
Abstract
Background
Pendred syndrome (PDS, MIM #274600) is an autosomal recessive disorder characterized by congenital sensorineural hearing loss and goiter. In this study, we describing the possible PDS causal mutations in a Malaysian family with 2 daughters diagnosed with bilateral hearing loss and hypothyroidism.
Methods and Results
Whole exome sequencing was performed on 2 sisters with PDS and their unaffected parents. Our results showed that both sisters inherited monoallelic mutations in the 2 known PDS genes, SLC26A4 (ENST00000265715:c.1343C > T, p.Ser448Leu) and GJB2 (ENST00000382844:c.368C > A, p.Thr123Asn) from their father, as well as another deafness-related gene, SCARB2 (ENST00000264896:c.914C > T, p.Thr305Met) from their mother. We postulated that these three heterozygous mutations in combination may be causative to deafness, and warrants further investigation. Furthermore, we also identified a compound heterozygosity involving the DUOX2 gene (ENST00000603300:c.1588A > T:p.Lys530* and c.3329G > A:p.Arg1110Gln) in both sisters which are inherited from both parents and may be correlated with early onset of goiter. All the candidate mutations were predicted deleterious by in silico tools.
Conclusions
In summary, we proposed that PDS in this family could be a polygenic disorder which possibly arises from a combination of heterozygous mutations in SLC26A4, GJB2 and SCARB2 which associated with deafness, as well as compound heterozygous DUOX2 mutations which associated with thyroid dysfunction.
Similar content being viewed by others


Background
Hearing loss is a multifactorial disease with nearly 50% of cases being heritable and attributable to genetic defects [1]. The annual incidence of congenital deafness is estimated to be 1:1000 newborns [2], hence, identification of the heritable causal genes is pivotal to reduce the incidence of childhood deafness. Pendred syndrome (PDS, MIM #274600) is among the most common types of syndromic hearing impairment, and accounts for approximately 10% of hereditary deafness [3]. PDS is clinically characterized by sensorineural deafness, enlargement of the vestibular aqueduct (EVA), goiter, and incomplete iodide organification [3, 4]. Even though PDS could be clinically confirmed with perchlorate discharge test in combination with temporal bone scan of the ear architecture, misdiagnosis with other deafness associated disease remains as the key challenge [5]. Late onset of goiter manifestation which usually develops after the age of 10 years and the presentation of only mild hypothyroidism have made definitive diagnosis of PDS difficult [6, 7]. Furthermore, the degree of hearing loss could vary from mild to profound, either contributed by physical malformation or genetic defects. The limitations to getting an accurate diagnosis will prevent early treatment and may lead to mental retardation which is preventable by thyroxine replacement therapy [8]. As such, identification of genes contributed to PDS is desirable to pave the way towards early detection of PDS as well as for carrier testing.
PDS is a complex genetic disease which may be inherited monogenically or digenically [4, 9–11]. It has been well documented that biallelic mutations in SLC26A4 (MIM #605646) is the hallmark of PDS, with a frequency of 25% [4, 9]. Clinically, SLC26A4 mutation has been used as genetic test to differentiate between PDS and non-syndromic familial EVA, which otherwise would not be possible to clinically distinguish, even with perchlorate discharge test [6, 12]. However, nearly 50% probands harboured only monoallelic mutation in SLC26A4, and for some patients, PDS is not due to SLC26A4 gene mutations [4]. The discovery of the involvement of other deafness genes, including FOXI1 (MIM #601093), KCNJ10 (MIM # 602208) and GJB2 (MIM #121011) [9–11] in combination with SLC26A4 monoallelic mutation has proposed the existence of digenic inheritance pattern in PDS and EVA. The complexity of the genetic defects attributed to PDS suggests that a comprehensive mutational screening is warranted to identify the disease causal genes.
In the past, limitations in genomic sequencing technologies have only permitted the identification of disease-causing mutations through the candidate gene screening approach. Now, with the advent of next generation sequencing technologies, genome wide screening can now be performed in a cost-effective manner. Among these, whole exome sequencing (WES) is preferable as it focuses only on coding regions in which ~85% disease-causing mutations are located [13]. WES has also successfully discovered genes for many rare diseases [14]. Given that the genetic makeup of PDS remains largely unknown and complex, we performed WES to identify the genes responsible for PDS in a family with 2 affected siblings and their unaffected parents. This study will enhance our understanding about the genetic aetiology underpinning PDS, and to identify candidate genes which may be useful for precise molecular diagnosis and to guide family planning for better management of heritable deafness.
Methods
Subjects
Two siblings diagnosed to have PDS were referred for molecular evaluation and confirmation of diagnosis. These sisters were the only children of a pair non-consanguineous parents. They were 15 and 9 years old respectively at the time of referral.
Elder sister
The elder sister first presented to her local doctor at the age of 10 months with progressively enlarging goiter. Investigation then showed hypothyroidism and L-thyroxin was started. At 3 years of age, her parents noted profound hearing impairment for which she required hearing aid. There was no other significant past medical or surgical history and she was not on any long term medications. Her parents reported that she had normal speech development and was able to attend normal school. On examination, she had a large, diffused multinodular goiter. Ultrasound of the thyroid gland revealed multiple complex cysts. Tc99m thyroid scan showed a hyperfunctioning multinodular goiter with increased total iodine uptake of 34.6%. These findings were consistent with dyshormonogenesis (goitrous hypothyroidism with increased radioactive iodine uptake by the thyroid). Unfortunately, perchlorate discharge test could not be done. Hearing test confirmed bilateral profound sensorineural hearing loss > 60 dB, worse for high frequency sounds. Magnetic resonance image (MRI) of the inner ear and temporal bones showed bilateral dilated vestibular aqueduct and presence of only 1 ½ turns of the cochlear (normal: 2 ¾ turns) consistent with cochlear hypoplasia. In view of the large goiter size, patient underwent total thyroidectomy at 13 years of age. Currently she is receiving full dose of thyroxin i.e. 100 mcg daily.
Younger sister
The younger sister presented with profound hearing loss at 10 months of age associated with delayed speech development. At 16 months, she was wearing hearing aid and receiving speech therapy. She underwent cochlear implantation at 5 years of age. Her parents also noticed progressively enlarging goiter then. She had delayed speech development. She used sign language for communication and attended special school. On examination, there was a large, diffused multinodular goiter. Ultrasound revealed multiple complex cysts within the thyroid gland. Tc99m thyroid scan showed a hyperfunctioning multinodular goiter with increased total iodine uptake of 30.4%. Hearing test showed bilateral profound sensorineural hearing loss > 60 dB, worse for high frequency sounds. MRI of the inner ear and temporal bones showed bilateral dilated vestibular aqueduct and presence of only 1 ½ turns of the cochlear (normal 2 ¾ turns) consistent with cochlear hypoplasia. Total thyroidectomy was anticipated.
Exome library construction and sequencing
Peripheral blood samples were collected from all the individuals included in this study with written informed consent. Genomic DNA was extracted from peripheral blood using salt extraction method and the DNA quality was assessed using agarose gel electrophoresis. The DNA samples were of good quality (A260/A280 > 2.0; A260/A230 > 2.0) as assessed by Nanodrop (Thermo Fisher Scientific, USA). The DNA concentration was measured using Qubit dsDNA BR Assay Kit (Thermo Fisher Scientific, USA). The DNA libraries were prepared employing the Ion AmpliSeq™ Exome RDY Kit (Thermo Fisher Scientific, USA) and were then sequenced by the Ion Proton™ System (Thermo Fisher Scientific, USA), according to the manufacturer’s protocol.
Bioinformatic data analysis
Read mapping and variant calling were performed by the Ion TorrentSuite™ v4.4.2 software (Thermo Fisher Scientific, USA) using default parameters setting. The reads were aligned to human reference genome hg19, followed by variant calling using TorrentSuite™ Variant Caller v4.4.2.1. Next, the variants with SNP quality scores ≤ 30 were filtered out using SnpSift [15], followed by annotation with ANNOVAR [16]. Only non-synonymous variants in the coding regions (exonic, splicing) with a read depth greater than 5X were retained for further analysis. Polymorphisms with allele frequencies > 0.01 reported in 1000 Genomes Project, NHLBI Exome Sequencing Project, and Maximum Population Frequency were filtered out. Subsequently, we identify the candidate disease causing mutation by comparing the variants detected in affected sisters with their parents based on monogenic (autosomal recessive), followed by digenic and polygenic inheritance traits. Variants which fulfilled the above criteria were manually inspected using Integrative Genomics Viewer to filter out false positive variants [17, 18]. The effect of the variants was assessed using several in silico prediction tools, including SIFT [19], Polyphen2 [20], MutationTaster [21], FATHMM [22], CADD [23], PROVEAN [24], and DANN [25]. Candidate mutations which predicted deleterious by one of the above tools were further studied by searching literature database.
Sanger validation
A total of 5 predicted pathogenic candidate mutations, i.e. SLC26A4 (ENST00000265715:c.1343C > T, p.Ser448Leu), GJB2 (ENST00000382844:c.368C > A, p.Thr123Asn), SCARB2 (ENST00000264896:c.914C > T, p.Thr305Met), DUOX2 (ENST00000603300:c.1588A > T, p.Lys530*), and DUOX2 (ENST00000603300:c.3329G > A, p.Arg1110Gln) were selected for validation by Sanger sequencing. The primers were designed using Primer3 (Additional file 1: Table S4). The regions were amplified by PCR using AmpliTaq Gold Polymerase (Thermo Fisher Scientific, USA), and the amplified products were purified using PCR Purification Kit (Qiagen, Germany), and sequenced using ABI BigDye Terminator v3.1 Cycle Sequencing Kit (Thermo Fisher Scientific, USA). The chromatograms were visualized using BioEdit software.
Results
Whole-exome sequencing
We sequenced the exomes of 4 individuals from a family with 2 daughters diagnosed with PDS and their unaffected parents. An average of 36 million reads were generated per sample, and the reads were mapped to the human reference genome hg19, with 91% of the bases covered at > 20X coverage. The mean depth of coverage was 104X with a uniformity of 91%. The variants were filtered as described in the Materials and Methods, and the summary of the sequencing results are as shown in Table 1.
Identification of disease causing genes in Pendred syndrome
After filtering out polymorphisms with allele frequency > 0.01 as reported in the 1000 Genomes Project, 6500 NHLBI exome and Maximum Population Frequency databases, we retained only non-synonymous variants for identifying PDS causative mutations. Subsequently, we filtered the variants based on monogenic autosomal recessive trait. Variants which present in both sisters were prioritized for further investigations. Our analysis identified DUOX2 (MIM #606759) compound heterozygous mutations (ENST00000603300: p.Lys530X, p.Arg1110Gln) were inherited by both sisters from their father and mother respectively, and may be causative to goiter manifestation. However, we did not find additional homozygous or compound heterozygous mutations which may be contributed to the deafness phenotype.
By taking into consideration that PDS could be inherited via digenic trait, we then look into heterozygous mutations which were inherited by both sisters from their unaffected parents. Interestingly, both sisters inherited missense mutations in 2 genes which known to be associated with PDS (i.e. SLC26A4: ENST00000265715:c.1343C > T,p.Ser448Leu; GJB2: ENST00000382844:c.368C > A, p.Thr123Asn) from their father, as well as another deafness gene, SCARB2 (ENST00000264896:c.914C > T,p.Thr305Met) from their mother. These 3 candidate mutations were predicted pathogenic by one of the variant effect prediction tools (i.e. SIFT, Polyphen-2, MutationTaster, FATHMM, CAAD, PROVEAN and DANN; Additional file 2: Table S1 and Additional file 3: Table S2), hence we postulated that the combination of these 3 candidate missense mutations may be contributed to PDS and associated with hearing loss phenotype. It is noteworthy to mention that SCARB2 could be a novel PDS candidate gene and needs further investigation.
Furthermore, we also looked into the autosomal recessive mutations which are present only in either probands. As depicted in Table 2, each sisters harboured additional non-shared compound heterozygous mutations, however none of these candidate genes are known to be implicated in PDS. Further analysis on heterozygous mutations (Additional file 2: Table S1 and Additional file 3: Table S2) found that the elder sister harboured additional mutations which may be contributed to the PDS phenotype (Additional file 4: Table S3), including hearing (inherited from father: DIAPH3, GPR171, LOXHD1; inherited from mother: MCOLN3, SYNE4), and thyroid function (inherited from father: C16orf89; inherited from mother: TXNDC11). However, these additional mutations have not exaggerated the manifestation of the hearing loss or goiter, in which both sisters were diagnosed with bilateral hearing loss and presented with goiter during 1 year old. Hence, we postulated that these additional mutations which present only in elder sister may not be relevant to PDS.
Sanger validation
Candidate genes predicted to be associated with PDS were selected for further validation by Sanger sequencing. Mutations which were present in both sisters and father (SLC26A4: c.1343C > T, p.Ser448Leu; GJB2:c.368C > A, p.Thr123Asn; DUOX2: c.1588A > T, p.Lys530*), and present in both sisters and mother (SCARB2: c.914C > T, p.Thr305Met; DUOX2: c.3329C > A, p.Arg1110Gln) were confirmed. The chromatograms are depicted in Fig. 1.

Sanger validation of DUOX2 compound heterozygous mutations (ENST00000603300:c.1588A > T:p.Lys530* and c.3329G > A:p.Arg1110Gln), SLC26A4 (ENST00000265715:c.1343C > T, p.Ser448Leu), GJB2 (ENST00000382844:c.368C > A, p.Thr123Asn), and SCARB2 (ENST00000264896:c.914C > T, p.Thr305Met) heterozygous mutations. The arrow shows the site of the changes
As summarized in Fig. 2, the father was a heterozygous carrier for SLC26A4, GJB2 and DUOX2, whereas mother was a heterozygous carrier for SCARB2 and DUOX2. Both of the sisters harboured the same heterozygous mutations in SLC26A4, GJB2 and SCARB2, hence suggesting that the combination of these 3 heterozygous mutations may led to hearing loss in these patients. Furthermore, DUOX2 compound heterozygous mutation may be associated with early onset of hypothyroidisms and goiter.

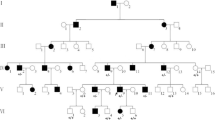
Pedigree of the family with autosomal recessive Pendred syndrome. Open symbols denote unaffected parents; filled black symbols denote affected siblings
Discussion
Clinically, PDS is characterized by the manifestation of a combination of severe to profound sensorineural hearing loss, inner ear anomalies such as Mondini’s dysplasia, EVA or vestibular anomalies, and goiter [26–28]. Also, deafness in PDS generally profound (>60 dB) with prelingual onset [29], and sometimes a fluctuating but worsening course [30–32], consistent with a progressive lesion of the sensory organ. In this case study, clinical diagnosis confirmed both sisters were PDS: (1) MRI examination of the inner ear confirmed both sisters had EVA, an essential prerequisite for the diagnosis of PDS [33, 34]; (2) both sisters had bilateral sensorineural hearing loss, with frequency > 60 dB; (3) both sisters are euthyroid and diagnosed with hypothyroidism at age of 1 year old; (4) the disease is potentially heritable via autosomal recessive or digenic/polygenic traits as both sisters are affected whilst their parents were unaffected.
It has been long considered that PDS is a monogenic disease attributed to SLC46A4 biallelic mutations [35, 36] or a digenic disease attributed to a combination of SLC46A4 and KCNJ10, FOXI1 or GJB2 [9–11]. Notably, our analysis did not detect homozygous or compound heterozygous in the known PDS genes (i.e. SLC26A4, KCNJ10, FOX1, GJB2) based on monogenic autosomal recessive trait, hence suggesting PDS in this family could be a more complex digenic or polygenic disorder. Interestingly, both sisters were found inherited SLC26A4 and GJB2 monoallellic mutation from their unaffected father. Loss of function in both SLC26A4 and GJB2 have been implicated in syndromic and non-syndromic hearing loss [10, 37, 38]. Whilst SLC26A4 defects mainly attributed to syndromic hearing loss, GJB2 mutations accounts for up to 50% of all recessive non-syndromic hearing loss based on ethnic background [39]. Essentially, SLC26A4 involves in maintaining the endocochlear potential [35, 36], whereas GJB2 play role in auditory transduction by recycling potassium ions back to the endolymph of the cochlear duct [40]. Given that both genes play pivotal roles in maintaining normal hearing function, we postulated that the SLC26A4 and GJB2 missense mutations are among the PDS driver mutations in this family. In addition, in contrast with earlier studies which have shown biallelic mutation of SLC26A4 to be correlated with bilateral EVA, while monoallelic mutation or zero mutation of SLC26A4 correlated with unilateral EVA [35, 39, 41], we did not observe the association of this monoallelic SLC26A4 mutation and the severity of cochlea anomalies. Both sisters with SLC26A4 monoallelic mutation had incomplete partition type II abnormalities and presented with bilateral hearing loss at the age of 13 and 8 years old respectively.
As the evidence showing SLC26A4 and/or GJB2 monoallelic mutation was not sufficient to cause PDS in this family, we explored the implication of other possible causal mutations. Our analysis discovered pathogenic heterozygous mutation in another deafness associated gene, SCARB2 (MIM #602257), in both siblings and mother. SCARB2 encodes for lysosomal integral membrane protein type 2, which is involved in membrane transportation and the reorganization of endosomal and lysosomal compartment. An earlier study has shown loss of function in SCARB2 being implicated in hearing loss, whereby the SCARB2 knockout mice manifested cochlear deafness, which is associated with massive spiral ganglion neuron losses, concomitant with loss of the inner and outer hair cells and a strongly impaired capacity to generate an endocochlear potential [42]. Beyond that, mutational analysis also identified that SCARB2 mutation was associated with hearing impairment [43, 44]. Given that both sisters inherited similar SCARB2, SLC26A4 and GJB2 mutations from their unaffected parents, our data supports the notion that a combination of these 3 heterozygous mutations may led to bilateral hearing loss in these 2 sisters.
In addition, we detected a compound heterozygous mutation in DUOX2 (p.Lys530* & p.Arg1110Gln) in both siblings. DUOX2 encodes for a key enzyme required to generate hydrogen peroxide (H2O2) which is essential for thyroid hormone synthesis and normal thyroid function [45, 46]. It has been well documented that mutations in DUOX2 are associated with congenital hypothyroidism [47–51]. For instance, biallelic and triallelic mutations in DUOX2 are associated with permanent congenital hypothyroidism, whilst mononoallelic mutation caused transient congenital hypothyroidism [49, 51]. Mutation p.Lys530* and p.Arg1110Gln in DUOX2 were found in patients with transient congenital hypothyroidism [49]. Earlier studies also have shown that p.Arg1110Glu in DUOX2 reduced H2O2 production (5–9%, P < 0.01), hence contributed towards transient congenital hypothyroidism [48, 52]. Our analysis suggested that the DUOX2 compound heterozygous mutations in both sisters may be involved in permanent congenital hypothyroidism, and correlated with significant goiter manifestation at a young age. DUOX2 mutational screening may be useful to detect thyroid dysfunction as compared to perchlorate discharge test, and to differentiate between PDS and other hearing loss diseases.
Taken together, our analysis suggested that PDS in this family could be a complex polygenic disorder which attributed to a combination of 3 heterozygous mutations implicated in deafness-related genes (SLC26A4:p.Ser448Leu; GJB2:p.Thr123Asn; SCARB2:p.Thr305Met), as well as a compound heterozygous mutation implicated in gene associated with thyroid function (DUOX2:p.Lys530* & p.Arg1110Gln).
Conclusions
In summary, our findings showed that exome sequencing has enabled the identification of new candidate causal genes underlying PDS, and suggested that PDS could be a complex heritable polygenic disorder. In this case study, we postulated that a combination of SLC26A4, GJB2 and SCARB2 heterozygous mutations may be implicated in deafness, whilst DUOX2 compound heterozygous mutations may be contributed towards thyroid dysfunction. Screening of additional family members and additional PDS cases may be required to strengthen the usefulness of SLC26A4, GJB2, SCARB2 and DUOX2 as candidate diagnostic biomarkers for PDS.
Abbreviations
- EVA:
-
Enlarged vestibular aqueduct
- H2O2 :
-
Hydrogen peroxide
- MRI:
-
Magnetic resonance image
- PDS:
-
Pendred syndrome
- WES:
-
Whole exome sequencing
References
Vele O, Schrijver I. Inherited hearing loss: molecular genetics and diagnostic testing. Expert Opin Med Diagn. 2008;2(3):231–48.
Yan D, Tekin M, Blanton SH, Liu XZ. Next-generation sequencing in genetic hearing loss. Genet Test Mol Biomarkers. 2013;17(8):581–7.
Everett LA, Glaser B, Beck JC, Idol JR, Buchs A, Heyman M, Adawi F, Hazani E, Nassir E, Baxevanis AD, et al. Pendred syndrome is caused by mutations in a putative sulphate transporter gene (PDS). Nat Genet. 1997;17(4):411–22.
Kopp P. Mutations in the Pendred Syndrome (PDS/SLC26A) gene: an increasingly complex phenotypic spectrum from goiter to thyroid hypoplasia. J Clin Endocrinol Metab. 2014;99(1):67–9.
O’Mahoney CF, Luxon LM, Chew SL, Wass JL. When the triad of congenital hearing loss, goitre and perchlorate positive is not Pendred syndrome. J Audiol Med. 2016;5(3):157–65.
Borck G, Napiontek U, Pfarr N, Müller-Forell W, Keilmann A, Pohlenz J. Congenital deafness and goiter: pendred syndrome. Dtsch Arztebl. 2006;103(46):A 3108–14.
Reardon W, Coffey R, Chowdhury T, Grossman A, Jan H, Britton K, Kendall-Taylor P, Trembath R. Prevalence, age of onset, and natural history of thyroid disease in Pendred syndrome. J Med Genet. 1999;36(8):595–8.
Rose SR, Brown RS, Foley T, Kaplowitz PB, Kaye CI, Sundararajan S, Varma SK. Pediatrics AAo, Section on Endocrinology and Committee on Genetics AeTA, Public Health Committee LwWPES: Update of newborn screening and therapy for congenital hypothyroidism. Pediatrics. 2006;117(6):2290–2303.21.
Pique LM, Brennan ML, Davidson CJ, Schaefer F, Greinwald J, Schrijver I. Mutation analysis of the SLC26A4, FOXI1 and KCNJ10 genes in individuals with congenital hearing loss. PeerJ. 2014;2, e384.
Ben Said M, Dhouib H, BenZina Z, Ghorbel A, Moreno F, Masmoudi S, Ayadi H, Hmani-Aifa M. Segregation of a new mutation in SLC26A4 and p.E47X mutation in GJB2 within a consanguineous Tunisian family affected with Pendred syndrome. Int J Pediatr Otorhinolaryngol. 2012;76(6):832–6.
Yang T, Vidarsson H, Rodrigo-Blomqvist S, Rosengren SS, Enerback S, Smith RJ. Transcriptional control of SLC26A4 is involved in Pendred syndrome and nonsyndromic enlargement of vestibular aqueduct (DFNB4). Am J Hum Genet. 2007;80(6):1055–63.
Reardon W, Coffey R, Pembrey ME, et al. Pitfalls in practice- diagnosis and misdiagnosis in Pendred syndrome. J Audiol Med. 1997;6:1–9.
Ng SB, Buckingham KJ, Lee C, Bigham AW, Tabor HK, Dent KM, Huff CD, Shannon PT, Jabs EW, Nickerson DA, et al. Exome sequencing identifies the cause of a mendelian disorder. Nat Genet. 2010;42(1):30–5.
Boycott KM, Dyment DA, Sawyer SL, Vanstone MR, Beaulieu CL. Identification of genes for childhood heritable diseases. Annu Rev Med. 2014;65:19–31.
Cingolani P, Patel VM, Coon M, Nguyen T, Land SJ, Ruden DM, Lu X. Using Drosophila melanogaster as a Model for Genotoxic Chemical Mutational Studies with a New Program, SnpSift. Front Genet. 2012;3:35.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16), e164.
Thorvaldsdóttir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14(2):178–92.
Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat Biotechnol. 2011;29(1):24–6.
Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073–81.
Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet. 2013; Chapter 7:Unit7.20.
Schwarz JM, Rödelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010;7(8):575–6.
Shihab HA, Gough J, Cooper DN, Stenson PD, Barker GL, Edwards KJ, Day IN, Gaunt TR. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum Mutat. 2013;34(1):57–65.
Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310–5.
Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the functional effect of amino acid substitutions and indels. PLoS One. 2012;7(10), e46688.
Quang D, Chen Y, Xie X. DANN: a deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics. 2015;31(5):761–3.
Reardon W, Trembath RC. Pendred syndrome. J Med Genet. 1996;33(12):1037–40.
Kopp P. Pendred’s syndrome and genetic defects in thyroid hormone synthesis. Rev Endocr Metab Disord. 2000;1:109–12.
Phelps PD, Coffey RA, Trembath RC, Luxon LM, Grossman AB, Britton KE, Kendall-Taylor P, Graham JM, Cadge BC, Stephens SG, et al. Radiological malformations of the ear in Pendred syndrome. Clin Radiol. 1998;53(4):268–73.
FRASER GR, MORGANS ME, TROTTER WR. The syndrome of sporadic goitre and congenital deafness. Q J Med. 1960;29:279–95.
Cremers CW, Admiraal RJ, Huygen PL, Bolder C, Everett LA, Joosten FB, Green ED, van Camp G, Otten BJ. Progressive hearing loss, hypoplasia of the cochlea and widened vestibular aqueducts are very common features in Pendred’s syndrome. Int J Pediatr Otorhinolaryngol. 1998;45(2):113–23.
FRASER GR. Association of congenital deafness with goitre (Pendred’s Syndrome) a study of 207 families. Ann Hum Genet. 1965;28:201–49.
Johnsen T, Larsen C, Friis J, Hougaard-Jensen F. Pendred’s syndrome: acoustic, vestibular and radiological findings in 17 unrelated patients. J Laryngol Otol. 1987;101:1187–92.
Coyle B, Coffey R, Armour JA, Gausden E, Hochberg Z, Grossman A, Britton K, Pembrey M, Reardon W, Trembath R. Pendred syndrome (goitre and sensorineural hearing loss) maps to chromosome 7 in the region containing the nonsyndromic deafness gene DFNB4. Nat Genet. 1996;12(4):421–3.
Fugazzola L, Mannavola D, Cerutti N, Maghine M, Pagella F, Bianchi P, Weber G, Persani L, Beck-Peccoz P. Molecular analysis of the Pendred’s syndrome gene and magnetic resonance imaging studies of the inner ear are essential for the diagnosis of true Pendred’s syndrome. J Clin Endocrinol Metab. 2000;85:2469–75.
Wu CC, Yeh TH, Chen PJ, Hsu CJ. Prevalent SLC26A4 mutations in patients with enlarged vestibular aqueduct and/or Mondini dysplasia: a unique spectrum of mutations in Taiwan, including a frequent founder mutation. Laryngoscope. 2005;115(6):1060–4.
Pryor SP, Madeo AC, Reynolds JC, Sarlis NJ, Arnos KS, Nance WE, Yang Y, Zalewski CK, Brewer CC, Butman JA, et al. SLC26A4/PDS genotype-phenotype correlation in hearing loss with enlargement of the vestibular aqueduct (EVA): evidence that Pendred syndrome and non-syndromic EVA are distinct clinical and genetic entities. J Med Genet. 2005;42(2):159–65.
Chang KW. Genetics of hearing loss--nonsyndromic. Otolaryngol Clin North Am. 2015;48(6):1063–72.
Albert S, Blons H, Jonard L, Feldmann D, Chauvin P, Loundon N, Sergent-Allaoui A, Houang M, Joannard A, Schmerber S, et al. SLC26A4 gene is frequently involved in nonsyndromic hearing impairment with enlarged vestibular aqueduct in Caucasian populations. Eur J Hum Genet. 2006;14(6):773–9.
Rabionet R, Zelante L, López-Bigas N, D’Agruma L, Melchionda S, Restagno G, Arbonés ML, Gasparini P, Estivill X. Molecular basis of childhood deafness resulting from mutations in the GJB2 (connexin 26) gene. Hum Genet. 2000;106(1):40–4.
Kikuchi T, Kimura RS, Paul DL, Adams JC. Gap junctions in the rat cochlea: immunohistochemical and ultrastructural analysis. Anat Embryol (Berl). 1995;191(2):101–18.
King KA, Choi BY, Zalewski C, Madeo AC, Manichaikul A, Pryor SP, Ferruggiaro A, Eisenman D, Kim HJ, Niparko J, et al. SLC26A4 genotype, but not cochlear radiologic structure, is correlated with hearing loss in ears with an enlarged vestibular aqueduct. Laryngoscope. 2010;120(2):384–9.
Gamp AC, Tanaka Y, Lüllmann-Rauch R, Wittke D, D’Hooge R, De Deyn PP, Moser T, Maier H, Hartmann D, Reiss K, et al. LIMP-2/LGP85 deficiency causes ureteric pelvic junction obstruction, deafness and peripheral neuropathy in mice. Hum Mol Genet. 2003;12(6):631–46.
Perandones C, Micheli FE, Pellene LA, Bayly MA, Berkovic SF, Dibbens LM. A case of severe hearing loss in action myoclonus renal failure syndrome resulting from mutation in SCARB2. Mov Disord. 2012;27(9):1200–1.
Rubboli G, Franceschetti S, Berkovic SF, Canafoglia L, Gambardella A, Dibbens LM, Riguzzi P, Campieri C, Magaudda A, Tassinari CA, et al. Clinical and neurophysiologic features of progressive myoclonus epilepsy without renal failure caused by SCARB2 mutations. Epilepsia. 2011;52(12):2356–63.
Ohye H, Sugawara M. Dual oxidase, hydrogen peroxide and thyroid diseases. Exp Biol Med (Maywood). 2010;235(4):424–33.
Maruo Y, Takahashi H, Soeda I, Nishikura N, Matsui K, Ota Y, Mimura Y, Mori A, Sato H, Takeuchi Y. Transient congenital hypothyroidism caused by biallelic mutations of the dual oxidase 2 gene in Japanese patients detected by a neonatal screening program. J Clin Endocrinol Metab. 2008;93(11):4261–7.
Park KJ, Park HK, Kim YJ, Lee KR, Park JH, Park HD, Lee SY, Kim JW. DUOX2 mutations are frequently associated with congenital hypothyroidism in the Korean population. Ann Lab Med. 2016;36(2):145–53.
Yoshizawa-Ogasawara A, Abe K, Ogikubo S, Narumi S, Hasegawa T, Satoh M. Transient congenital hypothyroidism caused by compound heterozygous mutations affecting the NADPH-oxidase domain of DUOX2. J Pediatr Endocrinol Metab. 2016;29(3):363–71.
Fu C, Zhang S, Su J, Luo S, Zheng H, Wang J, Qin H, Chen Y, Shen Y, Hu X, et al. Mutation screening of DUOX2 in Chinese patients with congenital hypothyroidism. J Endocrinol Invest. 2015;38(11):1219–24.
Jin HY, Heo SH, Kim YM, Kim GH, Choi JH, Lee BH, Yoo HW. High frequency of DUOX2 mutations in transient or permanent congenital hypothyroidism with eutopic thyroid glands. Horm Res Paediatr. 2014;82(4):252–60.
Wang F, Lu K, Yang Z, Zhang S, Lu W, Zhang L, Liu S, Yan S. Genotypes and phenotypes of congenital goitre and hypothyroidism caused by mutations in dual oxidase 2 genes. Clin Endocrinol (Oxf). 2014;81(3):452–7.
Ohye H, Fukata S, Hishinuma A, Kudo T, Nishihara E, Ito M, Kubota S, Amino N, Ieiri T, Kuma K, et al. A novel homozygous missense mutation of the dual oxidase 2 (DUOX2) gene in an adult patient with large goiter. Thyroid. 2008;18(5):561–6.
Acknowledgements
Not applicable.
Funding
This work was supported by Genomic Unit PPUKM-UMBI fund.
Availability of data and materials
Data are available with our restriction. All relevant data are included within the paper.
Authors’ contributions
CYP and NAAM and involved in the study design, data analysis and manuscript drafting. ZZMR and NAAM performed exome sequencing. ZZMR performed Sanger sequencing. KJS and CPS participated in the data analysis. WLL performed clinical evaluation of the patients. RJ involved in the study design and critical evaluation of the manuscript. All authors read and approved the final manuscript.
Authors’ information
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not Applicable.
Ethics approval and consent to participate
Informed consent was obtained from all subjects involved in this study.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1: Table S4.
List of PCR primers used. (XLSX 9 kb)
Additional file 2: Table S1.
List of non-synonymous heterozygous mutations inherited from father. (XLSX 94 kb)
Additional file 3: Table S2.
List of non-synonymous heterozygous mutations inherited from mother. (XLSX 91 kb)
Additional file 4: Table S3.
List of other possible PDS causal mutations which uniquely inherited by the eldest sister. (XLSX 11 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Chow, YP., Abdul Murad, N., Mohd Rani, Z. et al. Exome sequencing identifies SLC26A4, GJB2, SCARB2 and DUOX2 mutations in 2 siblings with Pendred syndrome in a Malaysian family. Orphanet J Rare Dis 12, 40 (2017). https://doi.org/10.1186/s13023-017-0575-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-017-0575-7