
Abstract
Background
Dubin-Johnson syndrome (DJS) is a rare autosomal recessive disorder characterized by predominantly conjugated hyperbilirubinemia that is caused by pathogenic mutations in the adenosine triphosphate-binding cassette subfamily C member 2 (ABCC2) gene, which encodes multidrug resistance-associated protein 2 (MRP2). However, little is known about the causative mutation of DJS in China. Recently, we have reported ABCC2 p.G693R mutation in two unrelated cases. In the present study, we investigated the pathogenicity of the ABCC2 p.G693R mutation in DJS in China.
Methods
Clinical and genetic analysis was conducted for the two patients with the ABCC2 p.G693R mutation. Whole exome sequencing for mutations in other known hyperbilirubinemia-related genes was conducted for the cases with ABCC2 p.G693R. Expression and cellular localization of the mutant MRP2 p.G693R were analyzed by Western blotting and immunofluorescence assay, respectively. Organic anion transport activity was evaluated by the analysis of glutathione-conjugated-monochlorobimane.
Results
The two DJS patients with ABCC2 p.G693R mutation, which was conserved among different species, showed typical hyperbilirubinemia phenotype. No pathogenic mutation was identified in the other known hyperbilirubinemia related genes. Functional studies in three cell lines showed that the expression, localization and the organic anion transport activity were significantly compromised by MRP2 p.G693R mutation compared with wild-type MRP2.
Conclusions
The recurrent ABCC2 p.G693R mutation is associated with loss of function of the MRP2 protein and may result in hyperbilirubinemia in DJS in China.
Similar content being viewed by others


Background
Dubin-Johnson syndrome (DJS) is an autosomal recessive disorder which was first described in 1954 [1]. As a rare disorder affecting both genders, DJS has been identified in all nationalities and races. Its incidence in Sephardic Jews is approximately 1 in 3000 [2]. The syndrome is characterized by predominantly conjugated hyperbilirubinemia, which is caused by impairment in the transfer of non-bile acid organic anions from hepatocytes into canaliculi [3].
The adenosine triphosphate-binding cassette subfamily C member 2 (ABCC2) gene, located on chromosome 10q24, encodes the multidrug resistance-associated protein 2 (MRP2). Comprised of 1545 amino acids, this protein belongs to an integral membrane glycoprotein family [4]. MRP2 traffic from the endoplasmic reticulum to the canalicular membrane of hepatocytes where it functions and then relocalizes back to endosomal vesicles for recycling [5]. This protein is a non-bile acid organic anion transporter and mediates the active transport of conjugate compounds with glutathione or glucuronate from the cytoplasm of hepatocytes into the canaliculi [5].
According to Human Gene Mutation Database (HGMD; www.hgmd.cf.ac.uk), a total of 68 variants in the ABCC2 gene including missense, nonsense, deletions and splice site mutations have been identified in DJS patients. However, no hotspot mutations have been identified in the ABCC2 gene. The majority of the DJS-causing mutations in ABCC2 are related to defects in MRP2 protein synthesis, localization or secretion activities. Some mutations may cause rapid degradation of the mRNA, mislocalization of protein or decreased organic anion transport activity [6]. ABCC2 mutations have been identified in DJS patients worldwide. However, less is known about the causative mutation of DJS in China.
Recently, we identified ABCC2 p.G693R mutation in our previous study [7]. Therefore, in the present study, we investigated the frequency of ABCC2 p.G693R in Chinese DJS patients and examined the pattern and biological consequences of the ABCC2 p.G693R mutation, focusing on their effects on protein maturation, localization and transport activity.
Methods
Study population
From the China Registry of Genetic/Metabolic Liver Diseases (ClinicalTrials.gov identifier, NCT03131427), a total of 14 patients suspected with DJS, who had biochemical evidence of fluctuating predominantly conjugated hyperbilirubinemia with or without family history, were initially included in the present study between June 2015 and December 2017. Whole blood samples from the 14 patients were collected and stored at − 20 °C for Sanger sequencing. 7 out of the 14 patients had ABCC2 gene mutations, and 2 of the 7 patients had ABCC2 p.G693R mutation [7].
The study was conducted in accordance with the Declaration of Helsinki. The Clinical Research Ethics Committee of Beijing Friendship Hospital, Capital Medical University approved the study protocol (No. 2019-P2–217-02). All patients provided written informed consent.
Clinical and genetic analysis of the two patients with the ABCC2 p.G693R
The clinical manifestation of the patients with the ABCC2 p.G693R, including age of onset, duration of jaundice, aggravating or relieving factors was recorded. Past medical history including drug or toxin exposure, alcohol intake, and family history of jaundice or other liver diseases were collected.
Relevant laboratory data of the two patients with the ABCC2 p.G693R were analyzed, including complete blood count, liver function tests, renal function tests and electrolytes, coagulation profile. Abdominal ultrasonography was done to exclude obstruction or dilation of the hepatobiliary tract exist and transient elastography (FibroScan) was conducted to evaluate the liver stiffness.
Conservative analysis was performed by http://genome.ucsc.edu/. Aligned amino acid sequences of human, rhesus, mouse, dog, elephant, chicken, xenopus tropicalis, zebrafish, and lamprey MRP2 with mutation p.G693R loci were analyzed.
Analysis of mutation in other known hyperbilirubinemia genes by whole exome sequencing
Approximately 1 μg of genomic DNA was used to construct a whole exome library with an insert size of 150–200 bp by an exome capture strategy using a GenCap custom exome enrichment kit (MyGenostics, Beijing, China). Paired-end 100 bp raw reads from each enriched library were generated with an Illumina HiSeq 2000 platform (Illumina, San Diego, USA) according to the manufacturer’s protocol. The paired-end reads were aligned against NCBI build 37 of the human genome using Burrows Wheeler Aligner. With the GenomeAnalysis Toolkit (GATK4.1.2.0, https://software.broadinstitute.org/gatk/download/), duplicate reads were marked; local indel realignment was performed, and base quality scores were recalibrated for each sample. The identified potential pathogenic variants were confirmed by Sanger sequencing.
Polyphen-2 (http://genetics.bwh.harvard.edu/pph2), SIFT (https://sift.bii.a-star.edu.sg/) and MutationTaster (http://www.mutationtaster.org/) were used to predict the biofunctional consequence of the identified variants.
Functional analysis of ABCC2 p.G693R mutant
Construction of the ABCC2 p.G693R mutant
To create the ABCC2 wild-type plasmid, we amplified ABCC2 from human cDNA and cloned it into Hind III/Not I sites of the pcDNA3.1 vector along with the N-terminal flag tag [8]. The ABCC2 p.G693R constructs were generated using the Gene Tailor Site-Directed Mutagenesis System (Invitrogen, Waltham, MA, USA).
Cell culture and transfection
Human embryonic kidney (HEK) 293A cells and human liver cancer cell lines Huh-7 and HepG2 were obtained from the Cell Resource Center of the Chinese Academy of Medical Science (Beijing, China). The Cell lines were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum, 100 units/ml penicillin and 100 units/ml streptomycin. Then the cells were transfected with plasmids expressing ABCC2 wild-type or ABCC2 p.G693R by Lipofectamine 3000 (Invitrogen, Carlsbad, USA) according to the manufacturer’s instructions; the culture medium was changed at 6 h after transfection. Finally, the cells were harvested 24 or 48 h after transfection.
Measurement of MRP2 expression by Western blotting
The cells were lysed in RIPA buffer with proteinase inhibitor. After centrifugation, the supernatant was used for western blot analysis. Proteins were separated by SDS-PAGE (8%) and transferred to nitrocellulose membranes; the membranes were incubated with anti-MRP2 monoclonal antibodies M2III-6 (1:200; sc-59,608; Santa Cruz Biotechnology, Dallas, TX; a mouse monoclonal antibody raised against a C-terminal region of MRP2 of human origin) or anti-GAPDH antibodies (1:5000; Santa Cruz Biotechnology) overnight at 4 °C, followed by incubation with horseradish peroxidase (HRP)-conjugated goat anti-mouse antibody (1:5000 dilution; Santa Cruz Biotechnology) for 1 h at 37 °C. Immunocomplexes on the membrane were visualized using Immobilon Western Chemiluminescent HRP Substrate (Millipore, Billerica, USA) and Image Lab Software (BIO-RAD, Hercules, USA).
Measurement of subcellular localization by indirect immunofluorescence staining
Immunofluorescence analysis was performed as described previously [9]. The cells were incubated with a primary antibody directed against rabbit anti-MRP2 (ab172630; Abcam) and mouse anti-flag (#8146; Cell Signaling technology) at 4 °C overnight. After three washes with phosphate-buffered saline for 5 min each, the cells were incubated with anti-rabbit Alex 647 and anti-mouse Alex 488-conjugated secondary antibodies (1:200; Invitrogen) for 1 h at room temperature. For F-actin staining, cells were incubated with FITC-conjugated phalloidin at 50 μg/ml (P5282; Sigma) for 40 min. Then cells were mounted on a slide in mounting medium (Molecular Probes). Finally, the cells were visualized and photographed using an FV 300 confocal microscope (Olympus, Tokyo, Japan).
Measurement of organic anion transport activity by export of glutathione conjugated monochlorobimane (GS-MCLB) assay
MCLB is an organic anion transport substrate for MRP2 and has absorption/emission maximal ~ 394/490 nm. MCLB (M1381MP, Thermo, USA) transport study was conducted as described by Terlouw et al [10]. The cells were pre-incubated with 0.2 mmol/L MCLB in medium for 30 min on ice. The medium was replaced with fresh Hank’s medium and incubated at 37 °C. At different time points, medium was collected and the GS-MCLB in the medium was measured by the fluorescence method with a spectrophotometer.
Statistical analysis
All experiments were carried out at least three times. Statistical analyses were performed using SPSS V12.0 software. Results were expressed as mean ± standard deviation (SD). Continuous variables were analyzed using the Student’s test. A two-sided P value of < 0.05 was considered statistically significant.
Results
Clinical and genetic profiles of the DJS patients with ABCC2 p.G693R mutation
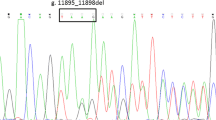
For the two unrelated patients with DJS, patient No. 1 (female, 21 years old) and patient No. 2 (male, 39 years old) harbored the heterozygous allelic variant c.2190G > A in exon 16 of ABCC2, which resulted in the substitution of arginine for glycine at position 693 (p.G693R) (Fig. 1a). Sequence comparison showed that amino acid 693 of MRP2 was conserved among different species (Fig. 1b). The two patients harbored the variant of p.G808V or p.R529Q in another allele respectively.

Mutation analysis of the two DJS cases with ABCC2 p.G693R. a Sequencing of the heterozygous missense mutation c.2190G > A, p.G693R in exon 16 in both cases. b Aligned amino acid sequences of human, rhesus, mouse, dog, elephant, chicken, xenopus tropicalis, zebrafish, and lamprey MRP2 with mutation p.G693R flanking regions. The position of the p.G693R mutation is indicated by the red box. Alignment was performed by http://genome.ucsc.edu/
The clinical features of these two patients are shown in Table 1. Total bilirubin and direct bilirubin were 94 μmol/L and 54 μmol/L in patient No.1, and 97 μmol/L and 49 μmol/L in patient No.2 respectively. The aminotransferase, alkaline phosphatase, γ-glutamyl transpeptidase, total bile acid and albumin in liver function tests were within the normal range. Abdominal ultrasonography showed no obstruction or dilation of the hepatobiliary tract in both patients. Liver stiffness by FibroScan was 3.5 Kpa in patient No.1, but not available in patient No. 2.
Results from whole exome sequencing revealed additional p.R110Q (c.329G > A) mutation in the gene 3β-hydroxysteroid dehydrogenase type 7 (HSD3B7), which is related with bile acid synthesis type I, in patient No. 2 with ABCC2 p.G693R mutation, with allele frequency of HSD3B7 p.R110Q is 5.80E-05 in East Asian and 3.00E-04 in total, respectively.. However, software prediction demonstrated that the missense mutation HSD3B7 p.R110Q is benign or tolerable. In patient No. 1 with ABCC2 p.G693R mutation, whole exome sequencing revealed no mutations in the other known hyperbilirubinemia-related genes.
MRP2 expression was decreased in cell lines expressing MRP2 p.G693R
Western blotting for MRP2 in HEK293, Huh-7 and HepG2 cell lines transfected with vector expressing ABCC2 with the p.G693R mutation demonstrated a decreased expression of the MRP2 p.G693R mutant, probably due to the degradation of the mutant protein, compared with wild-type MRP2 (p < 0.01) (Fig. 2a).

The MRP2 p.G693R mutant showed decreased expression and mislocalization in three cell lines in vitro. a Western blotting demonstrated significantly decreased expression of MRP2 in three cell lines expressing the p.G693R mutant compared with those expressing wild-type MRP2. **P < 0.05. b Immunofluorescence assay showed the mislocalization of the MRP2 p.G693R mutant, which was predominantly retained in the cytoplasm rather than the cell surface
MRP2 p.G693R mutant was predominantly retained in the cytoplasm rather than the cell surface
Indirect immunofluorescence staining in HEK293A, Huh-7 and HepG2 cell lines showed that wild-type MRP2 predominantly localized to the cell surface and cytoplasm. In contrast, we observed mislocalization of the MRP2 p.G693R mutant, which was predominantly retained in the cytoplasm rather than the cell surface (Fig. 2b). Immunofluorescence analysis of MRP2 fusion protein with N-terminal flag tag, and F-actin in Huh-7 cells showed wild-type MRP2 predominantly localized to the cell surface and cytoplasm, while MRP2 p.G693R mutant was mostly retained at the cytoplasm (Fig. 3a-b).

The MRP2 p.G693R mutant with flag tag showed mislocalization in Huh-7 cells in vitro. a Immunofluorescence analysis of MRP2 fusion protein with N-terminal flag tag in Huh-7 cells confirmed MRP2 expression in wild-type and p.G693R mutant MRP2 plasmids. b Immunofluorescence analysis of F-actin and MRP2 expression in Huh-7 cells showed wild-type MRP2 predominantly localized to the cell surface and cytoplasm, while MRP2 p.G693R mutant was mostly retained at the cytoplasm
MRP2 p.G693R mutant exhibited decreased organic anion transport activity
Following pre-incubation with MCLB, which was conjugated with glutathione to produce GS-MCLB, the GS-MCLB efflux in HEK293A, Huh-7 and HepG2 cells lines which expressed wild-type MRP2 was markedly increased compared with that in cells expressed MRP2 p.G693R (p < 0.05) (Fig. 4). These results indicated that the p.G693R mutant exhibited significantly decreased organic anion transport activity compared with the wild-type MRP2.

MRP2 p.G693R mutant exhibited decreased organic anion transport activity. Compared with wild-type MRP2, the MRP2 p.G693R mutant exhibited decreased organic anion transport activity in HEK293A, Huh-7 and HepG2 cell lines. The Student’s test was used to evaluate the differences of organic anion transport activity. *P < 0.05
Discussion
In the present study, we reported on the clinical manifestations and biological functional consequences of the ABCC2 p.G693R mutation, which was identified with a high frequency of 28.6% (2/7) in a small cohort of Chinese patients with DJS. Functional studies indicated significantly decreased expression, mislocalization, and decreased organic anion transport activity of mutant MRP2 compared with wild-type MRP2, indicating that this variant resulted in loss of function of the MRP2 protein.
DJS is characterized by predominantly conjugated hyperbilirubinemia in liver function tests and black liver in liver biopsies [11]. Urinary coproporphyrin levels, bromsulphalein excretion test and hepatobiliary scintigraphy with iodopanoic acid provide effective methods for identification and diagnosis on DJS [12, 13]. However, most of the diagnostic tests, including urinary corproporphyrin or bromsulphalein excretion test, are not widely available in clinical practice in China. Diagnose of DJS relies of ABCC2 gene Sanger sequencing or liver histopathology manifestations. Since liver biopsies are invasive, genetic analysis of peripheral blood samples is important. To date, a total of 68 ABCC2 variants have been reported in DJS according to HGMD, comprising 31 (45.6%) missense variants, 13 (19.1%) small deletions variants, 10 (14.7%) nonsense variants, 6 (8.8%) splice site variants and 3 (4.4%) gross deletions variants, 2 (2.9%) small indels variants, 2 (2.9%) insertions variants and 1 (1.5%) gross insertion variant, and the variants occur randomly throughout the gene (Supplementary Table 1). Of the 31 missense variants, 14 are pathogenic confirmed by previous functional study [14,15,16,17,18,19,20,21]. As the clinical data including serum level of total bilirubin were not available for the individual missense mutation carriers in the previous studies, the genotype and phenotype correlation remains to be explored. However, in the present study, the abnormal serum level of total bilirubin of 94 and 97 μmol/L were observed in the two cases with ABCC2 G693R mutation respectively. Therefore, identification and functional analysis of causative ABCC2 gene mutations for patients suspected with DJS are essential in diagnosis of DJS in China.
In the present study, whole exome sequencing revealed additional HSD3B7 p.R110Q mutation in a case with ABCC2 p.G693R mutation, but no mutations in the other known hyperbilirubinemia-related genes in the other case with ABCC2 p.G693R mutation. Gene HSD3B7, located on chromosome 16p11.2, encodes an enzyme which is a member of the short-chain dehydrogenase/reductase superfamily and involved in the initial stages of the synthesis of bile acids from cholesterol [22]. Pathogenic mutations in HSD3B7 are associated with congenital bile acid deficiency type I, which is a life-threatening liver disease and manifests as hyperbilirubinemia and neonatal cholestasis [23, 24]. However, in silico analysis showed this variant was benign, indicating it might be common variant but not a pathogenic mutation. Thus, the two cases with ABCC2 p.G693R heterozygous mutation showed that the ABCC2 mutation might be the main genetic factor of DJS in China.
In the present study, the p.G693R mutation was observed in 2 out of 7 patients, indicating that the p.G693R mutation might be a potential hotspot mutation in Chinese DJS patients. ABCC2 p.G693R is a missense mutation located in the first ATP-binding domain of MRP2 protein [25]. We demonstrated that while wild-type MRP2 predominantly localized to the cell surface and cytoplasm, the MRP2 p.G693R mutant was predominantly retained in the cytoplasm rather than the cell surface. The mislocalization of the p.G693R mutant may likely be due to deficient maturation and sorting, causing impaired insertion trafficking from the endoplasmic reticulum to the canalicular membrane in hepatocytes, similar to the p.R768W and p.W709R mutants reported in previous studies [26, 27].
Furthermore, efflux of GS-MCLB uptake into plasma from cells expressing the p.G693R mutant was markedly reduced compared with cells expressing wild-type MRP2, suggesting that the organic anion transport activity of p.G693R MRP2 on the canalicular membrane was defective [13]. Similarly, Hashimoto et al. reported that p.Q1382R MRP2 impairs substrate-induced ATP hydrolysis, which lead to the defect of organic anion transport activity [6].
We understand that the number of cases analyzed in the current study was limited, and thus more cases with DJS are required to confirm these conclusions. Using the China Registry of Genetic/Metabolic Liver Diseases, we will conduct further genetic and functional studies of DJS to strengthen the relationship between the genotype and phenotype of DJS.
Conclusions
In conclusion, here we show that the ABCC2 p.G693R mutation causes dysfunction of the MRP2 protein and may result in hyperbilirubinemia in DJS in China.
Availability of data and materials
All the data were collected from the hospital information system and can be available from the corresponding author upon reasonable request.
Abbreviations
- ABCC2 :
-
Adenosine triphosphate-binding cassette subfamily C member 2
- DJS:
-
Dubin-Johnson syndrome
- GS-MCLB:
-
Glutathione conjugated monochlorobimane
- HEK:
-
Human embryonic kidney
- HGMD:
-
Human Gene Mutation Database
- HRP:
-
Horseradish peroxidase
- HSD3B7 :
-
3β-hydroxysteroid dehydrogenase type 7
- MRP2:
-
Multidrug resistance-associated protein 2
- SD:
-
Standard deviation
References
Dubin IN, Johnson FB. Chronic idiopathic jaundice with unidentified pigment in liver cells; a new clinicopathologic entity with a report of 12 cases. Medicine (Baltimore). 1954;33:155–97.
Shani M, Seligsohn U, Gilon E, Sheba C, Adam A. Dubin-Johnson syndrome in Israel. I. Clinical, laboratory, and genetic aspects of 101 cases. Q J Med. 1970;39:549–67.
Togawa T, Mizuochi T, Sugiura T, Kusano H, Tanikawa K, Sasaki T, Ichinose F, et al. Clinical, pathologic, and genetic features of neonatal Dubin-Johnson syndrome: a multicenter study in Japan. J Pediatr. 2018. https://doi.org/10.1016/j.jpeds.2017.12.058.
Keppler D. The roles of MRP2, MRP3, OATP1B1, and OATP1B3 in conjugated hyperbilirubinemia. Drug Metab Dispos. 2014;42:561–5.
Gazzin S, Masutti F, Vitek L, Tiribelli C. The molecular basis of jaundice: an old symptom revisited. Liver Int. 2017;37:1094–102.
Hashimoto K, Uchiumi T, Konno T, Ebihara T, Nakamura T, Wada M, Sakisaka S, et al. Trafficking and functional defects by mutations of the ATP-binding domains in MRP2 in patients with Dubin-Johnson syndrome. Hepatology. 2002;36:1236–45.
Wu L, Zhang W, Jia S, Zhao X, Zhou D, Xu A, Duan W, et al. Mutation analysis of the ABCC2 gene in Chinese patients with Dubin-Johnson syndrome. Exp Ther Med. 2018;16:4201–6.
Zhang W, Xu A, Li Y, Zhao S, Zhou D, Wu L, Zhang B, et al. A novel SLC40A1 p.Y333H mutation with gain of function of ferroportin: A recurrent cause of haemochromatosis in China. Liver Int. 2019;39:1120–7.
Lv T, Zhang W, Xu A, Li Y, Zhou D, Zhang B, Li X, et al. Non- mutations in haemochromatosis in China: combination of heterozygous mutations involving signal peptide variants. J Med Genet. 2018;55:650–60.
Terlouw SA, Masereeuw R, van den Broek PH, Notenboom S, Russel FG. Role of multidrug resistance protein 2 (MRP2) in glutathione-bimane efflux from Caco-2 and rat renal proximal tubule cells. Br J Pharmacol. 2001;134:931–8.
Park SW, Jun CH, Choi SK, Kim HJ, Kim GE. Hepatobiliary and pancreatic: a black liver of Dubin-Johnson syndrome. J Gastroenterol Hepatol. 2018;33:562.
Gilibili RR, Chatterjee S, Bagul P, Mosure KW, Murali BV, Mariappan TT, Mandlekar S, et al. Coproporphyrin-I: a fluorescent, endogenous optimal probe substrate for ABCC2 (MRP2) suitable for vesicle-based MRP2 inhibition assay. Drug Metab Dispos. 2017;45:604–11.
Erlinger S, Arias IM, Dhumeaux D. Inherited disorders of bilirubin transport and conjugation: new insights into molecular mechanisms and consequences. Gastroenterology. 2014;146:1625–38.
Arlanov R, Porter A, Strand D, Brough R, Karpova D, Kerb R, Wojnowski L, et al. Functional characterization of protein variants of the human multidrug transporter ABCC2 by a novel targeted expression system in fibrosarcoma cells. Hum Mutat. 2012;33:750–62.
Hulot JS, Villard E, Maguy A, Morel V, Mir L, Tostivint I, William-Faltaos D, et al. A mutation in the drug transporter gene ABCC2 associated with impaired methotrexate elimination. Pharmacogenet Genomics. 2005;15:277–85.
Meyer zu Schwabedissen HE, Jedlitschky G, Gratz M, Haenisch S, Linnemann K, Fusch C, Cascorbi I, et al. Variable expression of MRP2 (ABCC2) in human placenta: influence of gestational age and cellular differentiation. Drug Metab Dispos. 2005;33:896–904.
Hirouchi M, Suzuki H, Itoda M, Ozawa S, Sawada J, Ieiri I, Ohtsubo K, Sugiyama Y. Characterization of the cellular localization, expression level, and function of SNP variants of MRP2/ABCC2. Pharm Res. 2004;21:742–8.
Mor-Cohen R, Zivelin A, Rosenberg N, Shani M, Muallem S, Seligsohn U. Identification and functional analysis of two novel mutations in the multidrug resistance protein 2 gene in Israeli patients with Dubin-Johnson syndrome. J Biol Chem. 2001;276:36923–30.
Floreani A, Carderi I, Variola A, Rizzotto ER, Nicol J, Bergasa NV. A novel multidrug-resistance protein 2 gene mutation identifies a subgroup of patients with primary biliary cirrhosis and pruritus. Hepatology. 2006;43:1152–4.
Megaraj V, Zhao T, Paumi CM, Gerk PM, Kim RB, Vore M. Functional analysis of nonsynonymous single nucleotide polymorphisms of multidrug resistance-associated protein 2 (ABCC2). Pharmacogenet Genomics. 2011;21:506–15.
Elens L, Tyteca D, Panin N, Courtoy P, Lison D, Demoulin JB, Haufroid V. Functional defect caused by the 4544G>a SNP in ABCC2: potential impact for drug cellular disposition. Pharmacogenet Genomics. 2011;21:884–93.
Cheng JB, Jacquemin E, Gerhardt M, Nazer H, Cresteil D, Heubi JE, Setchell KDR, et al. Molecular genetics of 3β-Hydroxy-Δ5-C27-steroid Oxidoreductase deficiency in 16 patients with loss of bile acid synthesis and liver disease. J Clin Endocrinol Metab. 2003;88:1833–41.
Huang HY, Zhou H, Wang H, Chen YX, Fang F. Novel mutations in the 3β-hydroxy-Δ5-C27-steroid dehydrogenase gene (HSD3B7) in a patient with neonatal cholestasis. Chin Med J. 2016;129:98–100.
Bossi G, Giordano G, Rispoli GA, Maggiore G, Naturale M, Marchetti D, Iascone M. Atypical clinical presentation and successful treatment with oral cholic acid of a child with defective bile acid synthesis due to a novel mutation in the gene. Pediatric Reports. 2017;9:7266.
Wen X, Joy MS, Aleksunes LM. In vitro transport activity and trafficking of MRP2/ABCC2 polymorphic variants. Pharm Res. 2017;34:1637–47.
Uchiumi T, Tanamachi H, Kuchiwaki K, Kajita M, Matsumoto S, Yagi M, Kanki T, et al. Mutation and functional analysis of ABCC2/multidrug resistance protein 2 in a Japanese patient with Dubin-Johnson syndrome. Hepatol Res. 2013;43:569–75.
Keppler D. Progress in the molecular characterization of Hepatobiliary transporters. Dig Dis. 2017;35:197–202.
Acknowledgements
We kindly acknowledge Dr. Zhao Xinyan (Liver Research Center, Beijing Friendship Hospital, Capital Medical University, Beijing, China) for histopathological analysis of the liver tissues of patients.
Funding
This work was supported by grants from the Digestive Medical Coordinated Development Center of Beijing Hospitals Authority (No.XXT06).
Author information
Authors and Affiliations
Contributions
LW collected and analyzed the clinical data, and drafted the article. YL performed the functional analysis of ABCC2 p.G693R mutant. XO, HJ and JJ conceived the study and revised the manuscript. YS, DZ, SJ, AX performed the polymerase chain reaction amplification analysis of the ABCC2 gene. WZ and HY collected the clinical data. All the authors approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Supplementary Table S1.
Known variants identified in the ABCC2 gene according to Human Gene Mutation Database.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wu, L., Li, Y., Song, Y. et al. A recurrent ABCC2 p.G693R mutation resulting in loss of function of MRP2 and hyperbilirubinemia in Dubin-Johnson syndrome in China. Orphanet J Rare Dis 15, 74 (2020). https://doi.org/10.1186/s13023-020-1346-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-020-1346-4