
Abstract
Background
Female-limited early-onset high myopia, also called Myopia-26 is a rare monogenic disorder characterized by severe short sightedness starting in early childhood and progressing to blindness potentially by the middle ages. Despite the X-linked locus of the mutated ARR3 gene, the disease paradoxically affects females only, with males being asymptomatic carriers. Previously, this disease has only been observed in Asian families and has not gone through detailed investigation concerning collateral symptoms or pathogenesis.
Results
We found a large Hungarian family displaying female-limited early-onset high myopia. Whole exome sequencing of two individuals identified a novel nonsense mutation (c.214C>T, p.Arg72*) in the ARR3 gene. We carried out basic ophthalmological testing for 18 family members, as well as detailed ophthalmological examination (intraocular pressure, axial length, fundus appearance, optical coherence tomography, visual field- testing) as well as colour vision- and electrophysiology tests (standard and multifocal electroretinography, pattern electroretinography and visual evoked potentials) for eight individuals. Ophthalmological examinations did not reveal any signs of cone dystrophy as opposed to animal models. Electrophysiology and colour vision tests similarly did not evidence a general cone system alteration, rather a central macular dysfunction affecting both the inner and outer (postreceptoral and receptoral) retinal structures in all patients with ARR3 mutation.
Conclusions
This is the first description of a Caucasian family displaying Myopia-26. We present two hypotheses that could potentially explain the pathomechanism of this disease.
Similar content being viewed by others


Background
Myopia or short-sightedness has become a serious world health issue recently [1]. This can be attributed to its extreme phenotypes on the „upper end of the scale”, namely high and pathologic myopia. Cases of high myopia with a rapid progression carry the risk of advancing into pathologic myopia, a condition that is associated with potentially blinding complications. There is an explicit increase in the prevalence of these conditions lately, therefore an urgent need for targeted treatments is recognized [1, 2]. To devise such treatment options however, we need to thoroughly understand the exact molecular mechanisms of refractive errors and myopia development. Albeit nearly 270 genes associated with myopia have been identified so far, the underlying pathways through which these genes influence refractive error development remain obscure in most of the cases [3].
Inheritance of late onset or common myopia and early onset high myopia (eoHM) was evidenced to differ basically yet earlier [4]. As opposed to common forms, eoHM is predominantly inherited in a Mendelian manner with one single causative, highly penetrant gene mutation, practically with minimal influence of environment or behaviour. The specific mode of inheritance of such diseases covers a wide range of forms including autosomal dominant, autosomal recessive or X-linked recessive [5]. One of the most curious and exceptional modes of transmission is that seen for Myopia-26, displaying X-linked dominant inheritance. This rare disease, described earlier only in three Asian families paradoxically affects females only, with male hemizygotes being asymptomatic (emmetropic) carriers [6]. The ARR3 gene, residing on the X-chromosome and encoding the cone-arrestin was found to be mutated in all affected patients. Associated symptoms were not reported for those cases, neither was a potential mechanism of pathogenesis provided.
Today, the general pathomechanism of refractive error development is assumed to be based on a retina-to-sclera signalling cascade guided locally by light stimuli in the retina [7]. All retinal cell types seem to participate in this retina-specific signal transduction and derailment of retinal cell physiology and light processing are the key mechanisms [3]. However, only recent advances allowed for deeper insight into the genetic background of these processes. There is still much to be discovered in this field, especially concerning the specific role of the mutated genes in pathogenesis to imply further treatment potentials. Promising is the fact that despite their different manners of inheritance, there is an overlap between eoHM and common myopia in both causative genes and pathways of pathogenesis [3].
In our study we investigated a large family of five generations displaying female-limited eoHM. Whole exome sequencing identified an early stop codon within the ARR3 gene, verifying the diagnosis of Myopia-26. In order to explore the clinical phenotype of this disease further, we accomplished thorough ophthalmological and electrophysiological testing. Electrophysiology test results altogether suggested a central macular retinal ganglion cell deficit besides the photoreceptoral disturbance, and permitted the formulation of the ganglion-cell hypothesis to explain the development of myopia, in addition to the hypothesis based on the cone-arrestin defect.
Results
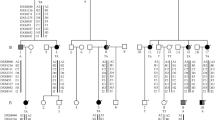
In the course of our routine ophthalmological work, we found multiple interrelated patients displaying eoHM. Precisely recording the personal and familial medical histories of the patients allowed the compilation of their pedigree (Fig. 1). This revealed a family of five generations comprising numerous affected patients, all of whom are females. Assuming a monogenic trait, this pattern seemed to be indicative of X-linked heredity where the mutant allele is dominant in females, but has no penetrance in males, i.e. it is female limited. We found only a single paper describing such transmission of eoHM, referred to as Myopia-26. All three reported families belonged to the East Asian ethnicity [6].

The pedigree of the investigated family. Dark shading indicates an eoHM phenotype. Dashed circles mark patients whose blood samples were obtained, the two arrows mark the two samples that went through whole exome sequencing
To identify the causative mutation, DNA prepared from the blood samples of patients III/3 and V/8 (a male carrier and a symptomatic female, respectively) were submitted to whole exome sequencing. We identified the same variant (NM_004312.2:c.214C>T NP_004303.2:p.Arg72Ter) in the X chromosome-based ARR3 gene in both individuals in hemizygous and heterozygous form, respectively. The presence of this candidate pathogenic variant was confirmed by conventional PCR amplification and Sanger sequencing as well. Segregation of this change with the disease was assessed for all available family members. We confirmed the presence of this nonsense variant in heterozygous state in all available symptomatic female members of the family (II/1, II/3, III/8, III/13, IV/1, IV/2, IV/6, IV/7, IV/10 and IV/18). We have also confirmed the absence of this ARR3 variant from all studied asymptomatic females (IV/4, IV/13, IV/14, IV/17 and V/5). Patient V/6, a healthy male was found to carry the wild type allele. To date, this variant has not been described in the Human Gene Mutation Database, the Exome Aggregation Consortium, the Exome Sequencing Project, ClinVar or the 1000 Genome Browser. Prediction programs Polyphen2, SIFT, and MutationTaster predicted pathogenicity of the nonsense variant. Overall, these results confirmed the diagnosis of Myopia-26.
Next, eight of our patients were exposed to a more thorough examination. Medical history revealed no other notable systemic or ophthalmological disorders relevant for this matter. The gender, age, best corrected visual acuities (BCVA), spherical equivalents (SE), intraocular pressures (IOP), axial lengths (available for patients who went through scleral reinforcement surgery), fundus appearance (classified according to the META-PM study [8]), OCT-, visual field and colour vision test results of these patients are shown in Table 1. Examples of our findings are shown in Fig. 2 and Additional file 1: Figures S2–S21.

a Ultra widefield (Optos® California) fundus image of the right eye of affected female patient IV/6 displaying a META-PM2 stage myopic fundus. The tesselated appearence of the retina along with peripapillary and diffuse chorioretinal atrophy is observable. b Macular OCT image of the right eye affected female IV/6 displaying thinner (incipient atrophic) sensory retina and posterior vitreous detachment characteristic of higher degrees of myopia. c Visual field of the right eye of affected female IV/6 (nasal loss + superior artefact)
Numerical values extracted from the electrophysiological test results are shown in Additional file 3: Tables S1, S2 and S3 of the Supplementary text. Examples of standard full-field electroretinography (ERG) recordings are shown in Fig. 3, pattern electroretinography (PERG) in Fig. 4, pattern visual evoked potentials (pVEP) in Fig. 5, and multifocal electroretinography (mfERG) in Fig. 6. All remaining recordings are available in Additional file 2: Figures S22–S55.

Normal photopic 3.0 ERGs in affected female IV/7. Despite prominent phenotypic signs of eoHM (SE: − 13.0/ − 9.0D, impaired BCVA, high myopic fundus alterations) in IV/7 individual, photopic 3.0 ERGs show no alterations, reflecting an overall normally functioning cone system

a Pattern ERG of carrier male III/3 is heavily affected. Despite no phenotypic sign of eoHM and visual impairment, pattern ERG of the carrier male patient is similarly subnormal as those of affected female patients. b Heavily affected PERG recordings of affected female IV/7. c Pattern ERG of unaffected male V/6. Physiological wave patterns are detected. In all sections, lines 1 and 3 and lines 2 and 4 represent pairs of replicate measurements

a Pattern VEP recordings of patient III/3 demonstrating increased implicit times and decreased amplitudes of P100 for 15′ (smaller checks) stimulation as compared to normal control. b Heavily affected pVEP recordings of affected female IV/7 demonstrating increased peak times and decreased amplitudes of P100. c Normal pattern VEP recordings of unaffected male V/6 (Note the change of the voltage scale). In all sections, lines 1 and 3 display responses to 60′ stimuli and lines 2 and 4 represent responses to 15′ stimuli

a MfERG recording of carrier male III/3, raw waveform. b MfERG recording and ring analysis of carrier male III/3
Some points of note:
-
1.
Fundus, OCT and visual field alterations showed no characteristics of cone dystrophy, such as „bull’s eye” appearance on the central fundus, outer retinal changes with OCT or a central scotoma with visual field testing. Rather they were characteristic of high myopia: META-PM1-2 fundus appearance (See Additional file 3: Supplementary text and Additional file 1) and thinner or incipient atrophic sensory retina on macular OCT scans (Fig. 2).
-
2.
Electrophysiology test results overall indicated a macular dysfunction in our patients with ARR3 mutation apparently affecting both the inner and outer retinal structures of the central retina (Figs. 3, 4, 5, 6), as opposed to a generalized cone dysfunction expected based on X-arrestin knockout animal models [9]. These electrophysiological alterations (detailed in the Additional file 3: Supplementary text) were detected in all patients with ARR3 mutation irrespective of their affected or carrier genetic status, and at the same time showed no correlation with either the VA, SE or the age of the patients. Accordingly, these alterations are most likely attributable to the genetic defect itself, and are not secondary consequences of the high myopic refractive error.
-
3.
Colour vision test results revealed a diffuse colour vision discrimination error with no specific axis in our patients tested with the Lanthony Desaturated D-15-hue Panel test. This is again consistent with the central macular deficit suggested by the electrophysiology tests of our patients (see Additional file 3: Supplementary text).
-
4.
Despite the fact that the possibility of an association of POAG with high myopia in our patients arose (detailed in the Additional file 3: Supplementary text), available data do not provide sufficient and inarguable evidence to support the diagnosis of POAG at present. Long- term follow-up will be necessary to reveal any evidence of potential progression of these parameters that could also be expected in glaucoma.
Discussion
In this study, we report a family displaying a heritable form of eoHM, where the disease is manifested only in females. Compilation of the pedigree permitted the identification of carrier males, and revealed that their female offspring are exclusively affected, which suggested an X-linked dominant, female-limited inheritance. Whole exome sequencing of two individuals indeed revealed a nonsense-mutation within the coding region of a gene on the X-chromosome, namely ARR3. Sanger sequencing of the respective locus in a total of 16 female family members unveiled a perfect correlation between the presence of the mutant allele and the high myopia phenotype. This is the first report of a mutation in ARR3 causing hereditary eoHM, called Myopia-26 in a Caucasian family. Three Chinese families have been reported earlier to display a similar, X-linked dominant, female-limited transmission of eoHM [6]. In those cases the ARR3 was found to carry c.893C>A (p.Ala298Asp), c.298C>T (p.Arg100*) and c.239T>C (p.Leu80Pro) mutations, respectively. The mutant allele identified in our study (c.214C>T, p.Arg72*) is therefore novel. The earlier publication on Myopia-26 lacked a detailed phenotypic description of the patients, and did not attempt to explain the pathomechanism of the disease. Our main goals from this point onwards were therefore to carry out a thorough ophthalmologic investigation of the family and use the acquired information, along with literature data to build reasonable hypotheses on the molecular mechanism of pathogenesis.
ARR3 encodes a 388 amino acid-long visual arrestin with multiple names (Arrestin 3, Arrestin 4, Cone-arrestin, Retinal cone arrestin-3, X-arrestin), we refer to it as X-arrestin. Besides its key role in the phototransduction process in retinal cones, it is also expressed in pinealocytes of the pineal gland [10]. Arrestins make up an important family of proteins, with the primary function of desensitizing phosphorylated G-protein coupled receptors (GPCRs). Arrestin 1 and X-arrestin bind to opsins (hence called visual arrestins), while β-arrestin 1 and 2 bind to numerous other types of GPCRs. Arrestin 1 has very high preference for opsins found in retinal rods and cones, whereas X-arrestin has a fairly high binding capacity to non-opsin binding partners as well, and therefore has more diverse synaptic roles [11].
Our knowledge about the function and cell type-specific expression of X-arrestin is, at this time based mostly on experimental data derived from animal models. X-arrestin is expressed in all cone types of the human retina [12], however it displays a weaker expression in the S-cones of mice [13]. Arrestin 1, on the other hand is detectable in rods and S-cones of baboons, but not in LM cones [14]. In the cones of knockout mice, Arrestin-1 seems to provide a functional replacement for X-arrestin [15]. This experimental dataset allows us to formulate two reasonable, albeit incomplete hypotheses on the pathogenesis of myopia in ARR3-mutant patients. We refer to these as the cone- and the ganglion cell-hypothesis, respectively. The cone-hypothesis assumes that Arrestin-1 expression in humans is present in S-cones, but not in LM cones, as seen in baboons [14], so an X-arrestin defect would lead to limited arrestin function in LM, but not in S cones. Since arrestins are responsible for the desensitization of opsins, decreased arrestin function in LM-cones would mean their increased activity, and the “sensitization” to red/green visual stimuli. Such selective cone dysfunction could explain the onset of myopia the following way. The physical phenomenon of chromatic aberration leads to shorter wavelengths forming an image in a more anterior, and longer wavelengths forming an image in a more posterior plane (Figure S1A). Normally, the measure of luminance contrast is maximized during accommodation, and long-wavelengths form an image behind the photoreceptors. In patients with a relatively increased sensitivity of L-cones, the posterior image will produce a stronger stimulus (Figure S1B). As a result, a higher luminance contrast will be attained upon increased accommodation and by ocular elongation, two hallmarks of myopia pathogenesis [16]. Although accommodation excess in itself may not be sufficient to cause myopia [17], the phenomenon of image-forming behind the retina, called hyperopic defocus has been shown to provoke ocular elongation in numerous animal studies [18, 19]. Briefly, since blue light is claimed to have a protective effect against myopia, the relative weakening of the blue light stimulus upon the loss of X-arrestin can explain the eventual development of myopia in these patients [20].
The selectively altered function of various cone types, however, cannot be tested with standard photopic 3.0 ERGs. Due to the quite extensively overlapping spectral sensitivities of different photopigments [21], these tests reflect the summed activity of all three retinal cone types. Photopic 3.0 ERGs indeed, were normal and showed no alteration in our patients (Fig. 3). L, M and S-cones responses can be isolated electrophysiologically by recording the light adapted ON/OFF-ERG and the S-cone ERG. Similar to the PhNR, these recordings are an extension of the full-field ERG [22] which enable characterisation of the different cone types, including bipolar cell interactions.
Our ganglion cell-hypothesis attributes the development of refractive error to the dysfunction of retinal ganglion cells (RGC). To better understand this connection, one must acknowledge that apart from their primary role of transmitting visual information from photoreceptors to higher cerebral visual centres, a subset of RGCs called intrinsically photosensitive retinal ganglion cells (ipRGCs) have an additional role [23]. As their name suggests, they can detect light directly through their photosensitive protein called melanopsin. At the same time, they also transduce the signal originating from rod and cone photoreceptor cells, analogously to classical RGCs [24]. Classical and ipRGCs are interconnected horizontally by amacrine cells, which allow them to influence the activity of one another [25]. IpRGCs and their light sensitive protein, melanopsin are primarily responsible for non-image forming visual functions such as circadian rhythms or pupil reactions [26,27,28]. They have recently been discovered to play a role in conscious, image-forming visual perception as well [27]. Eye development is connected to both image-forming and non-image forming light detection pathways and accordingly refractive error may be a consequence of the derailment of either.
There is an increasing body of evidence supporting that in the image-forming pathway, light plays a key role in emmetropization and refractive error development, and besides the intensity, the spectral composition of the light stimulus is just as crucial [29, 30]. As opposed to opsins, melanopsin is most sensitive to shorter wavelengths of the spectrum, i.e. blue light [31]. Besides the anti-myopic effect of blue light attributed to the myopic defocus it causes on the retina (discussed above) [20], it has a further protective effect mediated in part by dopamine through pre- and postsynaptic connections of ipRGCs to dopaminerg amacrin cells [32]. Dopamine has been long acknowledged as a retinal neurotransmitter acting against myopia development, and it has also been evidenced that blue light stimulates a larger amount of dopamine release than other wavelengths do [32]. Accordingly, a disruption of ipRGC function may result in the alteration of the wavelength composition of the perceived light with a chromatic aberration shifted towards longer wavelengths of the spectrum, along with decreased dopaminergic activity. Both issues reduce the protective effect of blue light against myopia, potentially leading to the development of a progressive refractive error.
The non-image forming visual functions of ipRGCs, such as circadian rhythm photoentrainment also play an important role in eye development [33]. IpRGCs and melanopsin mediate circadian cycles both endogenously in the retina (again, through dopamine release) and via a systemic route comprising the hypothalamic suprachiasmatic nucleus (SCN) and the pineal gland through the inhibition of melatonin release in pinealocytes [33]. The circadian clock influences ocular development, and disruption of the circadian cycle has been found to elongate eye components and yield myopia in various myopia models [34]. Therefore, either the primary defect of ipRGCs or the primary dysfunction of pinealocytes (or both) could cause the refractive error seen in our patients. Although the prior is difficult to explain (discussed below), the latter (pineal malfunction) is highly probable due to the fact that pinealocytes normally express the X-arrestin. Melatonin, the product of pinealocytes has been shown to inhibit retinal dopamine synthesis [35], modulate D2 dopamine-receptor expression in the retina of chicks [36] and abolish diurnal cycling of dopamine levels in goldfish retina [37]. These observations could strongly support the possibility that pinealocyte malfunction caused by ARR3 mutations lead to altered (probably increased) melatonin levels, which in turn cause myopia by impairing the diurnal rhythms of the eye.
Currently, the most obviously missing piece of both the cone- and the ganglion cell-hypothesis is the cause of RGC dysfunction displayed on the PERG recordings. Direct linkage to the ARR3 mutation would require ARR3 expression in RGCs, which was not detectable in mice [15]. However, the promoter of the human ARR3 and its murine orthologue are markedly different, which may result in disparate cell type specific expression as well [11]. Another possibility would be the secondary malfunction of RGCs, resulting from the altered activity of pinealocytes. This could be mediated by the humoral control of retinal dopaminerg transmission by the pineal gland (described above), or the direct effect of melatonin on RGCs via their MT1 and MT2 melatonin receptors [38]. The details of this control are currently missing, it is nevertheless noteworthy that myopes have higher melatonin levels than non-myopes [39]. Finally, altered cone function, resulting from reduced X-arrestin levels may also negatively influence RGC activity. We nevertheless have no reason to believe that the cone- and the ganglion cell hypotheses are mutually exclusive, or exclude other pathomechanisms.
Another major shortcoming of both the cone- and the ganglion cell hypothesis is the lack of explanation for the female-limited heredity pattern of myopia. It is especially curious that the central macular dysfunction seems to be present also in males, without leading to eoHM. We assume the presence of a “rescue mechanism” in males, or in other words, the lack of a pathological process that would lead to an axial length elongation in response to the central retinal dysfunction. Sex-dependent differences in retina function have been described in mice, and the risk of certain retinal diseases have been shown to be sex hormone-dependent in humans [40]. Further physiology and molecular biology studies are required however to unveil the exact mechanisms responsible for the observed female-limited phenotype. Such research may also shed light on why the mutant allele is dominant in females. In the course of molecular studies however, the limitations of animal models must always be kept in mind, despite their great value. For example, an age related cone dystrophy was suggested in Arr4−/− mice (Arr4 being the murine orthologue of ARR3) based on immune-histochemical findings and the pronounced diminishment in photopic flash and flicker ERGs [9]. In contrast, no generalized cone dysfunction could be evidenced in our patients carrying ARR3 mutation, either male or female, according to the electrophysiological and ophthalmological phenotypic characterization.
From the clinical point of view, our next investigative steps seem well defined: i) cone-specific ERGs (S-cone ERGs and ON/OFF ERGs) to isolate individual (L, M, or S) cone responses [41] and thus support or exclude our selective cone dysfunction hypothesis; ii) post-illumination pupil response (PIPR) to test melanopsin expressing ipRGC function [21] and thus shed light on the extent of ipRGC damage. iii) long-term follow-up of the progression of a potential POAG monitoring IOPs, visual field defects, optic nerve head appearances and RNFL OCTs.
Conclusions
Using whole exome sequencing, we identified the pathogenic mutation of the female-limited early onset high myopia observed in our patients to be a premature stop codon in the ARR3 gene. This illustrates that contrary to its current classification [42], female-limited eoHM, also referred to as Myopia-26 is not limited to the East Asian ethnicity.
Methods
Patients and ethical approval
In our genetic study of eoHM we investigated a five-generation family displaying numerous affected individuals in each generation. Blood samples were taken from 18 family members representing four generations, eight of whom went through comprehensive ophthalmological and electrophysiological testing. Written informed consent was obtained from all individual participants included in the study. This study was approved by the National Scientific and Research Ethics Committee of the Medical Research Council of Hungary (ETT TUKEB, registration number 58542-1/2017/EKU). All procedures performed in studies involving human participants were in accordance with the ethical standards of the National Scientific and Research Ethics Committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Genetic analyses
Whole exome sequencing (WES) of two family members (asymptomatic male III/3, and symptomatic female V/8) was performed. Human genomic DNA was prepared from blood samples using the MagCore Genomic Whole Blood Kit (RBC Bioscience, New Taipei City, Taiwan), according to manufacturer’s instructions. Genomic capture was carried out with SureSelect XT Human All Exon + UTRs v.5 Exome Kit (Agilent, Santa Clara, CA). Massively parallel sequencing was done using NextSeq500 Sequencer (Illumina, San Diego, CA) in combination with the NextSeq™ 500 High Output Kit (1 × 150 bp). Raw sequence data analyses, including base calling, de-multiplexing, alignment to the hg19 human reference genome (Genome Reference Consortium GRCh37), and variant calling, were performed using an in-house bioinformatics pipeline. For variant filtration, all disease-causing variants reported in HGMD®, ClinVar, or in CentoMD® as well as all variants with minor allele frequency (MAF) of less than 1% in ExAc database were considered. Variants that possibly impair the protein sequence, i.e., disruption of conserved splice sites, missense, nonsense, read-throughs, or small insertions/deletions, were prioritized. All relevant inheritance patterns were considered. The candidate pathogenic mutation (NM_004312.2:c.214C>T NP_004303.2:p.Arg72Ter) was verified by PCR amplification and Sanger sequencing for both individuals. Next, the same was done to test for the presence of this allele in all remaining DNA samples obtained from the family. The predicted pathogenicity of the variant identified in this study was tested with Polyphen2, SIFT, and MutationTaster.
Clinical investigation
Clinical assessment included comprehensive ophthalmological examination and electrophysiological testing. Patients’ own and family medical history was registered regarding other ophthalmological disorders than eoHM as well as any systemic diseases. Best corrected visual acuity (BCVA) was recorded (Snellen chart) and refractive error expressed as spherical equivalent (SE). High myopia was specified as SE > − 6.0 dioptres (D) on at least one of the eyes. Slit lamp biomicroscopy with applantion tonometry and fundus ophthalmoscopy in mydriasis was carried out (Topcon SL-D701, Topcon, Tokyo, Japan). Digital fundus photography (TRC-501X; Topcon, Tokyo, Japan) and in some cases also ultra-wide field (200°) fundus images (Optos® California, Optos, Marlborough, MA) were taken. Spectral domain optical coherence tomography (macular scan) (Heidelberg Engineering, Heidelberg, Germany) was performed where possible. Axial length measurements were executed with an optical biometry system (IOLMaster 700, Carl Zeiss, Jena, Germany). Automated kinetic full-field perimetry was carried out with Humphrey Field Analyzer (Carl Zeiss Meditec, Jena, Germany).
Electrophysiology
Pattern visual evoked potentials (VEPs), pattern-, standard full-field- and multifocal electroretinography (ERG) were carried out. All electrophysiology tests were performed according to the ISCEV standards [43,44,45,46] and using the Roland Electrophysiological Test Unit with the RETIport 32 software (Roland Consult, Brandenburg a.d. Havel, Germany). Please see the Additional file 3: Supplementary text for more details.
Colour vision testing
Colour vision deficiencies were assessed using the Lanthony Desaturated D-15-hue Panel tests where possible and the Isihara pseudoisochromatic plates (Isihara 24 plates edition, 2006) in the rest of the cases.
Availability of data and materials
The sequencing data used and analysed during the current study are available from the corresponding author on reasonable request. All other data generated or analysed during this study are included in this published article and its supplementary information files.
Abbreviations
- BCVA:
-
Best corrected visual acuity
- eoHM:
-
Early onset high myopia
- ERG:
-
Electroretinography
- IOP:
-
Intraocular pressure
- GPCR:
-
G-protein coupled receptor
- ipRGC:
-
Intrinsically photosensitive retinal ganglion cell
- mfERG:
-
Multifocal electroretinography
- OCT:
-
Optical coherence tomography
- ONH:
-
Optic nerve head
- PERG:
-
Pattern electroretinography
- POAG:
-
Primary open angle glaucoma
- RGC:
-
Retinal ganglion cell
- SE:
-
Spherical equivalent
- VA:
-
Visual acuity
- VEP:
-
Visual evoked potentials
- WES:
-
Whole exome sequencing
References
Holden BA, Fricke TR, Wilson DA, Jong M, Naidoo KS, Sankaridurg P, et al. Global prevalence of myopia and high myopia and temporal trends from 2000 through 2050. Ophthalmology. 2016;123(5):1036–42.
McFadden SA. Understanding and treating myopia: what more we need to know and future research priorities. Optom Vis Sci. 2016;93(9):1061–3.
Tedja MS, Haarman AEG, Meester-Smoor MA, Kaprio J, Mackey DA, Guggenheim JA, et al. IMI—myopia genetics report. Invest Ophthalmol Vis Sci. 2019;60(3):M89–105.
Guggenheim JA, Kirov G, Hodson SA. The heritability of high myopia: a reanalysis of Goldschmidt’s data. J Med Genet. 2000;37(3):227–31.
Zhang Q. Genetics of refraction and myopia. In: Hejtmancik JF, Nickerson JM, editors. Molecular biology of eye disease, vol. 134. London: Academic Press; 2015. p. 269–79.
Xiao X, Li S, Jia X, Guo X, Zhang Q. X-linked heterozygous mutations in ARR3 cause female-limited early onset high myopia. Mol Vis. 2016;22:1257–66.
Tedja MS, Wojciechowski R, Hysi PG, Eriksson N, Furlotte NA, Verhoeven VJM, et al. Genome-wide association meta-analysis highlights light-induced signaling as a driver for refractive error. Nat Genet. 2018;50(6):834–48.
Ohno-Matsui K, Kawasaki R, Jonas JB, Cheung CM, Saw SM, Verhoeven VJ, et al. International photographic classification and grading system for myopic maculopathy. Am J Ophthalmol. 2015;159(5):877-83e7.
Deming JD, Pak JS, Brown BM, Kim MK, Aung MH, Eom YS, et al. Visual cone arrestin 4 contributes to visual function and cone health. Invest Ophthalmol Vis Sci. 2015;56(9):5407–16.
Smith WC. Chapter ten: the role of arrestins in visual and disease processes of the eye. In: Luttrell LM, editor. Progress in molecular biology and translational science, vol. 118. Heidelberg: Academic Press; 2013. p. 243–65.
Craft CM, Deming JD. Cone arrestin: deciphering the structure and functions of arrestin 4 in vision. Handb Exp Pharmacol. 2014;219:117–31.
Zhang Y, Li A, Zhu X, Wong CH, Brown B, Craft CM. Cone arrestin expression and induction in retinoblastoma cells. In: Anderson RE, LaVail MM, Hollyfield JG, editors. New insights into retinal degenerative diseases. London: Kluwer Academic/Plenum Publishers; 2001. p. 309–19.
Haverkamp S, Wassle H, Duebel J, Kuner T, Augustine GJ, Feng G, et al. The primordial, blue-cone color system of the mouse retina. J Neurosci. 2005;25(22):5438–45.
Nir I, Ransom N. S-antigen in rods and cones of the primate retina: different labeling patterns are revealed with antibodies directed against specific domains in the molecule. J Histochem Cytochem. 1992;40(3):343–52.
Nikonov SS, Brown BM, Davis JA, Zuniga FI, Bragin A, Pugh EN, et al. Mouse cones require an arrestin for normal inactivation of phototransduction. Neuron. 2008;59(3):462–74.
Rucker FJ, Kruger PB. Cone contributions to signals for accommodation and the relationship to refractive error. Vis Res. 2006;46(19):3079–89.
Mutti DO, Zadnik K. Has near work’s star fallen? Optom Vis Sci. 2009;86(2):76–8.
Smith EL 3rd, Hung LF. The role of optical defocus in regulating refractive development in infant monkeys. Vis Res. 1999;39(8):1415–35.
Wildsoet C, Wallman J. Choroidal and scleral mechanisms of compensation for spectacle lenses in chicks. Vis Res. 1995;35(9):1175–94.
Rucker F, Britton S, Spatcher M, Hanowsky S. Blue light protects against temporal frequency sensitive refractive changes. Invest Ophthalmol Vis Sci. 2015;56(10):6121–31.
Spitschan M, Woelders T. The method of silent substitution for examining melanopsin contributions to pupil control. Front Neurol. 2018;9:941.
Sustar M, Holder GE, Kremers J, Barnes CS, Lei B, Khan NW, et al. ISCEV extended protocol for the photopic On-Off ERG. Doc Ophthalmol. 2018;136(3):199–206.
Do MT, Yau KW. Intrinsically photosensitive retinal ganglion cells. Physiol Rev. 2010;90(4):1547–81.
Graham DM, Wong KY. Melanopsin-expressing, intrinsically photosensitive retinal ganglion cells (ipRGCs). In: Kolb H, Fernandez E, Nelson R, editors. Webvision: the organization of the retina and visual system. Salt Lake City: John Moran Eye Center, University of Utah; 1995.
Vuong HE, Hardi CN, Barnes S, Brecha NC. Parallel inhibition of dopamine amacrine cells and intrinsically photosensitive retinal ganglion cells in a non-image-forming visual circuit of the mouse retina. J Neurosci. 2015;35(48):15955–70.
Berson DM. Strange vision: ganglion cells as circadian photoreceptors. Trends Neurosci. 2003;26(6):314–20.
Ecker JL, Dumitrescu ON, Wong KY, Alam NM, Chen SK, LeGates T, et al. Melanopsin-expressing retinal ganglion-cell photoreceptors: cellular diversity and role in pattern vision. Neuron. 2010;67(1):49–60.
Zaidi FH, Hull JT, Peirson SN, Wulff K, Aeschbach D, Gooley JJ, et al. Short-wavelength light sensitivity of circadian, pupillary, and visual awareness in humans lacking an outer retina. Curr Biol. 2007;17(24):2122–8.
Merle BM, Silver RE, Rosner B, Seddon JM. Dietary folate, B vitamins, genetic susceptibility and progression to advanced nonexudative age-related macular degeneration with geographic atrophy: a prospective cohort study. Am J Clin Nutr. 2016;103(4):1135–44.
Troilo D, Smith EL III, Nickla DL, Ashby R, Tkatchenko AV, Ostrin LA, et al. IMI—report on experimental models of emmetropization and myopia. Invest Ophthalmol Vis Sci. 2019;60(3):M31–88.
Lockley SW, Brainard GC, Czeisler CA. High sensitivity of the human circadian melatonin rhythm to resetting by short wavelength light. J Clin Endocrinol Metab. 2003;88(9):4502–5.
Wang M, Schaeffel F, Jiang B, Feldkaemper M. Effects of light of different spectral composition on refractive development and retinal dopamine in chicks. Invest Ophthalmol Vis Sci. 2018;59(11):4413–24.
Chakraborty R, Ostrin LA, Nickla DL, Iuvone PM, Pardue MT, Stone RA. Circadian rhythms, refractive development, and myopia. Ophthalmic Physiol Opt. 2018;38(3):217–45.
Stone RA, McGlinn AM, Chakraborty R, Lee DC, Yang V, Elmasri A, et al. Altered ocular parameters from circadian clock gene disruptions. PLoS ONE. 2019;14(6):e0217111.
Dubocovich ML. N-Acetyltryptamine antagonizes the melatonin-induced inhibition of [3H]dopamine release from retina. Eur J Pharmacol. 1984;105(1–2):193–4.
Ohngemach S, Feldkaemper M, Schaeffel F. Pineal control of the dopamine D2-receptor gene and dopamine release in the retina of the chicken and their possible relation to growth rhythms of the eye. J Pineal Res. 2001;31(2):145–54.
Ribelayga C, Wang Y, Mangel SC. A circadian clock in the fish retina regulates dopamine release via activation of melatonin receptors. J Physiol. 2004;554(Pt 2):467–82.
Huang H, Wang Z, Weng SJ, Sun XH, Yang XL. Neuromodulatory role of melatonin in retinal information processing. Prog Retin Eye Res. 2013;32:64–87.
Kearney S, O’Donoghue L, Pourshahidi LK, Cobice D, Saunders KJ. Myopes have significantly higher serum melatonin concentrations than non-myopes. Ophthalmic Physiol Opt. 2017;37(5):557–67.
Nuzzi R, Scalabrin S, Becco A, Panzica G. Gonadal hormones and retinal disorders: a review. Front Endocrinol (Lausanne). 2018;9:66.
Perlman I, Kondo M, Chelva E, Robson AG, Holder GE. ISCEV extended protocol for the S-cone ERG. Doc Ophthalmol. 2019;140:95–101.
Cai XB, Shen SR, Chen DF, Zhang Q, Jin ZB. An overview of myopia genetics. Exp Eye Res. 2019;188:107778.
McCulloch DL, Marmor MF, Brigell MG, Hamilton R, Holder GE, Tzekov R, et al. ISCEV Standard for full-field clinical electroretinography (2015 update). Doc Ophthalmol. 2015;130(1):1–12.
Hood DC, Bach M, Brigell M, Keating D, Kondo M, Lyons JS, et al. ISCEV standard for clinical multifocal electroretinography (mfERG) (2011 edition). Doc Ophthalmol. 2012;124(1):1–13.
Odom JV, Bach M, Brigell M, Holder GE, McCulloch DL, Tormene AP, et al. ISCEV standard for clinical visual evoked potentials (2009 update). Doc Ophthalmol. 2010;120(1):111–9.
Bach M, Brigell MG, Hawlina M, Holder GE, Johnson MA, McCulloch DL, et al. ISCEV standard for clinical pattern electroretinography (PERG): 2012 update. Doc Ophthalmol. 2013;126(1):1–7.
Acknowledgements
The authors thank Márta Széll for providing access to the Gene Bank of the University of Szeged, Gabriella Örsy for supporting the field work and Ibolya Lakatos for the help in compiling the pedigree.
Funding
This work was supported by the National Research, Development, and Innovation Office of Hungary (NKFIH) Grant No. K119298 (to TF) and the GINOP-2.3.2-15-2016-00001 (to TF). The funding bodies played no role in the design of the study, the collection, analysis, and interpretation of data or in writing the manuscript.
Author information
Authors and Affiliations
Contributions
NS discovered the patients and carried out their ophthalmologic investigation under the supervision of ZS, NS and TF acquired ethical approval and took the blood samples, ZM, TK, and IN designed the genetic analysis, DL and IN carried out DNA preparation and next generation sequencing, ZM and TK carried out sequence analysis, ZZO and MJ was responsible for the electrophysiology, AF coordinated the work and provided the institutional background, NS and TF drafted the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Written informed consent was obtained from all individual participants included in the study. This study was approved by the National Scientific and Research Ethics Committee of the Medical Research Council of Hungary (ETT TUKEB, registration number 58542-1/2017/EKU). All procedures performed in studies involving human participants were in accordance with the ethical standards of the National Scientific and Research Ethics Committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Consent for publication
Not applicable.
Competing interests
DL and IN are employees of Seqomics Biotechnologies Ltd. IN is an investor of Seqomics Biotechnologies Ltd.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Figure S1. The cone hypothesis. Figures S2–S21. Fundus images, macular OCTs, RNFLs and visual fields of patients III/8, IV/1, IV/2, IV/6, IV/7 and IV/10.
Additional file 2.
Figures S22–S55. Standard full field ERG, PERG, pVEP and mfERG of patients III/3, III/8, IV/1, IV/2, IV/6 and IV/10. (Not every patient went through the full list of electrophysiology analyses.)
Additional file 3.
Ophthalmology findings. Electrophysiology methods. Electrophysiology findings (Pattern VEP, Pattern ERG, Standard full field ERGs, Multifocal ERGs). Colour vision testing. Numerical electrophysiology data: Table S1, Table S2, Table S3.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Széll, N., Fehér, T., Maróti, Z. et al. Myopia-26, the female-limited form of early-onset high myopia, occurring in a European family. Orphanet J Rare Dis 16, 45 (2021). https://doi.org/10.1186/s13023-021-01673-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-021-01673-z