
Abstract
Background
Phenylalanine hydroxylase (PAH) deficiency is an autosomal recessive disorder that results in elevated concentrations of phenylalanine (Phe) in the blood. If left untreated, the accumulation of Phe can result in profound neurocognitive disability. The objective of this systematic literature review and meta-analysis was to estimate the global birth prevalence of PAH deficiency from newborn screening studies and to estimate regional differences, overall and for various clinically relevant Phe cutoff values used in confirmatory testing.
Methods
The protocol for this literature review was registered with PROSPERO (International prospective register of systematic reviews). Pubmed and Embase database searches were used to identify studies that reported the birth prevalence of PAH deficiency. Only studies including numeric birth prevalence reports of confirmed PAH deficiency were included.
Results
From the 85 publications included in the review, 238 birth prevalence estimates were extracted. After excluding prevalence estimates that did not meet quality assessment criteria or because of temporal and regional overlap, estimates from 45 publications were included in the meta-analysis. The global birth prevalence of PAH deficiency, estimated by weighting regional birth prevalences relative to their share of the population of all regions included in the study, was 0.64 (95% confidence interval [CI] 0.53–0.75) per 10,000 births and ranged from 0.03 (95% CI 0.02–0.05) per 10,000 births in Southeast Asia to 1.18 (95% CI 0.64–1.87) per 10,000 births in the Middle East/North Africa. Regionally weighted global birth prevalences per 10,000 births by confirmatory test Phe cutoff values were 0.96 (95% CI 0.50–1.42) for the Phe cutoff value of 360 ± 100 µmol/L; 0.50 (95% CI 0.37–0.64) for the Phe cutoff value of 600 ± 100 µmol/L; and 0.30 (95% CI 0.20–0.40) for the Phe cutoff value of 1200 ± 200 µmol/L.
Conclusions
Substantial regional variation in the birth prevalence of PAH deficiency was observed in this systematic literature review and meta-analysis of published evidence from newborn screening. The precision of the prevalence estimates is limited by relatively small sample sizes, despite widespread and longstanding newborn screening in much of the world.
Similar content being viewed by others

Background
Phenylalanine hydroxylase (PAH) deficiency is an autosomal recessive disorder that results in elevated concentrations of the amino acid phenylalanine (Phe) in the blood [1,2,3,4]. Over 1000 PAH variants exist [5], and depending on the inherited alleles, affected individuals may have very mild to pronounced elevation of Phe [4]. Phenylalanine hydroxylase catalyzes the conversion of Phe into tyrosine and is key to maintaining a stable concentration of Phe in the blood [7]. When PAH activity is decreased, blood Phe concentration increases from the typical mean of 60 μmol/L [3]. In addition, an estimated 1–2% of cases of hyperphenylalanemia (HPA) are secondary to a deficiency in tetrahydrobiopterin (BH4), a necessary cofactor for PAH and other amino acid-metabolizing enzymes [4, 6]. Cases of mutations in a heat shock co-chaperone family member, DNAJC12 have been also reported to result in HPA [8]. If left untreated, the accumulation of Phe can result in profound neurocognitive disability [2]. Early diagnosis and intervention are essential to preserve cognitive function [1, 3].
Treatment guidelines recommend initiation of treatment as early as possible upon diagnosis of PAH deficiency [3]. Treatment options include dietary and pharmaceutical management. Dietary management involves severely restricted intake of Phe (and protein)-rich foods based on each individual’s maximum Phe tolerance [9, 10] in combination with medical foods to supplement inadequate intake of protein and other essential nutrients due to the Phe-restricted diet. Approved pharmaceutical treatments for PAH deficiency include pegvaliase and sapropterin. While pegvaliase, a Phe-metabolizing enzyme composed of pegylated recombinant phenylalanine ammonia lyase, is approved for use only in adults (United States) and persons aged 16 years and above (Europe) who have uncontrolled Phe in blood (> 600 uM/L) with current treatment [11, 12], sapropterin dihydrochloride, a synthetic form of BH4, is indicated for use in children (> 1 month of age) and adults with BH4-responsive PKU in conjunction with a Phe-restricted diet [2, 13, 14].
Phenylalanine hydroxylase deficiency is classified into mild HPA, mild phenylketonuria (PKU), moderate PKU, and classical PKU based on blood Phe concentration obtained in the neonatal period (Table 1); however, concentrations determined in this period are unlikely to reflect peak untreated levels, as neonates vary in their dietary exposure to Phe before the blood sample is taken, and early treatment often precludes obtaining more definitive Phe concentrations [1].
Because of the severe consequences of untreated phenylalanine hydroxylase deficiency, many countries currently perform routine newborn screening for elevated blood Phe concentration [15,16,17]. Methods for measuring Phe have evolved over time, with increasing accuracy, initiating with the bacterial inhibition assay (Guthrie test) in 1963 [18] to the current state-of-the-art tandem mass spectrometry [19]. The Guthrie test has been suggested to miss as many as 1 in 25 affected newborns screened at or before 3 days of age [20].
The accumulation of data from newborn screening programs with varied screening methods employed across the world provides an opportunity to evaluate the birth prevalence of HPA and PKU at the regional and global levels. Here, we systematically review the published literature and analyze regional differences in HPA and PKU birth prevalence, overall and for various clinically relevant blood Phe concentration cutoff values used in confirmatory testing.
Methods
The protocol for this literature review was registered with PROSPERO (International prospective register of systematic reviews: https://www.crd.york.ac.uk/PROSPERO/display_record.php?RecordID=156377; ID 156377).
Birth prevalence
For the purpose of this review and to ensure consistent methodology in calculation of birth prevalence estimates across studies, birth prevalence was defined as cases identified during newborn screening divided by the number of newborns screened. This method was most frequently described in studies reporting birth prevalence of PAH deficiency from newborn screening programs.
Literature search
PubMed and Embase were searched using a strategy based on the PICOS (population, intervention, comparison, outcomes, study design) framework (Additional file 1: Table A-1) [21]. The search strategy included terms to identify newborns, prevalence, incidence, newborn screening, Guthrie and other tests, PKU, HPA, and PAH deficiency (Additional file 1: Table A-2 and Table A-3). No language or time limits were implemented. Animal studies, editorials, and commentaries were excluded.
Study selection
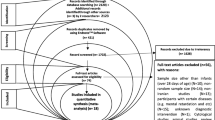
Entries retrieved from PubMed and Embase were screened in two steps (Fig. 1): in level 1 screening, two researchers independently reviewed titles and abstracts; in level 2 screening, two researchers independently reviewed full-text articles. Lack of agreement on inclusion was resolved by discussion and consensus within the research team.

Study selection process. PRISMA chart modeled after Moher et al. [21]. BH4 = tetrahydrobiopterin; PAH = phenylalanine hydroxylase deficiency; PKU = phenylketonuria
In level 1 screening (Additional file 1: Table A-4), conference abstracts, studies reporting exclusively on BH4 deficiency but not on PAH deficiency, and studies that focused primarily on assay development and/or validation were excluded. Publications were eligible if the abstract or title indicated that the paper presented original research and contained numeric reports on the birth prevalence of PAH deficiency. Birth prevalence must have been reported on an unselected population (e.g., studies on institutionalized patients were not eligible) and was required to be directly measured (rather than estimated from models). When duplicate records reporting on one study were identified, only one was retained; in this circumstance, records published in English were preferred.
The following additional criteria were applied in level 2 screening (Additional file 1: Table A-5): articles were required to be written in English and birth prevalence was required to be based on confirmed cases. When two or more publications on any given region were identified, both were included if the research had been conducted by different groups, or if both the geography and time frame did not overlap. For reports with geographic and temporal overlap conducted by the same institution, the study covering the largest population was eligible.
Data extraction and quality assessment
Extracted data elements included country and region, dates of data collection, study design, assay method for screening and for case confirmation (when diagnostic methods varied among sites or over time, scoring for the estimate was based on the lowest scoring diagnostic method, per the list in Table 2), diagnosis as reported in the publication (“nominal diagnosis”), Phe concentration used as a positive cutoff value, whether patients with BH4 deficiency were included in the number of cases reported, number of newborns screened, number of cases, and reported birth prevalence. For publications that reported birth prevalence stratified by multiple variables, values for each variable were extracted separately (herein referred to as “estimates”).
Data were extracted by one researcher using a form specifically designed for this study; extracted data were verified by a second researcher. Each estimate was assessed for quality as strong, moderate, or weak in each of five scoring domains (Table 2). The quality assessment tool used in this study was based on existing tools for assessing the quality of studies that report the prevalence of conditions assessed by surveillance [22] or conditions of genetic origin [23].
Meta-analyses
To mitigate errors that may arise from using early, less reliable assays, such as the Guthrie test, only estimates derived from confirmatory diagnostic assays that were assessed as strong in the quality assessment tool (Table 2) were eligible for meta-analysis. Inclusion in the meta-analysis also required that the number of cases and the number of screened newborns were reported. For each region and Phe concentration cutoff value category, at least 2 birth prevalence estimates were required to conduct a meta-analysis. For regions and Phe concentration cutoff value categories with only one published birth prevalence estimate, the single published estimate was used to represent the region (or Phe concentration cutoff value) in the global prevalence estimates. Once the eligible estimates for each planned meta-analysis were identified, estimates with both temporal and geographic overlap were assessed, and the estimate representing the largest geographic coverage or time period was included.
Meta-analyses were performed to determine aggregated regional birth prevalence (Europe, North America, Middle East/North Africa, Latin America, South Pacific, and West Pacific; Additional file 1: Table A-6) and a global birth prevalence. The global birth prevalence was estimated by using two approaches. A “regionally weighted” global prevalence was calculated, in which results from each region were weighted by the region’s relative numerical contribution to the total population of the regions for each analysis. For this determination, country-specific population counts were obtained from 2020 United Nations population estimates [24] and were summed within each region to determine regional totals (weights for analyses incorporating results from six regions: Europe, 0.126; Latin America, 0.097; Middle East/North Africa, 0.125; North America, 0.055; Southeast Asia, 0.303; West Pacific, 0.293). A non-regionally weighted global prevalence was also calculated for comparison to other recently published PKU global birth prevalence estimates that were not regionally-weighted.
For both regional and global birth prevalence determinations, birth prevalence was calculated and stratified by three confirmatory Phe concentration cutoff values (360 ± 100 μmol/L, 600 ± 100 μmol/L, 1200 ± 200 μmol/L). When a publication reported birth prevalence by Phe cutoff interval (e.g., separate birth prevalence values for ≥ 360 ± 100 to 600 μmol/L, ≥ 600 ± 100 μmol/L to 1200 μmol/L and ≥ 1200 ± 200 μmol/L), the sum of all values above the cutoff value was used. Finally, an unstratified meta-analysis was conducted, which additionally included estimates from studies in which Phe cutoff values were not reported, to determine overall (regionally weighted and non-regionally weighted) birth prevalence.
To provide appropriate weights for meta-analysis, birth prevalence estimates were transformed using the double arcsine method [25]; meta-analysis was conducted using a random-effects model with inverse variance weighting. Transformation and calculations were performed using MetaXL (version 5.3, EpiGear International). Heterogeneity was assessed using the I2 statistic [26, 27].
Results
Literature search and review
Searches in PubMed and Embase identified 1112 entries (Fig. 1). Screening of 997 unique PubMed and Embase entries and an additional 28 publications identified from reference lists of screened entries identified 85 publications meeting eligibility criteria, resulting in 238 birth prevalence estimates (Additional file 2).
These 85 publications were published from 1964 [28] to 2019 [29] and reported on data from 1960 [30] to 2018 [29] from 59 countries. Newborn blood or urine samples for screening were taken between the first day of life [31] and age 3–8 weeks [32]; 25 publications (125 birth prevalence estimates) did not report age at screening. Phe concentration used for confirmatory testing ranged from 120 μmol/L [33] to over 2600 μmol/L [34]. Forty-three publications (135 birth prevalence estimates) did not report the cutoff value for confirmatory testing. Nominal diagnoses were inconsistent. For example, classical PKU was defined using confirmatory Phe cutoff values ranging from 726 μmol/L [35] to 1816 μmol/L [36]. Cases with BH4 deficiency were included in 5 publications (6 birth prevalence estimates) and the presence or absence of BH4 deficiency was not reported in 58 publications (186 birth prevalence estimates).
The only domains of the quality assessment tool on which > 50% of the estimates scored strong were statistical methods and study setting/source population. Sixty percent of the estimates scored moderate or weak on precision, and 53% scored moderate or weak on the method for case confirmation (Fig. 2A).

a–e Quality of evidence assessments of birth prevalence estimates
Meta-analysis results
A total of 112 birth prevalence estimates (54 publications) scored strong in the quality assessment domain diagnostic method used for case confirmation and were therefore potentially eligible for meta-analysis. One publication (18 estimates) with strong scores in the diagnostic method used for case confirmation reported birth prevalence (in the format 1:8000), but did not provide the number of cases or screened newborns [37] and was not deemed eligible. No birth prevalence estimates from the African region were included in the meta-analysis, and the only estimates eligible for inclusion in Southeast Asia were from Thailand.
Birth prevalence estimates ranged from 0 (Estonia [38], Finland [39], and Thailand [40]) to 2.46 per 10,000 births (Macedonia) [41] (Table 3).
Estimates from 45 publications were included in at least one meta-analysis, and the rest were excluded due to temporal and regional overlap. Meta-analysis results are summarized in Table 4 and Additional file 3: Figures A2–A5. The regionally weighted global birth prevalence of PAH deficiency (N = 44 publications, 1 estimate per publication) was 0.64 (95% confidence interval [CI] 0.53–0.75) per 10,000 births (Table 4; quality assessment results shown in Fig. 2E). The lowest regional birth prevalence was observed in Southeast Asia, with 0.03 cases per 10,000 births (95% CI 0.02–0.05); the highest was observed in the Middle East/North Africa, with 1.18 (95% CI 0.64–1.87) cases per 10,000 births.
Eleven publications reported birth prevalence estimates (1 estimate per publication) with a confirmatory test Phe concentration cutoff value of 360 ± 100 µmol/L. The regionally weighted global birth prevalence was 0.96 (95% CI 0.50–1.42) per 10,000 births (Table 4 and Fig. 2B). The lowest regional birth prevalence was observed in North America, with 0.49 cases per 10,000 births (95% CI 0.38–0.61), based on two publications that presented very similar results [42, 43], as reflected in the heterogeneity statistic I2 value of 0. The highest birth prevalence was observed in the Middle East/North Africa, 1.60 (95% CI 1.06–2.31) per 10,000 births, based on a single estimate [44].
Ten publications (1 estimate each) reported birth prevalence estimates using a confirmatory test Phe concentration cutoff value of 600 ± 100 µmol/L. The regionally weighted global birth prevalence was 0.50 (95% CI 0.37–0.64) per 10,000 births (Table 4 and Fig. 2C) for this cutoff value.
For the 1200 ± 200 µmol/L cutoff value for a Phe concentration confirmatory test, 20 publications (1 estimate each) were eligible and the regionally weighted global birth prevalence was 0.30 (95% CI 0.20–0.40) per 10,000 births (Table 4 and Fig. 2D).
Discussion
The overall meta-analysis conducted in this systematic review provides a regionally weighted global birth prevalence of PAH deficiency of 0.64 (95% CI 0.53–0.75) per 10,000 births. It is important to weight birth prevalence estimates by region so that the global PAH deficiency birth prevalence reflects both the birth prevalence and population size of each region rather than just the inverse variance (primarily driven by the sample size) of the individual studies (as was done for the calculation of non–regionally weighted birth prevalence). The highest regional birth prevalence in the overall analysis was reported in the Middle East/North Africa, where consanguineous marriages are among the most frequent in the world, with frequencies up to 42% in Saudi Arabia [45].
Among estimates with a confirmatory test Phe concentration cutoff value of 360 ± 100 µmol/L, the regionally weighted global birth prevalence was 0.96 (95% CI 0.50–1.42) per 10,000 births. On the basis of recent European and American College of Medical Genetics and Genomics guidelines (Table 1), this would represent the population for which treatment in children is recommended. Based on the single estimate for Middle East/North Africa, the birth prevalence was again highest in this region [44].
In the meta-analyses based on Phe concentration cutoff values of 600 µmol/L and 1200 µmol/L, the regionally weighted global prevalences were 0.50 (95% CI 0.37–0.64) and 0.30 (95% CI 0.20–0.40), respectively, per 10,000 births. Regional variation in the prevalence of PAH deficiency defined by these cutoff values was observed, with higher prevalences in Europe, Latin America, North America, and the Middle East than was observed globally. In a recent analysis of global variations in PAH genotype [46], genotypes associated with classical PKU (Phe ≥ 1200 µmol/L) tended to be the most common in the Middle East.
As might be expected, in this meta-analysis we observed decreasing pooled birth prevalence as confirmatory test Phe cutoff values increased (Table 4). The decreasing prevalence we observed with increasing Phe cutoff values should be interpreted cautiously. Specifically, this finding does not necessarily reflect differences in the relative frequencies of classical, moderate, mild PKU and HPA, but rather the fact that individuals with higher Phe levels are included in the estimates with lower cutoff values (e.g., the pooled prevalence for the 360 µmol/L cutoff value includes individuals that would be diagnosed as having classical and severe PKU per Table 1). This approach was taken to ascertain the birth prevalence of all individuals whose Phe levels were within the treatable range and the impact different confirmatory Phe cutoff thresholds have on PAH deficiency birth prevalence estimates. The confidence intervals for the various Phe cutoff thresholds had substantial overlap, likely due to heterogeneity of estimates from individual studies.
As evidenced by the high I2 values, heterogeneity of birth prevalence estimates was generally high, even among estimates stratified by region and Phe concentration cutoff values for case confirmation. Heterogeneity may be partly explained by random variation related to sampling, which is supported by the fact that many included studies were small (35% of the 238 estimates scored weak on precision of the prevalence estimate [Fig. 2]). Other reasons for heterogeneity include variations in age at screening and confirmatory testing, and dietary intake prior to sampling.
We found that data elements that are key to understanding the reported birth prevalence estimates were often missing: 30% of the 238 estimates scored weak on case definition (i.e., failed to provide Phe cutoff values for both screening and for case confirmation), and 66% scored moderate on this domain (failed to report on either screening or confirmatory Phe cutoff values); 11% did not report the study setting/source population or derived the information from personal communications. In addition, 126 of 238 reported birth prevalence estimates (53%) scored moderate or weak in diagnostic method used for case confirmation. Thirteen percent of the 238 estimates lacked information on the time period assessed, 3% on the assay used for screening, and 38% on the assay used for case confirmation. Although the frequency of BH4 deficiency is very low (1–2% of HPA cases) [6], it was not reported or not excluded from the reported birth prevalence estimates in 81% of the 238 birth prevalence estimates included in this review.
Substantial inconsistencies were observed in the nominal diagnoses reported, even in recent publications, with poor or inaccurate distinction between PKU, moderate PKU, classical PKU, and HPA (Additional file 2).
We have not found published papers estimating the global prevalence of PAH deficiency. However, two recently published reviews estimated the global prevalence of PKU. Shoraka et al. [47] identified studies reporting the birth prevalence of classical PKU in newborns and meta-analyzed them by region and overall (non-regionally weighted, with no stratification by case confirmation Phe cut off value). Hillert et al. [46] used unpublished information from national screening centers and reports identified through a literature search to estimate a global prevalence of PKU in newborns. Table 5 provides a comparison of the birth prevalence estimates from our analysis with the results from the studies by Shoraka et al. [46] and Hillert et al. [45].
The largest differences between the current study and the study by Shoraka et al. were seen in Europe, the Americas, and the West Pacific regions. The similarity between the overall estimate by Shoraka et al. and the currently reported regionally weighted global birth prevalence is likely largely due to chance, as substantially different inclusion criteria and methodologies were employed in the two studies (Additional file 1: Figure A-1). Shoraka et al. excluded publications considered to have a high risk of bias as assessed using an existing 10-point checklist [48], which has some similar elements to the quality of evidence tool used in this publication. There was no requirement that cases be confirmed. The reported prevalence was described as relating to classical PKU, even though the Phe cutoff for confirmatory tests of the included studies ranged from 1.65 mg/dL (equivalent to 100 µmol/L) to 20 mg/dL (1211 µmol/L).
The current study provides a higher estimate of the global birth prevalence of PAH deficiency than Hillert et al. Unfortunately the inclusion and exclusion criteria and the method(s) for combining estimates from individual studies are not fully described in that paper, nor are the sources fully described; the global estimate included data from countries that the study describes as lacking newborn screening programs in parts of Africa, Asia, South America, and the Caribbean [46].
The current findings confirm that regional differences exist in the birth prevalence of PAH deficiency, with higher frequencies of inheritance of this autosomal recessive disease in areas with higher frequencies of consanguineous marriages, as has also been noted by others [46, 47].
Limitations of this study include incomplete reporting of key data elements in many of the included publications. In addition, the precision of the reported prevalence was low for most of the included estimates due to small sample sizes. No articles were identified reporting on the birth prevalence of PAH deficiency in Sub-Saharan Africa and birth prevalence estimates from countries in Southeast Asia were limited, lacking representation of some of the most populous countries in the region such as India. Absence of estimates could be attributed to absence of newborn screening programs for PAH deficiency in specific countries and regions [15], or lack of published estimates from newborn screening programs meeting the inclusion criteria for this review, such as the requirement that the full-text article be written in English. Strengths of this study include the fact that only confirmed cases were included in the qualitative synthesis, and that the meta-analysis only included estimates based on higher quality confirmatory assays. In addition, meta-analyses were undertaken based on clinically relevant diagnostic cutoff values.
Conclusions
In this systematic literature review and meta-analysis, we estimated the regionally weighted global birth prevalence of PAH deficiency to be 0.64 (95% CI 0.53–0.75) per 10,000 births (overall). The estimated regionally weighted global birth prevalence among newborns with Phe level ≥ 360 ± 100 µmol/L at diagnosis was 0.96 (95% CI 0.50–1.42), which is the population for whom treatment is recommended. Substantial regional variation was observed with an elevated birth prevalence of this autosomal recessive disease in regions with higher frequencies of consanguineous births. Despite the fact that newborn screening has been widely implemented in much of the world for decades, the precision of the estimates is limited by the unavailability of publications on large population samples. This observation underscores the need for more comprehensive and systematic data collection as well as improved standards for reporting results. Only with more widespread availability of data from newborn screening programs from large populations will it be possible to obtain robust estimates and truly understand the magnitude of this serious and treatable condition.
Availability of data and materials
The dataset supporting the conclusions of this article is within the published manuscript and its appendices.
Abbreviations
- ACMG:
-
American College of Medical Genetics and Genomics
- BH4:
-
Tetrahydrobiopterin
- CI:
-
Confidence interval
- HPA:
-
Hyperphenylalaninemia
- NA:
-
Not available
- NR:
-
Not reported
- PAH:
-
Phenylalanine hydroxylase
- Phe:
-
Phenylalanine
- PICOS:
-
Population, intervention, comparison, outcomes, study design
- PKU:
-
Phenylketonuria
- US:
-
United States
References
van Spronsen FJ, van Wegberg AM, Ahring K, Belanger-Quintana A, Blau N, Bosch AM, et al. Key European guidelines for the diagnosis and management of patients with phenylketonuria. Lancet Diabetes Endocrinol. 2017;5(9):743–56.
van Wegberg AMJ, MacDonald A, Ahring K, Belanger-Quintana A, Blau N, Bosch AM, et al. The complete European guidelines on phenylketonuria: diagnosis and treatment. Orphanet J Rare Dis. 2017;12(1):162.
Vockley J, Andersson HC, Antshel KM, Braverman NE, Burton BK, Frazier DM, et al. Phenylalanine hydroxylase deficiency: diagnosis and management guideline. Genet Med. 2014;16(2):188–200.
Blau N. Genetics of phenylketonuria: then and now. Hum Mutat. 2016;37(6):508–15.
PAHvdb: Phenylalanine Hydroxylase Gene Locus-Specific Database. 2020. http://www.biopku.org/home/pah.asp. Accessed 4 Sept 2020.
Opladen T, López-Laso E, Cortès-Saladelafont E, Pearson TS, Sivri HS, Yildiz Y, et al. Consensus guideline for the diagnosis and treatment of tetrahydrobiopterin (BH(4)) deficiencies. Orphanet J Rare Dis. 2020;15(1):126.
Williams RA, Mamotte CD, Burnett JR. Phenylketonuria: an inborn error of phenylalanine metabolism. Clin Biochem Rev. 2008;29(1):31–41.
Anikster Y, Haack TB, Vilboux T, Pode-Shakked B, Thony B, Shen N, et al. Biallelic mutations in DNAJC12 cause hyperphenylalaninemia, dystonia, and intellectual disability. Am J Hum Genet. 2017;100(2):257–66.
MacDonald A, van Wegberg AMJ, Ahring K, Beblo S, Bélanger-Quintana A, Burlina A, et al. PKU dietary handbook to accompany PKU guidelines. Orphanet J Rare Dis. 2020;15(1):171.
MacDonald A, van Wegberg AMJ, Ahring K, Beblo S, Bélanger-Quintana A, Burlina A, et al. Correction to: PKU dietary handbook to accompany PKU guidelines. Orphanet J Rare Dis. 2020;15(1):230.
European Medicines Agency. Palynziq—Summary of product characteristics. 2019. https://www.ema.europa.eu/en/documents/product-information/palynziq-epar-product-information_en.pdf. Accessed 4 Sept 2020.
FDA. Palynziq—US package insert. 2018. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/761079s000lbl.pdf.
European Medicines Agency. Kuvan—Summary of product characteristics. 2020. https://www.ema.europa.eu/en/documents/product-information/kuvan-epar-product-information_en.pdf. Accessed 4 Sept 2020.
FDA. Kuvan - US package insert. 2007. https://www.accessdata.fda.gov/drugsatfda_docs/label/2007/022181lbl.pdf. Accessed 4 Sep 2020.
Therrell BL, Padilla CD, Loeber JG, Kneisser I, Saadallah A, Borrajo GJ, et al. Current status of newborn screening worldwide: 2015. Semin Perinatol. 2015;39(3):171–87.
Zerjav Tansek M, Groselj U, Angelkova N, Anton D, Baric I, Djordjevic M, et al. Phenylketonuria screening and management in southeastern Europe—survey results from 11 countries. Orphanet J Rare Dis. 2015;30(10):68.
Borrajo GJC. Newborn screening for phenylketonuria: Latin American consensus guidelines. J Inborn Errors Metab Screen. 2016 2016/01/01;4:2326409816682764.
Guthrie R, Susi A. A simple phenylalanine method for detecting phenylketonuria in large populations of newborn infants. Pediatrics. 1963;32:338–43.
Chace DH, Hannon WH. Technological journey from colorimetric to tandem mass spectrometric measurements in the diagnostic investigation for phenylketonuria. J Inborn Errors Metab Screen. 2016 2016/01/01;4:2326409816671733.
Blumenfeld CM, Wallace MJ, Anderson R. Phenylketonuria-the guthrie screening test-a method of quantitation, observations on reliability and suggestions for improvement. Calif Med. 1966;105(6):429–34.
Moher D, Liberati A, Tetzlaff J, Altman DG, Group P. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. BMJ. 2009;21(339):b2535.
Silva LC, Ordunez P, Paz Rodriguez M, Robles S. A tool for assessing the usefulness of prevalence studies done for surveillance purposes: the example of hypertension. Rev Panam Salud Publica. 2001;10(3):152–60.
Al-Jader LN, Newcombe RG, Hayes S, Murray A, Layzell J, Harper PS. Developing a quality scoring system for epidemiological surveys of genetic disorders. Clin Genet. 2002;62(3):230–4.
United Nations. World population prospects 2019. File POP/1–1: total population (both sexes combined) by region, subregion and country, annually for 1950–2100 (thousands). 2019. https://population.un.org/wpp/. Accessed 28 May 28 2020.
Barendregt JJ, Doi SA, Lee YY, Norman RE, Vos T. Meta-analysis of prevalence. J Epidemiol Commun Health. 2013;67(11):974–8.
Higgins J, Thomas J, Chandler J, Cumpston M, Li T, Page M. Cochrane handbook for systematic reviews of interventions, version 6.0. 2019. https://training.cochrane.org/handbook. Accessed 5 March 2020.
Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta-analyses. BMJ. 2003;327(7414):557–60.
Maccready RA, Hussey MG. Newborn phenylketonuria detection program in Massachusetts. Am J Public Health Nations Health. 1964;54:2075–81.
Lin Y, Zheng Q, Zheng T, Zheng Z, Lin W, Fu Q. Expanded newborn screening for inherited metabolic disorders and genetic characteristics in a southern Chinese population. Clin Chim Acta. 2019;494:106–11.
Carson NA, Carre IJ, Neill DW. Results of routine screening for phenylketonuria in early infancy, Northern Ireland (1960–1967). Arch Dis Child. 1968;43(228):145–6.
Arnopp JJ, Lorey FW, Currier RJ, Eastman JW, Velazquez KB, Morales DR, et al. Results of screening for phenylketonuria using a lower cutoff value in early collected specimens. Screening. 1995;3(4):193–9.
Martin PH. Six years of newborn PKU screening. J Indiana State Med Assoc. 1968;61(8):1107–8.
Yang LL, Mao HQ, Zhang WF, Zhao ZY, Yang RL, Zhou XL, et al. Pitfalls in the management of phenylketonuria in China. Hong Kong J Paediatr. 2012;17(3):143–7.
Boneh A, Francis DE, Humphrey M, Upton HJ, Peters HL. Three-year audit of the hyperphenylalaninaemia/phenylketonuria spectrum in Victoria. J Paediatr Child Health. 2006;42(9):496–8.
Read KS, Allen RJ, Haddy TB. Phenylketonuria in newborns. Mich Med. 1969;68(13):691–7.
Ervin EN. Hyperphenylalanemia. Incidence in Maine since 1964. J Maine Med Assoc. 1970;61(2):30–2.
Somers DG, Favreau L. Newborn screening for phenylketonuria: incidence and screening procedures in North America. Can J Public Health. 1982;73(3):206–7.
Ounap K, Lilleväli H, Metspalu A, Lipping-Sitska M. Development of the phenylketonuria screening programme in Estonia. J Med Screen. 1998;5(1):22–3.
Visakorpi JK, Palo J, Renkonen OV. The incidence of PKU in Finland. Acta Paediatr Scand. 1971;60(6):666–8.
Ratrisawadi V, Horpaopan S, Chotigeat U, Sangtawesin V, Kanjanapattanakul W, Ningsanond V, et al. Neonatal screening program in Rajavithi Hospital, Thailand. Southeast Asian J Trop Med Public Health. 1999;30(Suppl 2):28–32.
Kocova M, Anastasovska V. Phenylketonuria screening in the Republic of Macedonia. Orphanet J Rare Dis. 2016;11(1):112.
Frazier DM, Millington DS, McCandless SE, Koeberl DD, Weavil SD, Chaing SH, et al. The tandem mass spectrometry newborn screening experience in North Carolina: 1997–2005. J Inherit Metab Dis. 2006;29(1):76–85.
Wainer SC, Sideman L. Nine years of PKU screening in Pennsylvania. Health Lab Sci. 1974;11(4):306–11.
Habib A, Fallahzadeh MH, Kazeroni HR, Ganjkarimi AH. Incidence of phenylketonuria in Southern Iran. Iran J Med Sci. 2010;35(2):137–9.
Hamamy H, Antonarakis SE, Cavalli-Sforza LL, Temtamy S, Romeo G, Kate LP, et al. Consanguineous marriages, pearls and perils: Geneva International Consanguinity Workshop Report. Genet Med. 2011;13(9):841–7.
Hillert A, Anikster Y, Belanger-Quintana A, Burlina A, Burton BK, Carducci C, et al. The genetic landscape and epidemiology of phenylketonuria. Am J Hum Genet. 2020;107(2):234–50.
Shoraka HR, Haghdoost AA, Baneshi MR, Bagherinezhad Z, Zolala F. Global prevalence of classic phenylketonuria based on Neonatal Screening Program data: systematic review and meta-analysis. Clin Exp Pediatr. 2020;63(2):34–43.
Hoy D, Brooks P, Woolf A, Blyth F, March L, Bain C, et al. Assessing risk of bias in prevalence studies: modification of an existing tool and evidence of interrater agreement. J Clin Epidemiol. 2012;65(9):934–9.
Thalhammer O. Distribution and frequency of PKU and hyperphenylalaninemia in eastern and western Austria. Ir Med J. 1976;69(15):396–7.
Lindner M, Gramer G, Haege G, Fang-Hoffmann J, Schwab KO, Tacke U, et al. Efficacy and outcome of expanded newborn screening for metabolic diseases–report of 10 years from South-West Germany. Orphanet J Rare Dis. 2011;6:44.
Mathias D, Bickel H. Follow-up study of 16 years neonatal screening for inborn errors of metabolism in West Germany. Eur J Pediatr. 1986;145(4):310–2.
Schulze A, Lindner M, Kohlmuller D, Olgemoller K, Mayatepek E, Hoffmann GF. Expanded newborn screening for inborn errors of metabolism by electrospray ionization-tandem mass spectrometry: results, outcome, and implications. Pediatrics. 2003;111(6 Pt 1):1399–406.
Loukas YL, Soumelas GS, Dotsikas Y, Georgiou V, Molou E, Thodi G, et al. Expanded newborn screening in Greece: 30 months of experience. J Inherit Metab Dis. 2010;33(Suppl 3):S341–8.
Missiou-Tsagaraki S, Soulpi K, Loumakou M. Phenylketonuria in Greece: 12 years’ experience. J Ment Defic Res. 1988;32(Pt 4):271–87.
Mehes K, Juhasz E, Ruszinko V. Chromatographic screening of 70,328 neonates for inborn errors of amino acid metabolism. Acta Paediatr Hung. 1985;26(2):147–9.
Antonozzi I, Dominici R, Andreoli M, Monaco F. Neonatal screening in Italy for congenital hypothyroidism and metabolic disorders: hyperphenylalaninemia, maple syrup urine disease and homocystinuria. J Endocrinol Invest. 1980;3(4):357–63.
Zaffanello M, Zamboni G, Tatò L. Neonatal screening program for inborn errors of metabolism: a retrospective study from 1978 to 1997 in Northeastern Italy. Ital J Pediatr. 2002;28:479–83.
Cabalska B, Nowaczewska I, Duczynska N, Laskowska-Klita T. Twenty-five years experience with newborn screening for phenylketonuria (PKU) in Poland. Screening. 1993;2(1):29–32.
Vilarinho L, Rocha H, Sousa C, Marcao A, Fonseca H, Bogas M, et al. Four years of expanded newborn screening in Portugal with tandem mass spectrometry. J Inherit Metab Dis. 2010;33(Suppl 3):S133–8.
Dluholucký S, Knapková M. Newborn screening in Slovakia—from 1985 till today/Novorodencký skríning na Slovensku—od roku 1985 doposiaľ. Acta Facul Pharm Univ Comen. 2013;60(Suppl VIII):32–6.
Smon A, Groselj U, Zerjav Tansek M, Bicek A, Oblak A, Zupancic M, et al. Newborn screening in Slovenia. Zdr Varst. 2015;54(2):86–90.
Fernández-Iglesias C, Flórez IG, Rodríguez-González MC, Gascón S. Neonatal screening for phenylketonuria and congenital hypothyroidism in Principado de Asturias (Spain) using two types of blood samples. Screening. 1995;4(3):131–8.
Gerasimova NS, Samutin AA, Steklova IV, Tuuminen T. Phenylketonuria screening in Moscow using a microplate fluorometric method. Screening. 1992;1(1):27–35.
Walker V, Clayton BE, Ersser RS, Francis DE, Lilly P, Seakins JW, et al. Hyperphenylalaninaemia of various types among three-quarters of a million neonates tested in a screening programme. Arch Dis Child. 1981;56(10):759–64.
Mardesic D, Gjuric G, Jancikovic J. Screening for phenylketonuria in Yugoslavia (SR Crotia) 1979–1984. J Inherit Metab Dis. 1986;9(Suppl 2):234–6.
Botler J, Camacho LA, Cruz MM. Phenylketonuria, congenital hypothyroidism and haemoglobinopathies: public health issues for a Brazilian newborn screening program. Cad Saude Publica. 2012;28(9):1623–31.
Ramalho AR, Ramalho RJ, Oliveira CR, Magalhaes MM, Santos EG, Sarmento PM, et al. Evaluation of effectiveness and outcome of PKU screening and management in the state of Sergipe. Brazil Arq Bras Endocrinol Metabol. 2014;58(1):62–7.
Cornejo V, Raimann E, Cabello JF, Valiente A, Becerra C, Opazo M, et al. Past, present and future of newborn screening in Chile. J Inherit Metab Dis. 2010;33(Suppl 3):S301–6.
Abbaskhanian A, Zamanfar D, Afshar P, Asadpoor E, Rouhanizadeh H, Jafarnia A, et al. Incidence of neonatal hyperphenylalaninemia based on high-performance liquid chromatography confirmatory technique in Mazandaran province, northern Iran (2007–2015). Int J Prev Med. 2017;8:93.
Karamifar H, Ordoei M, Karamizadeh Z, Amirhakimi G. Incidence of neonatal hyperphenylalaninemia in Fars province, south Iran. Iran J Pediatr. 2010;20(2):216–20.
Motamedi N, Godarzi E, Pordanjani SR, Valizadeh R, Moradi Y, Sohrabivafa M, et al. Incidence of phenylketonuria in Lorestan province, west of Iran (2006–2016). Int J Pediatr. 2017;5(4):4713–21.
Alfadhel M, Al Othaim A, Al Saif S, Al Mutairi F, Alsayed M, Rahbeeni Z, et al. Expanded newborn screening program in Saudi Arabia: incidence of screened disorders. J Paediatr Child Health. 2017;53(6):585–91.
Ozalp I, Coskun T, Tokatli A, Kalkanoglu HS, Dursun A, Tokol S, et al. Newborn PKU screening in Turkey: at present and organization for future. Turk J Pediatr. 2001;43(2):97–101.
Al Hosani H, Salah M, Osman HM, Farag HM, El-Assiouty L, Saade D, et al. Expanding the comprehensive national neonatal screening programme in the United Arab Emirates from 1995 to 2011. East Mediterr Health J. 2014;20(1):17–23.
Hansen H, Shahidi A, Stein ZA. Screening for phenylketonuria in New York City Threshold values reconsidered. Public Health Rep. 1978;93(3):246–51.
Kelly S, Palombi J. Phenylketonuria in New York State. Public Health Rep. 1967;82(10):921–4.
Zytkovicz TH, Fitzgerald EF, Marsden D, Larson CA, Shih VE, Johnson DM, et al. Tandem mass spectrometric analysis for amino, organic, and fatty acid disorders in newborn dried blood spots: a two-year summary from the New England Newborn Screening Program. Clin Chem. 2001;47(11):1945–55.
Charoensiriwatana W, Janejai N, Boonwanich W, Krasao P, Chaisomchit S, Waiyasilp S. Neonatal screening program in Thailand. Southeast Asian J Trop Med Public Health. 2003;34(Suppl 3):94–100.
Pangkanon S, Ratrisawadi V, Charoensiriwatana W, Techasena W, Boonpuan K, Srisomsap C, et al. Phenylketonuria detected by the neonatal screening program in Thailand. Southeast Asian J Trop Med Public Health. 2003;34(Suppl 3):179–81.
Pangkanon S, Charoensiriwatana W, Janejai N, Boonwanich W, Chaisomchit S. Detection of phenylketonuria by the newborn screening program in Thailand. Southeast Asian J Trop Med Public Health. 2009;40(3):525–9.
Sutivijit Y, Banpavichit A, Wiwanitkit V. Prevalence of neonatal hypothyroidism and phenylketonuria in Southern Thailand: a 10-year report. Indian J Endocrinol Metab. 2011;15(2):115–7.
Chen RG, Pan XS, Qian DL, Guo H. Twenty-one cases of phenylketonuria out of 358,767 newborns in Shanghai, China. J Inherit Metab Dis. 1989;12(4):485.
Liu SR, Zuo QH. Newborn screening for phenylketonuria in eleven districts. Chin Med J (Engl). 1986;99(2):113–8.
Maitusong R, Japaer R, Zhao ZY, Yang RL, Huang XL, Mao HQ. Newborn screening in Zhejiang. China Chin Med J (Engl). 2012;125(4):702–4.
Shi XT, Cai J, Wang YY, Tu WJ, Wang WP, Gong LM, et al. Newborn screening for inborn errors of metabolism in mainland china: 30 years of experience. JIMD Rep. 2012;6:79–83.
Su Y, Wang H, Rejiafu N, Wu B, Jiang H, Chen H, et al. The molecular epidemiology of hyperphenylalaninemia in Uygur population: incidence from newborn screening and mutational spectra. Ann Transl Med. 2019;7(12):258.
Tu WJ, Cai J, Shi XD. Newborn screening for inborn errors of metabolism in Beijing, China: 22 years of experience. J Med Screen. 2011;18(4):213–4.
Wang X, He Y, Jiang Y, Feng X, Zhang G, Xia Z, et al. Screening and mutation analysis of hyperphenylalaninemia in newborns from Xiamen. China Clin Chim Acta. 2019;498:161–6.
Zhan JY, Qin YF, Zhao ZY. Neonatal screening for congenital hypothyroidism and phenylketonuria in China. World J Pediatr. 2009;5(2):136–9.
Yoon HR, Lee KR, Kang S, Lee DH, Yoo HW, Min WK, et al. Screening of newborns and high-risk group of children for inborn metabolic disorders using tandem mass spectrometry in South Korea: a three-year report. Clin Chim Acta. 2005;354(1–2):167–80.
Niu DM, Chien YH, Chiang CC, Ho HC, Hwu WL, Kao SM, et al. Nationwide survey of extended newborn screening by tandem mass spectrometry in Taiwan. J Inherit Metab Dis. 2010;33(Suppl 2):S295-305.
Acknowledgements
Editorial assistance was provided by John Forbes (RTI Health Solutions), medical writing support was provided by Kate Lothman (RTI Health Solutions), and graphical services were provided by Jason Mathes (RTI Health Solutions).
Funding
This work was funded by BioMarin Pharmaceutical Inc. RTI’s authors received no compensation other than annual salary from employer.
Author information
Authors and Affiliations
Contributions
Substantial contributions to the conception or design of the work; or the acquisition, analysis, or interpretation of data for the work: PKF, SL, RS, KA, AVM, BC, MP, AH. Drafting the work or revising it critically for important intellectual content: PKF, SL, RS, KA, AVM, BC, MP, AH. Final approval of the version to be published: PKF, SL, RS, KA, AVM, BC, MP, AH. Agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved: PKF, SL, RS, KA, AVM, BC, MP, AH. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
RS, KA, and SL are employees of BioMarin Pharmaceutical Inc. PKF was a consultant for BioMarin Pharmaceutical Inc. when this research was conducted. Research team members AH, AVM, BC and MP are full-time employees of RTI Health Solutions. RTI Health Solutions is a unit of RTI International, an independent, nonprofit organization that conducts work for government, public, and private organizations, including pharmaceutical companies. RTI authors participate in this work in the course of employment as work for hire, pursuant to a contract to conduct an independent research study for a client (BioMarin Pharmaceutical Inc.).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Literature search strategy, quality assessment, and country region classification. Description of the literature search strategy, how quality assessments were calculated for precision, and how country regions were classified.
Additional file 2.
Data extraction table. 238 birth prevalence estimates from 85 publications.
Additional file 3.
Meta-analysis forest plot figures. Figure A-2. Meta-Analysis Results by Region: Confirmatory Test Phenylalanine Cutoff Value of 360 ± 100 μmol/L. Figure A-3. Meta-Analysis Results by Region: Confirmatory Test Phenylalanine Cutoff Value of 600 ± 100 μM/L. Figure A-4. Meta-Analysis Results by Region: Confirmatory Test Phenylalanine Cutoff Value of 1200 ± 200 μM/L. Figure A-5. Meta-Analysis Results by Region: Overall Analysis.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Foreman, P.K., Margulis, A.V., Alexander, K. et al. Birth prevalence of phenylalanine hydroxylase deficiency: a systematic literature review and meta-analysis. Orphanet J Rare Dis 16, 253 (2021). https://doi.org/10.1186/s13023-021-01874-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-021-01874-6