decline and lower motor neuron degeneration. Brain. 2008;131:772–84. https://doi.org/10.1093/brain/awm293.
Abdel Aleem A, Abu-Shahba N, Swistun D, Silhavy J, Bielas SL, Sattar S, et al. Expanding the clinical spectrum of SPG11 gene mutations in recessive hereditary spastic paraplegia with thin corpus callosum. Eur J Med Genet. 2011;54:82–5. https://doi.org/10.1016/j.ejmg.2010.10.006.
Kara E, Tucci A, Manzoni C, Lynch DS, Elpidorou M, Bettencourt C, et al. Genetic and phenotypic characterization of complex hereditary spastic paraplegia. Brain. 2016;139:1904–18. https://doi.org/10.1093/brain/aww111.
Hanein S, Martin E, Boukhris A, Byrne P, Goizet C, Hamri A, et al. Identification of the SPG15 gene, encoding spastizin, as a frequent cause of complicated autosomal-recessive spastic paraplegia, including Kjellin syndrome. Am J Hum Genet. 2008;82:992–1002. https://doi.org/10.1016/j.ajhg.2008.03.004.
Schule R, Wiethoff S, Martus P, Karle KN, Otto S, Klebe S, et al. Hereditary spastic paraplegia: clinicogenetic lessons from 608 patients. Ann Neurol. 2016;79:646–58. https://doi.org/10.1002/ana.24611.
Pensato V, Castellotti B, Gellera C, Pareyson D, Ciano C, Nanetti L, et al. Overlapping phenotypes in complex spastic paraplegias SPG11, SPG15, SPG35 and SPG48. Brain. 2014;137:1907–20. https://doi.org/10.1093/brain/awu121.
Martin E, Schule R, Smets K, Rastetter A, Boukhris A, Loureiro JL, et al. Loss of function of glucocerebrosidase GBA2 is responsible for motor neuron defects in hereditary spastic paraplegia. Am J Hum Genet. 2013;92:238–44. https://doi.org/10.1016/j.ajhg.2012.11.021.
Boukhris A, Schule R, Loureiro JL, Lourenco CM, Mundwiller E, Gonzalez MA, et al. Alteration of ganglioside biosynthesis responsible for complex hereditary spastic paraplegia. Am J Hum Genet. 2013;93:118–23. https://doi.org/10.1016/j.ajhg.2013.05.006.
Tesson C, Nawara M, Salih MA, Rossignol R, Zaki MS, Al Balwi M, et al. Alteration of fatty-acid-metabolizing enzymes affects mitochondrial form and function in hereditary spastic paraplegia. Am J Hum Genet. 2012;91:1051–64. https://doi.org/10.1016/j.ajhg.2012.11.001.
Bouhouche A, Birouk N, Azzedine H, Benomar A, Durosier G, Ente D, et al. Autosomal recessive axonal Charcot-Marie-Tooth disease (ARCMT2): phenotype-genotype correlations in 13 Moroccan families. Brain. 2007;130:1062–75. https://doi.org/10.1093/brain/awm014.
Bouhouche A, Benomar A, Bouslam N, Ouazzani R, Chkili T, Yahyaoui M. Autosomal recessive mutilating sensory neuropathy with spastic paraplegia maps to chromosome 5p15.31–14.1. Eur J Hum Genet 2006;14:249–52. doi: https://doi.org/10.1038/sj.ejhg.5201537.
Boukhris A, Stevanin G, Feki I, Denora P, Elleuch N, Miladi MI, et al. Tunisian hereditary spastic paraplegias: clinical variability supported by genetic heterogeneity. Clin Genet. 2009;75:527–36. https://doi.org/10.1111/j.1399-0004.2009.01176.x.
Ribai P, Depienne C, Fedirko E, Jothy AC, Viveweger C, Hahn-Barma V, et al. Mental deficiency in three families with SPG4 spastic paraplegia. Eur J Hum Genet. 2008;16:97–104. https://doi.org/10.1038/sj.ejhg.5201922.
Hentati A, Deng HX, Zhai H, Chen W, Yang Y, Hung WY, et al. Novel mutations in spastin gene and absence of correlation with age at onset of symptoms. Neurology. 2000;55:1388–90. https://doi.org/10.1212/wnl.55.9.1388.
Landoure G, Dembele K, Cisse L, Samassekou O, Diarra S, Bocoum A, et al. Hereditary spastic paraplegia type 35 in a family from Mali. Am J Med Genet A. 2019;179:1122–5. https://doi.org/10.1002/ajmg.a.61179.
Guinto CO, Diarra S, Diallo S, Cisse L, Coulibaly T, Diallo SH, et al. A novel mutation in KIF5A in a Malian family with spastic paraplegia and sensory loss. Ann Clin Transl Neurol. 2017;4:272–5. https://doi.org/10.1002/acn3.402.
Landoure G, Zhu PP, Lourenco CM, Johnson JO, Toro C, Bricceno KV, et al. Hereditary spastic paraplegia type 43 (SPG43) is caused by mutation in C19orf12. Hum Mutat. 2013;34:1357–60. https://doi.org/10.1002/humu.22378.
de Bot ST, Burggraaff RC, Herkert JC, Schelhaas HJ, Post B, Diekstra A, et al. Rapidly deteriorating course in Dutch hereditary spastic paraplegia type 11 patients. Eur J Hum Genet. 2013;21:1312–5. https://doi.org/10.1038/ejhg.2013.27.
Orlacchio A, Montieri P, Babalini C, Gaudiello F, Bernardi G, Kawarai T. Late-onset hereditary spastic paraplegia with thin corpus callosum caused by a new SPG3A mutation. J Neurol. 2011;258:1361–3. https://doi.org/10.1007/s00415-011-5934-z.
Rossor AM, Evans MR, Reilly MM. A practical approach to the genetic neuropathies. Pract Neurol. 2015;15:187–98. https://doi.org/10.1136/practneurol-2015-001095.
Centner CM, Bateman KJ, Heckmann JM. Manifestations of HIV infection in the peripheral nervous system. Lancet Neurol. 2013;12:295–309. https://doi.org/10.1016/S1474-4422(13)70002-4.
Previtali SC, Zhao E, Lazarevic D, Pipitone GB, Fabrizi GM, Manganelli F, et al. Expanding the spectrum of genes responsible for hereditary motor neuropathies. J Neurol Neurosurg Psychiatry. 2019;90:1171–9. https://doi.org/10.1136/jnnp-2019-320717.
Nouioua S, Hamadouche T, Funalot B, Bernard R, Bellatache N, Bouderba R, et al. Novel mutations in the PRX and the MTMR2 genes are responsible for unusual Charcot-Marie-Tooth disease phenotypes. Neuromuscul Disord. 2011;21:543–50. https://doi.org/10.1016/j.nmd.2011.04.013.
Finsterer J, Loscher WN, Wanschitz J, Iglseder S. Orphan peripheral neuropathies. J Neuromuscul Disord. 2021;8:1–23. https://doi.org/10.3233/JND-200518.
Tazir M, Azzedine H, Assami S, Sindou P, Nouioua S, Zemmouri R, et al. Phenotypic variability in autosomal recessive axonal Charcot-Marie-Tooth disease due to the R298C mutation in lamin A/C. Brain. 2004;127:154–63. https://doi.org/10.1093/brain/awh021.
De Sandre-Giovannoli A, Chaouch M, Kozlov S, Vallat JM, Tazir M, Kassouri N, et al. Homozygous defects in LMNA, encoding lamin A/C nuclear-envelope proteins, cause autosomal recessive axonal neuropathy in human (Charcot-Marie-Tooth disorder type 2) and mouse. Am J Hum Genet. 2002;70:726–36. https://doi.org/10.1086/339274.
Hamadouche T, Poitelon Y, Genin E, Chaouch M, Tazir M, Kassouri N, et al. Founder effect and estimation of the age of the c.892C>T (p.Arg298Cys) mutation in LMNA associated to Charcot-Marie-Tooth subtype CMT2B1 in families from North Western Africa. Ann Hum Genet 2008;72:590–7. doi: https://doi.org/10.1111/j.1469-1809.2008.00456.x.
Baxter RV, Ben Othmane K, Rochelle JM, Stajich JE, Hulette C, Dew-Knight S, et al. Ganglioside-induced differentiation-associated protein-1 is mutant in Charcot-Marie-Tooth disease type 4A/8q21. Nat Genet. 2002;30:21–2. https://doi.org/10.1038/ng796.
Boubaker C, Hsairi-Guidara I, Castro C, Ayadi I, Boyer A, Kerkeni E, et al. A novel mutation in FGD4/FRABIN causes Charcot Marie Tooth disease type 4H in patients from a consanguineous Tunisian family. Ann Hum Genet. 2013;77:336–43. https://doi.org/10.1111/ahg.12017.
Delague V, Jacquier A, Hamadouche T, Poitelon Y, Baudot C, Boccaccio I, et al. Mutations in FGD4 encoding the Rho GDP/GTP exchange factor FRABIN cause autosomal recessive charcot-marie-tooth type 4H. Am J Hum Genet. 2007;81:1–16. https://doi.org/10.1086/58428==].
De Sandre-Giovannoli A, Delague V, Hamadouche T, Chaouch M, Krahn M, Boccaccio I, et al. Homozygosity mapping of autosomal recessive demyelinating Charcot-Marie-Tooth neuropathy (CMT4H) to a novel locus on chromosome 12p11.21-q13.11. J Med Genet 2005;42:260–5. doi: https://doi.org/10.1136/jmg.2004.024364.
Baudot C, Esteve C, Castro C, Poitelon Y, Mas C, Hamadouche T, et al. Two novel missense mutations in FGD4/FRABIN cause Charcot-Marie-Tooth type 4H (CMT4H). J Peripher Nerv Syst. 2012;17:141–6. https://doi.org/10.1111/j.1529-8027.2012.00405.x.
Azzedine H, Bolino A, Taieb T, Birouk N, Di Duca M, Bouhouche A, et al. Mutations in MTMR13, a new pseudophosphatase homologue of MTMR2 and Sbf1, in two families with an autosomal recessive demyelinating form of Charcot-Marie-Tooth disease associated with early-onset glaucoma. Am J Hum Genet. 2003;72:1141–53. https://doi.org/10.1086/375034.
Birouk N, Azzedine H, Dubourg O, Muriel MP, Benomar A, Hamadouche T, et al. Phenotypical features of a Moroccan family with autosomal recessive Charcot-Marie-Tooth disease associated with the S194X mutation in the GDAP1 gene. Arch Neurol. 2003;60:598–604. https://doi.org/10.1001/archneur.60.4.598.
Azzedine H, Ruberg M, Ente D, Gilardeau C, Perie S, Wechsler B, et al. Variability of disease progression in a family with autosomal recessive CMT associated with a S194X and new R310Q mutation in the GDAP1 gene. Neuromuscul Disord. 2003;13:341–6. https://doi.org/10.1016/S0960-8966(02)00281-X.
Azzedine H, Ravise N, Verny C, Gabreels-Festen A, Lammens M, Grid D, et al. Spine deformities in Charcot-Marie-Tooth 4C caused by SH3TC2 gene mutations. Neurology. 2006;67:602–6. https://doi.org/10.1212/01.wnl.0000230225.19797.93.
Bouhouche A, Birouk N, Benomar A, Ouazzani R, Chkili T, Yahyaoui M. A novel GDAP1 mutation P78L responsible for CMT4A disease in three Moroccan families. Can J Neurol Sci. 2007;34:421–6. https://doi.org/10.1017/S0317167100007290.
Dubourg O, Azzedine H, Yaou RB, Pouget J, Barois A, Meininger V, et al. The G526R glycyl-tRNA synthetase gene mutation in distal hereditary motor neuropathy type V. Neurology. 2006;66:1721–6. https://doi.org/10.1212/01.wnl.0000218304.02715.04.
Antonellis A, Ellsworth RE, Sambuughin N, Puls I, Abel A, Lee-Lin SQ, et al. Glycyl tRNA synthetase mutations in Charcot-Marie-Tooth disease type 2D and distal spinal muscular atrophy type V. Am J Hum Genet. 2003;72:1293–9. https://doi.org/10.1086/375039.
Kakar R, Ma W, Dutra A, Seltzer WK, Grewal RP. Clinical and genetic analysis of CMT1B in a Nigerian family. Muscle Nerve. 2003;27:628–30. https://doi.org/10.1002/mus.10344.
Villar-Quiles RN, Le VT, Leonard-Louis S, Trang NT, Huong NT, Laddada L, et al. Leukoencephalopathy and conduction blocks in PLEKHG5-associated intermediate CMT disease. Neuromuscul Disord. 2021;31:756–64. https://doi.org/10.1016/j.nmd.2021.06.004.
Yalcouye A, Diallo SH, Coulibaly T, Cisse L, Diallo S, Samassekou O, et al. A novel mutation in the GARS gene in a Malian family with Charcot-Marie-Tooth disease. Mol Genet Genomic Med. 2019;7: e00782. https://doi.org/10.1002/mgg3.782.
Nel M, Mahungu AC, Monnakgotla N, Botha GR, Mulder NJ, Wu G, et al. Revealing the mutational spectrum in Southern Africans with amyotrophic lateral sclerosis. Neurology Genetics. 2022;8:1–11. https://doi.org/10.1212/NXG.0000000000000654.
Plante-Bordeneuve V, Said G. Familial amyloid polyneuropathy. Lancet Neurol. 2011;10:1086–97. https://doi.org/10.1016/S1474-4422(11)70246-0.
McColgan P, Viegas S, Gandhi S, Bull K, Tudor R, Sheikh F, et al. Oculoleptomeningeal Amyloidosis associated with transthyretin Leu12Pro in an African patient. J Neurol. 2015;262:228–34. https://doi.org/10.1007/s00415-014-7594-2.
Jacobson DR, Pastore RD, Yaghoubian R, Kane I, Gallo G, Buck FS, et al. Variant-sequence transthyretin (isoleucine 122) in late-onset cardiac amyloidosis in black Americans. N Engl J Med. 1997;336:466–73. https://doi.org/10.1056/NEJM199702133360703.
Cassereau J, Lavigne C, Letournel F, Ghali A, Verny C, Dubas F, et al. Hereditary amyloid neuropathy by transthyretin Val107 mutation in a patient of African origin. J Peripher Nerv Syst. 2008;13:251–4. https://doi.org/10.1111/j.1529-8027.2008.00185.x.
Monani UR, Lorson CL, Parsons DW, Prior TW, Androphy EJ, Burghes AH, et al. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum Mol Genet. 1999;8:1177–83. https://doi.org/10.1093/hmg/8.7.1177.
Farrar MA, Kiernan MC. The genetics of spinal muscular atrophy: progress and challenges. Neurotherapeutics. 2015;12:290–302. https://doi.org/10.1007/s13311-014-0314-x.
Lorson CL, Hahnen E, Androphy EJ, Wirth B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci U S A. 1999;96:6307–11.
Wirth B, Herz M, Wetter A, Moskau S, Hahnen E, Rudnik-Schoneborn S, et al. Quantitative analysis of survival motor neuron copies: identification of subtle SMN1 mutations in patients with spinal muscular atrophy, genotype-phenotype correlation, and implications for genetic counseling. Am J Hum Genet. 1999;64:1340–56.
Sbiti A, Ratbi I, Kriouile Y, Sefiani A. Spinal muscular atrophy: frequent cause of congenital hypotonia in Morocco. Arch Pediatr. 2011;18:1261–4. https://doi.org/10.1016/j.arcped.2011.09.025.
They-They TP, Nadifi S, Dehbi H, Bellayou H, Brik H, Slassi I, et al. Phenotype-genotype correspondence in spinal muscular atrophy in a Moroccan family. Arch Pediatr. 2008;15:1201–5. https://doi.org/10.1016/j.arcped.2008.04.015.
Essawi ML, Effat LK, Shanab GM, Al-Ettribi GM, El-Haronui AA, Karim AM. Molecular analysis of SMN1 and NAIP genes in Egyptian patients with spinal muscular atrophy. Bratisl Lek Listy. 2007;108:133–7.
Bouhouche A, Benomar A, Birouk N, Bouslam N, Ouazzani R, Yahyaoui M, et al. High incidence of SMN1 gene deletion in Moroccan adult-onset spinal muscular atrophy patients. J Neurol. 2003;250:1209–13. https://doi.org/10.1007/s00415-003-0186-1.
Mrad R, Dorboz I, Ben Jemaa L, Maazoul F, Trabelsi M, Chaabouni M, et al. Molecular analysis of the SMN1 and NAIP genes in 60 Tunisian spinal muscular atrophy patients. Tunis Med. 2006;84:465–9.
Sifi Y, Sifi K, Boulefkhad A, Abadi N, Bouderda Z, Cheriet R, et al. Clinical and genetic study of Algerian patients with spinal muscular atrophy. J Neurodegener Dis. 2013;2013: 903875. https://doi.org/10.1155/2013/903875.
Shawky RM, Abd el-Aleem K, Rifaat MM, Moustafa A. Molecular diagnosis of spinal muscular atrophy in Egyptians. East Mediterr Health J 2001;7:229–37.
Lumaka A, Bone D, Lukoo R, Mujinga N, Senga I, Tady B, et al. Werdnig-Hoffmann disease: report of the first case clinically identified and genetically confirmed in central Africa (Kinshasa-Congo). Genet Couns. 2009;20:349–58.
Sangare M, Hendrickson B, Sango HA, Chen K, Nofziger J, Amara A, et al. Genetics of low spinal muscular atrophy carrier frequency in sub-Saharan Africa. Ann Neurol. 2014;75:525–32. https://doi.org/10.1002/ana.24114.
Stevens G, Yawitch T, Rodda J, Verhaart S, Krause A. Different molecular basis for spinal muscular atrophy in South African black patients. Am J Med Genet. 1999;86:420–6. https://doi.org/10.1016/j.nmd.2007.05.005.
Wilmshurst JM, Reynolds L, Van Toorn R, Leisegang F, Henderson HE. Spinal muscular atrophy in black South Africans: concordance with the universal SMN1 genotype. Clin Genet. 2002;62:165–8. https://doi.org/10.1034/j.1399-0004.2002.620210.x.
Labrum R, Rodda J, Krause A. The molecular basis of spinal muscular atrophy (SMA) in South African black patients. Neuromuscul Disord. 2007;17:684–92. https://doi.org/10.1016/j.nmd.2007.05.005.
Vorster E, Essop FB, Rodda JL, Krause A. Spinal muscular atrophy in the black South African population: a matter of rearrangement? Front Genet. 2020;11:54. https://doi.org/10.3389/fgene.2020.00054.
Moisse M, Zwamborn RAJ, van Vugt J, van der Spek R, van Rheenen W, Kenna B, et al. The effect of SMN gene dosage on ALS risk and disease severity. Ann Neurol. 2021;89:686–97. https://doi.org/10.1002/ana.26009.
Tullio-Pelet A, Salomon R, Hadj-Rabia S, Mugnier C, de Laet MH, Chaouachi B, et al. Mutant WD-repeat protein in triple-A syndrome. Nat Genet. 2000;26:332–5. https://doi.org/10.1038/81642.
Barat P, Goizet C, Tullio-Pelet A, Puel O, Labessan C, Barthelemy A. Phenotypic heterogeneity in AAAS gene mutation. Acta Paediatr. 2004;93:1257–9. https://doi.org/10.1080/08035250410027706.
Albertyn CH, Sonderup M, Bryer A, Corrigall A, Meissner P, Heckmann JM. Acute intermittent porphyria presenting as progressive muscular atrophy in a young black man. S Afr Med J. 2014;104:283–5. https://doi.org/10.7196/samj.7785.
Acknowledgements
This work was supported by the National Research Foundation of South Africa (113416) (ACM, JMH) and University of Cape Town Faculty of Health Sciences fellowships (NM, ACM) and made possible (in part) by a Carnegie Developing Emerging Academic Leaders (DEAL) award from the Carnegie Corporation of New York (MN).
Funding
None.
Author information
Authors and Affiliations
Contributions
ACM performed the literature search and wrote the first draft, NM assisted with data extraction and collation, JMH supervised data extraction and edited the draft, MN provided editorial assistance. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
. Table A: Frequencies of genetic variants in African populations with HSP reviewed in gnomAD database. Table B: Frequencies of genetic variants in African populations with CMT reviewed in gnomAD database
Additional file 2
. Table C: Reports of autosomal recessive SMN1-SMA identified in African populations
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Mahungu, A.C., Monnakgotla, N., Nel, M. et al. A review of the genetic spectrum of hereditary spastic paraplegias, inherited neuropathies and spinal muscular atrophies in Africans. Orphanet J Rare Dis 17, 133 (2022). https://doi.org/10.1186/s13023-022-02280-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-022-02280-2