
Abstract
Background
Costello syndrome (CS) and cardio-facio-cutaneous syndrome (CFCS) belong to the RASopathies, a group of neurodevelopmental disorders with skeletal anomalies. Due to their rarity, the characterization of the musculo-skeletal phenotype in both disorders has been poorly characterized.
Patients and methods
Herein we reported data on orthopedic findings and functional status of a large sample of CS and CFCS patients. Thirty-four patients (CS = 17 and CFCS = 17) were recruited. Functional and disability evaluations were performed by assessing the 6-min walking test (6MWT) and Pediatric Outcomes Data Collection Instrument (PODCI). Genotype/phenotype correlation was also provided.
Results
Orthopedic manifestations are highly prevalent in CS and CFCS and overlap in the two disorders. Overall, patients with CS harboring the recurrent HRAS Gly12Ser substitution show a more severe skeletal phenotype compared to patients carrying the Gly12Ala and Gly13Cys variants. Among CFCS patients, those with the MAP2K1/2 variant show different skeletal characteristics compared to BRAF variants, with a higher prevalence of orthopedic abnormalities. Functional assessment showed that patients with CS and CFCS reached lower values compared to the general population, with CFCS patients displaying the lowest scores.
Conclusions
Orthopedic manifestations appear universal features of CS and CFCS and they can evolve across patients’ life. Longitudinal assessment of disability status by using 6MWT and PODCI could be useful to evaluate the functional impact of orthopedic manifestations on patients’ outcome and help planning a tailored treatment of these comorbidities.
Similar content being viewed by others
Background
Costello syndrome (CS, OMIM #218040) and cardio-facio-cutaneous syndrome (CFCS, OMIM PS115150) are neurodevelopmental disorders caused by gain-of-function mutations in genes encoding components of the RAS/mitogen-activated protein kinase (MAPK) pathway, an intracellular signaling cascade playing a pivotal role in cell cycle regulation, differentiation, growth and senescence [1]. These conditions are grouped under the term RASopathies together with Noonan syndrome (NS) and neurofibromatosis type 1 (NF1) and other emerging clinically related disorders, due to a common pathogenetic mechanism resulting in dysregulation of the RAS/MAPK pathway [2]. Both CS and CFCS are ultra-rare conditions with approximately 300 individuals each reported worldwide [3, 4]. CS is caused by heterozygous activating mutations in the HRAS gene, with a missense change resulting in the Gly12Ser substitution, which represents the most common event underlying the disorder. As a consequence, CS is characterized by a relatively homogeneous phenotype [3]. Conversely, CFCS is genetically heterogeneous and caused by heterozygous activating mutations in the BRAF, MAP2K1, MAP2K2 and KRAS genes, leading to a more variable clinical presentation [4]. Overall, key-features affecting RASopathies include a distinctive facial appearance, postnatal growth failure, a wide spectrum of cardiac defects, variable intellectual disability, skin manifestations and different signs of musculo-skeletal involvement. The musculo-skeletal profiling of NS and NF1 syndrome has previously been outlined in the medical literature [5,6,7], while incidence rates are lacking for both CS and CFCS, possibly because their overall rarity. Indeed, available data on skeletal anomalies in CS and CFCS have been collected during the International Costello Syndrome Conferences [8,9,10,11,12,13,14], but poor information is available about the function and disability level [8, 15] or daily living activities in these patients [16].
The present study aimed to improve our general knowledge about the musculo-skeletal phenotype of CS and CFCS thorough the assessment of a single, unselected and relatively large monocentric cohort of patients, and to add information about the functional status of such patients. Moreover, we discuss on the genotype/phenotype correlations found.
Patients and methods
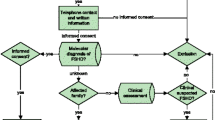
A prospective study was performed between May 2018 and January 2020. Patients with molecularly confirmed diagnosis of CS and CFCS managed in our Institution were consecutively recruited. A written informed consent was obtained from all participating individuals and families. The study was approved by the local Ethics Committee. All individuals underwent pediatric, genetic, neurological and orthopedic evaluations by physicians experienced in the management of RASopathies. Patients’ past medical history was reviewed in order to check for: (1) congenital orthopedic malformations; (2) achievement of neuro-motor developmental milestones; (3) use of orthosis; (4) physical activity; (5) congenital heart disease (CHD). Information about the cardiac status was collected to exclude CHD as a potential confounder for the poor results in functional assessments tests.
A standardized musculo-skeletal physical examination was carried out by filling in a comprehensive checklist, including 100 items (Additional file 1: Table S1). Coxa valga subluxans (CVS) was radiologically diagnosed when femoral neck-shaft angle was > 140°. Functional and disability evaluations were performed by assessing the 6-min walking test (6MWT) [17] in all patients with independent walking and by using the Pediatric Outcomes Data Collection Instrument (PODCI) [15, 18, 19]. The 6MWT was used to measure the distance covered on a flat and hard surface in a total of 6 min to monitor overtime worsening of general status and deambulation [17]. 6MWT data were normalized taking into account sex, age, body mass index and height [20, 21]. PODCI was used to assess the global (functional and psychological) profile of the orthopedic patient. Based on the global level of intellectual disability, PODCI was filled in by patients’ parents or caregivers; data on all PODCI domains (physical functions, transfer and basic mobility, sports, comfort, pain, happiness and satisfaction, global function) were collected.
Study data were analyzed with GraphPad PRISM software (version 5.1; GraphPad Software. Inc., San Diego CA, USA). Descriptive analysis was expressed as mean ± standard deviation (SD) and percentages. According to the cut-off age identified by PODCI, the whole sample was divided into two groups: children (when < 11 years), adolescents and young adults (if ≥ 11 years).
The nonparametric Mann Whitney test was used to compare orthopedic findings in CS and CFCS, PODCI scores between CS and CFCS and with normative values [19]. A p value < 0.05 was considered statistically significant.
Results
A total of 34 patients with molecularly confirmed diagnosis of CS (N = 17) and CFCS (N = 17) were recruited in the study (CS: age range 4–36 years; CFCS: age range 5–35 years); most patients were older than 8 years (15/17 with CS and 12/17 with CFCS) (Table 1). No one patient reported in the present paper was ever enrolled in previous studies about orthopedic anomalies in CS and CFCS. Prevalence of congenital malformations detected at birth was very low (hip dysplasia 3/34, 9%; clubfoot 2/34, 6%) (Table 2). All patients experienced a delay in the neuromotor development milestones, which was treated with dedicated physical therapy, requiring the use of orthosis (mostly insoles, orthopedic shoes and ankle foot orthosis or AFO) in 15 (88%) CS patients and 13 (76%) CFCS patients (Table 2). At the time of clinical evaluation, all patients with CS reached head and trunk control, and 16/17 achieved an independent walking. In contrast, 6 (35%) patients with CFCS needed some kind of support for standing upright and/or start deambulation (orthosis and/or wheelchair for long distances). Most of patients played sport and/or performed physical therapy at the time of clinical evaluation (14/17, 82% CS; 13/17, 76% CFCS) (Table 2).
All patients showed a mild CHD which did not interfere with PODCI scales and 6MWT results. Only one patient with CS and a severe form of HCM underwent a surgical intervention of miectomy at 11 years. At the time of the present study, this patient was 19-year-old and she regularly performed sport three times per week without showing any clinical symptom.
The standardized checklist to detect musculo-skeletal manifestations revealed that most patients were affected by a variable combination of deformities. The most frequent osteo-articular abnormalities are shown in Table 3. The most frequent skeletal findings in CS were scoliosis, anterior chest anomalies, ulnar deviation of fingers, elbows contractures, cubitus valgus, feet anomalies (metatarsus varus, pes planus or cavus), Achille’s tight heel cord and small joint laxity. Age of onset of the syndromic scoliosis was after 10 years in 3/5 of patients (60%), and this was slowly progressive. Conversely, 2/5 of patients (40%) showed a moderate dystrophic scoliosis started at the age of 6 that did not require any surgical intervention. Both groups were regularly followed-up with yearly monitoring of overnight saturation and pulmonary function tests, which showed no significantly abnormal results during sleeping and a mild restrictive lung disease.
Overall, patients with CS harboring the Gly12Ser variant showed a more severe skeletal phenotype compared to patients carrying other HRAS pathogenic variants (e.g., Gly12Ala and Gly13Cys).
Scoliosis in CFCS was more prevalent compared to CS. Age of onset was after 8 years in 2/6 of patients (34%) with a slow progression over the years; whereas 4/6 of patients (66%) showed an early onset-scoliosis associated with hypotonia. Three of them were treated with a corset, and they were not able to walk. Other frequent skeletal findings were pectus anomalies and pterigium colli. Ulnar deviation of fingers, elbow contractures and cubitus valgus were also present, but they were less prevalent than in CS. Hip and knees contractures and feet anomalies (mostly characterized by pes valgus and planus) were also present. In the CFCS group, patients with MAP2K1/2 pathogenic variants showed different skeletal characteristics compared to those carrying BRAF variants, with a higher prevalence of scoliosis, pectus anomalies, upper limbs anomalies, hip contractures and pes valgus compared to the BRAF variant. Statistical analysis to compare orthopedic findings detected in CS vs CFCS (full cohorts) did not reach any significance for any item. At the time of clinical evaluation, coxa valga subluxans was radiologically documented in 7/8 patients with CS (88%) and 3/11 patients with CFCS and BRAF pathogenic variant (27%). However, one patient with CS (Gly12Ser) was radiologically negative for hip subluxation at the time of recruitment, though she was surgically treated at the age of 5 for this problem. Generalized muscular hypotrophy (small and flabby muscles both in upper al lower limbs) was detected in most of patients with CS and CFCS, with higher prevalence in CS (Table 3).
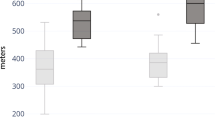
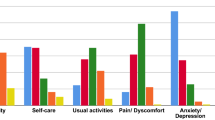
Functional and disability evaluations were performed by using the 6MWT and PODCI scale, respectively. A total of 24 patients were able to perform the 6MWT (12/17 CS and 12/17 with CFCS). Among the 5 patients with CS who did not perform the test, 1/17 did not reached independent walking at the time of clinical evaluation, 2/17 had undergone surgical interventions and they were in the post-operative period, and 2/17 patients complained of hip pain due to hip dislocation. In the CFCS subcohort, the 5 patients who were not able to perform the 6MWT showed multiple musculo-skeletal deformities (particularly, a severe scoliosis was present in 4 of them) associated with loss of walking ability and severe epilepsy. Overall, the CS and CFCS groups covered on average 49% and 46% of the expected distance, respectively. No significant differences occurred between genetic analysis, phenotypes, gender or age of assessment (Fig. 1). The average PODCI scores for CFCS (both children and adolescents/young adults) were constantly lower compared to CS population (Figs. 2, 3). Statistically significant differences between CFCS and CS patients were recorded in the following domains: upper-extremities mobility (children p = 0.04, adolescents and young adults p = 0.01), transfer and basic mobility (children p = 0.04, adolescents and young adults p = 0.03) and global functioning domains (adolescents and young adults p = 0.02). When compared to normative values, the CFCS group (both children and adolescents/young adults’ sub-groups) reached significantly lower scores in all domains, with the exception of comfort and pain in the group of children (p = 0.2). Significant differences between CS and normative values were detected in all domains, with the exception of happiness both in children (p = 0.3) and adolescents/young adults (p = 0.08) (Fig. 3). The prevalence of main orthopedic findings and the comparison with the literature data are shown in Table 4.

Six minute walk test in CS and CFCS

PODCI scores in Children (population under 11 years of age)

PODCI scores in Adolescents and young adults (population over 11 years of age)
Discussion
In the present study, the musculo-skeletal phenotype and functional status of CS and CFCS were assessed considering a single unselected and relatively large monocentric cohort of patients, and genotype/phenotype correlations were explored. We have found that orthopedic manifestations are highly prevalent in both disorders. Compared with previously published observations, the different prevalence of these features could be related to the different ages of patients recruited in the cohorts [8,9,10,11,12,13]. As demonstrated by the need of orthosis and surgical intervention, the musculo-skeletal phenotype of CS and CFCS evolves with age; for this reason, the routinely use of functional tests could help physicians to plan a timely treatment before worsening of autonomy skills.
According to the medical literature, approximately 80% of mutations in CS result in a Gly12Ser missense change, which is associated with the classic CS phenotype [3]. Based on current evidence and on our analysis, we speculate that CS patients carrying the Gly12Ser variant show a higher prevalence of orthopedic abnormalities compared to subjects with other pathogenic variants in HRAS. The Gly13Cys mutation shows a milder orthopedic phenotype. This adds further information about this cohort of patients with CS. Moreover, this finding is in line with previous observations demonstrating a milder neurodevelopmental impairment, a better growth outcome, and a lower risk for malignant tumors compared to the classical Gly12Ser variant [3, 22]. On the same way, our study documents that patients with CFCS harboring the MAP2K1/2 pathogenic variants generally show a more severe skeletal phenotype compared to other CFCS patients, with a higher prevalence of scoliosis and multiple joint contractures.
Limitations of this study were certainly the relatively small sample of patients with uncommon pathogenic variants and different ages of subjects recruited, even though most patients were older than 8 years. In order to confirm these data, we recommend future multicentric studies, including larger cohorts of patients with the less frequent HRAS and MAP2K1/2 variants.
The prevalence of congenital bone deformities detected in our sample was low; although the high prevalence of coxa valga subluxans in our CS population suggests that orthopedic phenotype may evolve in the long-term, highlighting the importance of performing hip X-ray during infancy to rule out this possible picture. In our sample, some deformities, such as pes planus and tight Achille’s heel cord, were surgically treated, and therefore some specific items could be underestimated.
Both CFCS and CS patients frequently required use of orthosis and postural/mobility devices to improve posture and facilitate acquisition of neurodevelopmental milestones. In particular, CFCS patients reported a higher need of stroller and corset, as they had a higher incidence of scoliosis and lower limb contractures (especially knee and hip contractures), as already reported in the medical literature [11]. In our cohort, these deformities showed a progressive and worsening pattern, needing surgical intervention in some cases and compromising the independent walking.
Generalized muscular hypotrophy and hypotonia were detected in most patients. Skeletal muscle hypotonia of varying degree is a universal finding in CS and CFCS, and some authors speculated that this could be related to myopathy [23]. Tidyman et al. reported muscle biopsies in a limited number of CS and CFCS patients, describing the presence of abnormal muscle fiber size and variability as one of the potential mechanisms leading to hypotonia. Due to the small number of biopsies available, the same authors concluded that since overall muscle tissue architecture was relatively intact, experiments on animal models would be essential to better define this myopathy in vivo [23]. As it has been demonstrated the vital role of the RAS/MAPK pathway in the myogenesis regulation, mainly in myoblast differentiation and proliferation [23, 24], we hypothesize that decreased muscle fiber size and hypotonia in CS and CFCS could result from inhibition of myoblast differentiation during early muscle development, and this could explain both muscle hypotrophy found in our patients and their functional status.
As expected, CS and CFCS patients showed a worse functional performance and level of disability assessed by both 6MWT and PODCI scales, compared to normative values. All patients able to walk performed 6MWT, covering less than 50% of the expected distance. Overall, the walked distance decreased proportionally to patient’s age and, in particular, patients with CFCS showed worse performances compared to CS. This finding has been also confirmed by the PODCI scale. Global functioning items between children with CS and CFCS were not significantly different, and we speculate that this could be related to the high level of global care assistance needed by children with these genetic conditions. The lower PODCI scale scores collected in CS and CFCS compared to normative data suggest how cognitive impairment, complex clinical history, neurological condition, and musculo-skeletal deformities have a great impact on the disability in RASopathies. Data on functional and disability status published by Johnson et al. detected lower scores in CS compared to CFCS [15]. Interestingly, both Johnson’s and our study did not detect a significant difference in the comfort and pain domains between syndromic patients and normative values. We believe that parents’ report could represent a crucial bias, since detection of pain in patients with intellectual disability is quite challenging [25]. On the same way, the absence of significant differences between happiness domain in CS vs normative values could be related to the friendly behavior shown by patients with CS [26].
Overall, the musculo-skeletal manifestations in CS and CFCS should be included in a more comprehensive clinical assessment, in which growth and neurodevelopmental delay have to be considered as universal features. We suggest that worsening of the neurological profile in terms of behavioral pattern and epilepsy in CFCS [27] and the abnormal posture and bone health in CS [28, 29] may contribute to the definite outcome, justifying the lower PODCI scores found in our patients compared to normatives. Results of the 6MWT and PODCI scales underscore how the routinely assessment of disability status could be useful to evaluate the functional impact of orthopedic and neurological manifestations on patients’ outcome, monitor their worsening and plan a tailored treatment of comorbidities.
The adequate classification of musculo-skeletal deformities and their overtime monitoring together with disability status assessment could represent issues to assess in the frame of future clinical trials related to CS and CFCS.
Conclusions
Previous studies focused on the prevalence of musculo-skeletal phenotype in CS and CFCS, highlighting the wide clinical variability of orthopedic malformations in these syndromes and their frequent overlap. This paper adds further information about the impact that multiple orthopedic abnormalities have on the functional status. The routinely use of both 6MWT and PODCI scale together with the orthopedic evaluation would be relevant to monitor the progression and impact of orthopedic malformations on disability along life and to plan a timely treatment (conservative or surgical). Early use of the orthosis is also suggested to reduce the progressive onset and worsening of contractures, especially in the lower limbs, and consequently reduce surgical indications. When necessary, surgical treatment should be performed as early as possible to allow minimally invasive or low impact surgical techniques. In fact, the severe progressive worsening of such deformities may lead to complex orthopedic surgery often unsustainable for these fragile patients. In this view, the precocious classification of musculo-skeletal deformities and their careful monitoring over time is of crucial relevance.
Availability of data and materials
Data available from the corresponding author on request from physicians.
Abbreviations
- CS:
-
Costello syndrome
- CFCS:
-
Cardio-facio-cutanoeus syndrome
- NS:
-
Noonan syndrome
- 6MWT:
-
6-Min walking test
- PODCI:
-
Pediatric Outcomes Data Collection Instrument
References
Tartaglia M, Gelb BD. Disorders of dysregulated signal traffic through the RAS-MAPK pathway: phenotypic spectrum and molecular mechanisms. Ann N Y Acad Sci. 2010;1214:99–121.
Tartaglia M, Gelb BD, Zenker M. Noonan syndrome and clinically related disorders. Best Pract Res Clin Endocrinol Metab. 2011;25:161–79.
Gripp KW, Morse LA, Axelrad M, Chatfield KC, Chidekel A, Dobyns W, et al. Costello syndrome: clinical phenotype, genotype, and management guidelines. Am J Med Genet A. 2019;179:1725–44.
Pierpont ME, Magoulas PL, Adi S, Kavamura MI, Neri G, Noonan J, et al. Cardio-facio cutaneous syndrome: clinical features, diagnosis, and management guidelines. Pediatrics. 2014;134:e1149–62.
Stevenson DA, Yang FC. The musculoskeletal phenotype of the RASopathies. Am J Med Genet C Semin Med Genet. 2011;157:90–103.
Rhodes SD, Zhang W, Yang D, Yang H, Chen S, Wu X, et al. Dystrophic spinal deformities in a neurofibromatosis type 1 murine model. PLoS ONE. 2015;10:e0119093.
Gutmann DH, Ferner RE, Listernick RH, Korf BR, Wolters PL, Johnson KJ. Neurofibromatosis type 1. Nat Rev Dis Primers. 2017;3:17004.
Yassir WK, Grottkau BE, Goldberg MJ. Costello syndrome: orthopaedic manifestations and functional health. J Pediatr Orthop. 2003;23:94–8.
White SM, Graham JM Jr, Kerr B, Gripp K, Weksberg R, Cytrynbaum C, et al. The adult phenotype in Costello syndrome. Am J Med Genet A. 2005;136:128–35.
Armour CM, Allanson JE. Further delineation of cardio-facio-cutaneous syndrome: clinical features of 38 individuals with proven mutations. J Med Genet. 2008;45:249–54.
Reinker KA, Stevenson DA, Tsung A. Orthopaedic conditions in Ras/MAPK related disorders. J Pediatr Orthop. 2011;31:599–605.
Stevenson DA, Allen S, Tidyman WE, Carey JC, Viskochil DH, Stevens A, et al. Peripheral muscle weakness in RASopathies. Muscle Nerve. 2012;46:394–9.
Detweiler S, Thacker MM, Hopkins E, Conway L, Gripp KW. Orthopedic manifestations and implications for individuals with Costello syndrome. Am J Med Genet Part A. 2013;161:1940–9.
Stevenson DA, Schill L, Schoyer L, Andresen BS, Bakker A, Bayrak-Toydemir P, et al. The fourth international symposium on genetic disorders of the Ras/MAPK pathway. Am J Med Genet A. 2016;170:1959.
Johnson B, Goldberg-Strassler D, Gripp K, Thacker M, Leoni C, Stevenson D. Function and disability in children with Costello syndrome and Cardiofaciocutaneous syndrome. Am J Med Genet A. 2014;167:40–4.
Axelrad ME, Schwartz DD, Katzenstein JM, Hopkins E, Gripp KW. Neurocognitive, adaptive and behavioral functioning of individuals with Costello syndrome: a review. Am J Med Genet C Semin Med Genet. 2011;157:115–22.
American Thoracic Society. ATS statement: guidelines for the six-minute walk test. Am J Respir Crit Care Med. 2002;166:111–7.
Daltroy LH, Liang MH, Fossel AH, Goldberg MJ. The POSNA pediatric musculoskeletal functional health questionnaire: report on reliability, validity, and sensitivity to change. Pediatric Outcomes Instrument Development Group. Pediatric Orthopaedic Society of North America. J Pediatr Orthop. 1998;18:561–71.
Haynes RJ, Sullivan E. The Pediatric Orthopaedic Society of North America pediatric orthopaedic functional health questionnaire: an analysis of normal. J Pediatr Orthop. 2001;21:619621.
Geiger R, Strasak A, Treml B, Gasser K, Kleinsasser A, Fischer V, et al. Six-minute walk test in children and adolescents. J Pediatr. 2007;150:395–9.
Jenkins S, Cecins N, Camarri B, Williams C, Thompson P, Eastwood P. Regression equations to predict 6-minute walk distance in middle-aged and elderly adults. Physiother Theory Pract. 2009;25:516–22.
Leoni C, Giorgio V, Onesimo R, Kuczynska E, Zampino G. Impact of Costello syndrome on growth patterns. Am J Med Genet Part A. 2020. https://doi.org/10.1002/ajmg.a.61812.
Tidyman WE, Lee HS, Rauen KA. Skeletal muscle pathology in Costello and Cardio-Facio Cutaneous Syndromes: developmental consequences of germline Ras/MAPK activation on myogenesis. Am J Med Genet Part C. 2011;157:104–14.
Jo C, Jang BG, Jo SA. MEK1 plays contrary stage-specific roles in skeletal myogenic differentiation. Cell Signal. 2009;21:1910–7.
Leoni C, Triumbari EKA, Vollono C, Onesimo R, Podagrosi M, Giorgio V, et al. Pain in individuals with RASopathies: prevalence and clinical characterization in a sample of 80 affected patients. Am J Med Genet A. 2019;179:940–7.
Alfieri P, Piccini G, Caciolo C, Perrino F, Gambardella ML, Mallardi M, et al. Behavioral profile in RASopathies. Am J Med Genet A. 2014;164:934–42.
Yoon G, Rosenberg J, Blaser S, Rauen KA. Neurological complications of cardio-facio cutaneous syndrome. Dev Med Child Neurol. 2007;49:894–9.
Dileone M, Zampino G, Profice P, Pilato F, Leoni C, Ranieri F, et al. Dystonia in Costello syndrome. Parkinsonism Relat Disord. 2012;18:798–800.
Leoni C, Stevenson DA, Martini L, De Sanctis R, Mascolo G, Pantaleoni F, et al. Decreased bone mineral density in Costello syndrome. Mol Genet Metab. 2014;111:41–5.
Acknowledgements
We would like to thank all patients and their families for consenting to participate in this study. We greatly thank the family support group (Associazione Italiana Sindromi Costello e CFC). We finally thank A. Santoro e M. Malagisi for supporting the medical and paramedical staff.
Funding
M.T. is funded by the Italian Ministry of Health (Ricerca Corrente 2019 and 2020) and European Joint Program on Rare Diseases (NSEuroNet).
Author information
Authors and Affiliations
Contributions
Dr. Leoni conceptualized and designed the study and data collection instruments, interpreted data, wrote the manuscript, and revised the manuscript for important intellectual content. Dr. Romeo analysed and interpreted data, reviewed results and drafted the article. Drs. Pelliccioni, Di Già, Tedesco, Onesimo and Giorgio collected data. Drs. Flex and Tartaglia interpreted clinical data and drafted the article. Dr. Rigante analysed data and thoroughly reviewed the study for both content and significance. Dr. Valassina conceptualized the study and reviewed the manuscript for its content. Dr. Zampino conceptualized the study and critically revised the manuscript. All authors approved the final version of this manuscript as submitted and they agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethical approval and consent to participate
The study was approved by our Institutional Ethic Committee. Parents’ patients signed a written informed consent.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Musculo-skeletal items evaluated in our sample.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Leoni, C., Romeo, D.M., Pelliccioni, M. et al. Musculo-skeletal phenotype of Costello syndrome and cardio-facio-cutaneous syndrome: insights on the functional assessment status. Orphanet J Rare Dis 16, 43 (2021). https://doi.org/10.1186/s13023-021-01674-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-021-01674-y