
Abstract
Background
Cerebellar atrophy and developmental delay are commonly associated features in large numbers of genetic diseases that frequently also include epilepsy. These defects are highly heterogeneous on both the genetic and clinical levels. Patients with these signs also typically present with non-specific neuroimaging results that can help prioritize further investigation but don’t suggest a specific molecular diagnosis.
Methods
To genetically explore a cohort of 18 Egyptian families with undiagnosed cerebellar atrophy identified on MRI, we sequenced probands and some non-affected family members via high-coverage whole exome sequencing (WES; >97 % of the exome covered at least by 30x). Patients were mostly from consanguineous families, either sporadic or multiplex. We analyzed WES data and filtered variants according to dominant and recessive inheritance models.
Results
We successfully identified disease-causing mutations in half of the families screened (9/18). These mutations are located in seven different genes, PLA2G6 being the gene most frequently mutated (n = 3). We also identified a recurrent de novo mutation in the KIF1A gene and a molybdenum cofactor deficiency caused by the loss of the start codon in the MOCS2A open-reading frame in a mildly affected subject.
Conclusions
This study illustrates the necessity of screening for dominant mutations in WES data from consanguineous families. Our identification of a patient with a mild and improving phenotype carrying a previously characterized severe loss of function mutation also broadens the clinical spectrum associated with molybdenum cofactor deficiency.
Similar content being viewed by others


Background
Atrophy and hypoplasia of the cerebellum are neuroradiological findings identified in pediatric-onset cerebellar ataxias and generally associated with imbalance, poor coordination and developmental delay. Hereditary cerebellar atrophy in childhood is a clinically and genetically heterogeneous group of conditions. They include a vast number of differential diagnoses with overlapping clinical findings such as intellectual disability and epilepsy [1, 2].
The specificity of cerebellar atrophy as a neuroradiological finding has been formerly discussed [2]. In association with a careful initial clinical evaluation, the characterization of cerebellar atrophy and/or hypoplasia by MRI is currently used to prioritize specialized investigations and potential diagnosis [1, 3].
The most frequent causes of early-onset cerebellar atrophy include mitochondrial disorders, neuronal ceroid lipofuscinosis, congenital disorders of glycosylation, ataxia telangiectasia and infantile neuroaxonal dystrophy [1]. The reason for the exquisite sensitivity of the cerebellum to defects in general cellular processes involving mitochondria, protein glycosylation or lysosomes is not known. The loss of Purkinje cells could play a central role in the pathology of many cerebellar ataxias as they are highly metabolic cells and these cells are the only out-put of the cerebellum.
This group of disorders includes an ever-increasing number of very rare conditions and currently over 169 OMIM clinical synopses are associated with cerebellar atrophy (OMIM December 2015). The overlapping clinical features of these numerous conditions often prevent a rapid and accurate clinical and genetic diagnosis and for more than half of the patients with childhood-onset cerebellar atrophy, a molecular diagnosis is not available [3].
Additionally, many of these conditions start with motor and cognitive delay or deterioration but some diagnostic criteria appear later during the developmental course of the disease. For example, in the case of PLA2G6-associated neurodegeneration (PLAN), brain iron accumulation is generally not detected in the early stages of the disease [4, 5].
In such situations whole exome sequencing (WES) has been able to accelerate molecular diagnosis, better delineate the clinical spectrum associated with specific genetic defects and improve patient management [6, 7].
In this study, we assessed a cohort of 18 families of Egyptian origin with childhood-onset cerebellar atrophy and developmental delay. We analyzed them for both recessive and dominant variants and successfully identified disease-causing mutations in half of the families (9/18). This study highlights the importance of searching for dominant mutations in consanguineous families and broadens the clinical spectrum associated with some cerebellar conditions, including molybdenum cofactor deficiency.
Methods
Subject information
We studied 18 families including 12 sporadic cases (67 %) and 16 families with reported consanguinity (89 %). For half of these families only the proband was exome sequenced and for the other half we performed trio exome sequencing. The mean age was 4.6 years (Standard deviation 3.2 years) at the time of the study. Patients had various degrees of cerebellar atrophy identified on MRI, involving mainly the vermis or the entire cerebellum. Both static and progressive conditions were included. Written informed consent was obtained from all families, and the study was approved by the ethics committee of the National Research Center in Cairo.
Whole exome sequencing
DNA was extracted from blood and the sequencing core facility at the Imagine Institute performed WES. Briefly, WES libraries were prepared from 3 μg of genomic DNA sheared by ultrasonication (Covaris S220 Ultrasonicator). Exome capture was performed with the 51 Mb SureSelect Human All Exon kit V5 (Agilent technologies). Sequencing of the WES libraries was carried out on a HiSeq2500 (Illumina). Paired-end reads were generated and mapped on the human genome reference using Burrows-Wheeler Aligner (BWA) [8]. The mean depth of coverage obtained for each sample was > 160x with >97 % of the exome covered at least 30x. SNP and indel calling was made using GATK tools. Families CIE3, 4, 5, 8, 11, 12, 13, 18 and 19 (n = 9; 50 %) were investigated by trio WES and for families CIE1, 2, 7, 9, 14, 16, 17, 21, 29 (n = 9; 50 %) WES was only performed for the proband.
Bioinformatics, databases
A variant filtering pipeline was systematically applied to narrow down the number of putative causative variants. All the possible inheritance patterns were tested. Briefly, common (>1 % minor allele frequency) variants were filtered out by using dbSNP, 1000 genomes databases and our in house exome collection, which includes more than 7000 exomes. Functional (protein-altering) alleles were prioritized versus non-functional. Potentially pathogenic variants in known disease genes were identified if flagged as damaging by polyphen2 (http://genetics.bwh.harvard.edu/pph2/), Sift (http://sift.jcvi.org/) or mutation taster (http://www.mutationtaster.org/). Remaining variants were compared with those in the public databases EXAC (http://exac.broadinstitute.org/) and EVS (http://evs.gs.washington.edu/EVS/) exome database. The presence of candidate recessive variants in homozygous intervals was checked by identifying predicted regions of SNP homozygosity from exome data with the unifiedgenotyper tool from GATK (https://www.broadinstitute.org/gatk/). In order to identify fully penetrant dominant mutation in singleton WES data we used the following method. We filtered out variants that were present in control individuals from our in house exome database and not predicted to be pathogenic by at least two prediction programs: PolyPhen, SIFT or Mutation-Taster. When a single rare variant predicted to be deleterious was observed in a single known cerebellar atrophy gene and associated with a dominant mode of inheritance, it was considered as potential candidate. When the same rare variant has been previously associated with disease, this was considered strong evidence that it was likely to be pathogenic. We validated potential de novo mutations by using Sanger sequencing on patients and parents DNA. However considering the large number of variants generated by this method, it is considered efficient only for the identification of mutations in known disease genes.
Results
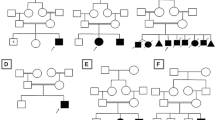
Homozygous, compound heterozygous and potential de novo mutations were investigated in all families. We identified pathological mutations in 7 genes within nine families (9/18; 50 %): a homozygous TPP1 mutation c.790C > T (NM_000391.3) p.Gln264* in family CIE7; a homozygous EXOSC3 mutation c.395A > C (NM_016042.3) p.Asp132Ala in CIE9; a homozygous PLA2G6 c.2070_2072del (NM_003560.2) p.Val691del mutation in families CIE11 and CIE13; a homozygous MOCS2 c.3G > A (NM_176806.3) p.Met1? mutation in CIE12; a homozygous SURF1 mutation c.237G > A (NM_003172.3) p.Trp79* in CIE16; a homozygous MFSD8 c.1213C > T (NM_152778.2) p.Gln405* mutation in family CIE17; a de novo KIF1A mutation c.173C > T (NM_001244008.1) p.Ser58Leu in family CIE21 and a homozygous PLA2G6 mutation c.1613G > A (NM_003560.2) p.Arg538His in family CIE29. Mutation segregation was checked by Sanger sequencing in all available samples (indicated by a star on Fig. 1).

Pedigrees of families with identified disease-causing genes. A star indicates individuals with DNA available for segregation test and arrowheads point to the samples sequenced by WES
Clinical findings in the affected individuals with molecular diagnosis are summarized in Table 1. Previously described mutations were identified in six genes TPP1, EXOSC3, PLA2G6, MOCS2 and SURF1 and KIF1A. The PLA2G6 gene was the most frequently mutated gene and the c.2070_2072del mutation was identified in two families in our WES data. The identification of this recurrent mutation during the course of the project, led us to implement Sanger sequencing of this specific allele and only samples negative for this prescreen where submitted to WES. Using this method, we could identify a third family with the same c.2070_2072del mutation. In total, this 3 bp deletions accounts for 15 % of all the families tested in this study. The identification of MFSD8 mutations in family CIE17 was consistent with an early onset of ceroid lipofuscinosis, neuronal 7 (CLN7) associated with cortical and cerebellar atrophy and an enlargement of the cerebral ventricles in the proband (Fig. 2a-c), a phenotype that usually manifests between 2 and 6 years old [9]. In family CIE21, we identified a recurrent de novo mutation in the KIF1A gene. This mutation is predicted to disrupt some hydrogen bonds in the ATP binding domain (Additional file 1: Figure S1) and results in cerebellar atrophy (Fig. 2d-f) and epilepsy (Table 1). In family CIE12, we found biallelic mutations in the initiation codon of the MOCS2A open reading frame of the bicistronic MOCS2 gene. Mutations in this gene cause Molybdenum Cofactor (MoCo) deficiency, a severe neonatal inborn error of metabolism usually associated with subcortical multicystic lesions and death during early childhood stage [10]. Patient CIE12-A2 was born to consanguineous parents and was affected since birth. At 3.8 years, she presented with delayed speech and delayed walking. She had wide based gait, hyperreflexia and hypertonia with the left side of her body being more affected than the right. The mother had an uncomplicated pregnancy although she was exposed to X-Rays when she was one month pregnant. This patient has two unaffected siblings, one deceased brother and an older brother who suffered from global development delay since birth and mild cortical brain atrophy on MRI associated with abnormal EEG. At 6.0 years, patient CIE12-A2 appeared to improve gradually. Despite some residual unsteadiness, she can walk, read and memorize short sentences. Her initial investigations included CT and MRI, which revealed right frontal arachnoid cyst, mild right frontal lobe brain atrophy and cerebellar atrophy (Fig. 2g-i). A follow up CT two years latter indicated that the arachnoid cyst had slightly resolved. Her EEG revealed left fronto-temporal epileptogenic dysfunction. The follow up EEGs showed improvement in the epileptogenic bursts probably as a result of the antiepileptic therapy she is receiving. The molecular diagnosis was supported by the blood tests results, consistent with Molybdenum Cofactor deficiency. Plasma S-Sulphocysteine level was 309 micromol/mmol (Control value <10) and Plasma Xanthine 1291 micromol/mmol (Control value <40). She is currently receiving sodium valproate (30mh/kg), Levitiracetam (20 mg/kg), omega 3 supplements, and IM (intra-muscular) B complex.

Brain MRI scans from families CIE17, CIE21 and CIE12. a-c MRIs of case CIE17-A1 (MFSD8 mutation) at 3 years. a Midline Sagittal T2-weighted image demonstrating enlarged cerebellar folia (arrow head) and thin corpus callosum. Ventricular system dilatation (star) is visible on coronal T1 image (b). d-f MRIs of patient CIE-21-A1 (KIF1A mutation) at 5 years showing cerebellar atrophy (arrowhead) on midline sagittal T2 (d), coronal T1 (e) and axial T1 (f) images. g-i MRIs of patient CIE12-A2 (MOCS2 mutation) with evidence of cerebellar vermis atrophy (arrowhead) on sagittal T1 image (g) and Right frontal arachnoid cyst (red arrow) visible on axial T2 scan (h)
Discussion
In this study, we explored sporadic and multiplex Egyptian families with early-onset cerebellar atrophy associated with developmental delay by using high-coverage whole exome sequencing. Our analysis included the investigation of recessive and dominant variants and we identified pathogenic mutations in 50 % of the 18 families studied. Previous reports that focused on sporadic and inherited cerebellar atrophies [11] or ataxias [12] and obtained comparable success rates of 39.1 and 41 % respectively, in the identification of pathological mutations.
The absences of diagnosis for half of the cohort could be explain with the following reasons. Despite high depth of sequencing (mean depth of coverage > 160x), exome capture does not provide complete coverage of all coding regions of the genome, particularly those with GC-rich regions. Moreover, large genomic rearrangements and trinucleotide repeat sequences are not reliably detected from exome-capture data. It is also possible that some causal variants will reside within non-coding regulatory regions. Some of these issues will be resolved by whole genome sequencing, although not without substantial additional cost and bioinformatics analyses. However it is likely that mutation in yet unknown cerebellar atrophy genes contribute significantly to these diseases. In this study, half of the patients were explored by using trio WES and the other half by singletons WES. Trio WES did not improve the diagnosis rate but considerably decreased the number of Sanger cosegregation analyses. It impacted specially de novo mutations analysis but singleton WES did not prevent de novo mutation identification as illustrated with the KIF1A gene mutation. One of the clear advantages of trio WES is its ability to point to a limited number of variants located in novel candidate disease-causing genes when mutations located in known disease gene are absent. The identification of KIF1A de novo mutation highlights the importance of considering not only recessive inheritance patterns when analyzing consanguineous exomes despite the presence of extensive regions of homozygosity (data not shown). This finding identifies p.Ser58Leu as a recurrent KIF1A mutation [13] and indicates that cerebellar atrophy is the main MRI feature associated with this allele. Several typical symptoms of infantile-onset PLA2G6-associated neurodegeneration (PLAN) are not always observed before 4 years, such as optic atrophy, electroencephalogram fast rhythms and amyotrophy [4], making very early diagnosis more difficult. PLA2G6 is the most frequently mutated gene in this cohort; 3 families were identified with the same homozygous mutation previously described as a founder mutation in Mediterranean countries [4]. This observation suggests that the p.Val691del allele is an especially common cause of PLAN in the Egyptian population. Among other clinical symptoms, patients with this mutation shared the association of development delay or regression with cerebellar atrophy, epilepsy and nystagmus. Mutations in the MOCS2 genes cause MoCo deficiency type B, which is currently untreatable [14]. These mutations can occur in one of the two open-reading frames (i.e., MOCS2A and MOCS2B) of this bi-cistronic gene and are generally associated with untreatable seizures, multiple cystic cavities on MRI and death at early age [10]. Milder cases have been reported to be associated with a hypomorphic allele [15] or mutations involving a non-constitutively spliced exon [16]. Strikingly, the p.Met1? mutation identified in MOCS2A has been tested in vitro and shown to abolish translation [17]. This result highlights the existence of mild presentations of MoCo syndrome, potentially detectable with plasma sulfite and xanthine screening of patients with undiagnosed cerebellar atrophy, developmental delay and isolated arachnoid cyst.
Conclusions
Our study emphasizes the benefits of whole exome sequencing to efficiently diagnose early-onset cerebellar atrophy defects, to better delineate the clinical spectrum associated with these disorders and to open the way for the identification of new disease genes.
Ethics approval and consent to participate and for publication
Written informed consent was obtained from all families, and the study was approved by the ethics committee of the National Research Center in Cairo.
Abbreviations
- MoCo:
-
molybdenum cofactor
- PLAN:
-
PLA2G6-associated neurodegeneration
- WES:
-
whole exome sequencing
References
Boddaert N, Desguerre I, Bahi-Buisson N, Romano S, Valayannopoulos V, Saillour Y, et al. Posterior fossa imaging in 158 children with ataxia. Journal of neuroradiology Journal de neuroradiologie. 2010;37(4):220–50.
Poretti A, Wolf NI, Boltshauser E. Differential diagnosis of cerebellar atrophy in childhood. Eur J Paediatr Neurol. 2008;12(3):155–67. doi:10.1016/j.ejpn.2007.07.010.
Al-Maawali A, Blaser S, Yoon G. Diagnostic approach to childhood-onset cerebellar atrophy: a 10-year retrospective study of 300 patients. J Child Neurol. 2012;27(9):1121–32. doi:10.1177/0883073812448680.
Romani M, Kraoua I, Micalizzi A, Klaa H, Benrhouma H, Drissi C, et al. Infantile and childhood onset PLA2G6-associated neurodegeneration in a large North African cohort. Eur J Neurol. 2015;22(1):178–86. doi:10.1111/ene.12552.
Gregory A, Westaway SK, Holm IE, Kotzbauer PT, Hogarth P, Sonek S, et al. Neurodegeneration associated with genetic defects in phospholipase A(2). Neurology. 2008;71(18):1402–9. doi:10.1212/01.wnl.0000327094.67726.28.
Deciphering Developmental Disorders S. Large-scale discovery of novel genetic causes of developmental disorders. Nature. 2015;519(7542):223–8. doi:10.1038/nature14135.
Dixon-Salazar TJ, Silhavy JL, Udpa N, Schroth J, Bielas S, Schaffer AE, et al. Exome sequencing can improve diagnosis and alter patient management. Sci Transl Med. 2012;4(138):138ra78. doi:10.1126/scitranslmed.3003544.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–60. doi:10.1093/bioinformatics/btp324.
Kousi M, Lehesjoki AE, Mole SE. Update of the mutation spectrum and clinical correlations of over 360 mutations in eight genes that underlie the neuronal ceroid lipofuscinoses. Hum Mutat. 2012;33(1):42–63. doi:10.1002/humu.21624.
Reiss J, Johnson JL. Mutations in the molybdenum cofactor biosynthetic genes MOCS1, MOCS2, and GEPH. Hum Mutat. 2003;21(6):569–76. doi:10.1002/humu.10223.
Ohba C, Osaka H, Iai M, Yamashita S, Suzuki Y, Aida N, et al. Diagnostic utility of whole exome sequencing in patients showing cerebellar and/or vermis atrophy in childhood. Neurogenetics. 2013;14(3–4):225–32. doi:10.1007/s10048-013-0375-8.
Pyle A, Smertenko T, Bargiela D, Griffin H, Duff J, Appleton M, et al. Exome sequencing in undiagnosed inherited and sporadic ataxias. Brain. 2015;138(Pt 2):276–83. doi:10.1093/brain/awu348.
Lee JR, Srour M, Kim D, Hamdan FF, Lim SH, Brunel-Guitton C, et al. De novo mutations in the motor domain of KIF1A cause cognitive impairment, spastic paraparesis, axonal neuropathy, and cerebellar atrophy. Hum Mutat. 2015;36(1):69–78. doi:10.1002/humu.22709.
Schwahn BC, Van Spronsen FJ, Belaidi AA, Bowhay S, Christodoulou J, Derks TG et al. Efficacy and safety of cyclic pyranopterin monophosphate substitution in severe molybdenum cofactor deficiency type A: a prospective cohort study. Lancet. 2015. doi:10.1016/S0140-6736(15)00124-5.
Leimkuhler S, Freuer A, Araujo JA, Rajagopalan KV, Mendel RR. Mechanistic studies of human molybdopterin synthase reaction and characterization of mutants identified in group B patients of molybdenum cofactor deficiency. J Biol Chem. 2003;278(28):26127–34. doi:10.1074/jbc.M303092200.
Arenas M, Fairbanks LD, Vijayakumar K, Carr L, Escuredo E, Marinaki AM. An unusual genetic variant in the MOCS1 gene leads to complete missplicing of an alternatively spliced exon in a patient with molybdenum cofactor deficiency. J Inherit Metab Dis. 2009;32(4):560–9. doi:10.1007/s10545-009-1151-7.
Stallmeyer B, Drugeon G, Reiss J, Haenni AL, Mendel RR. Human molybdopterin synthase gene: identification of a bicistronic transcript with overlapping reading frames. Am J Hum Genet. 1999;64(3):698–705. doi:10.1086/302295.
Acknowledgments
This project was funded by grant ANR-12-PDOC-0026 that supported also VC and MN salary and by grant ANR-10-IAHU-01. We are grateful to Campus France that allowed the French-Egyptian collaboration and supported some travel and accommodation fees for HM. We acknowledge the assistance and expert advice from collaborators at the genomic and bioinformatics cores of the Imagine institute. We thank L. S. Nguyen and C. Gordon for critical reading of the manuscript.
Funding
The project was funded by grant ANR-12-PDOC-0026, ANR-10-IAHU-01 and Campus France.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
HM ascertained all the Egyptian patients and participated in the WES analysis. MN coordinated samples preparation, analyzed and validated the data with the help of KSP and DMC; CBF coordinated the exome sequencing. CM and PN designed and adjusted the bioinformatics pipeline to analyze sequence data. GB, MR, ID, NBB and AM reviewed clinical data in regard of the WES results. NB reviewed brain imaging. VC analyzed the data, directed the study and wrote the manuscript with the help of LC. All authors read and approved the final manuscript.
Additional file
Additional file 1: Figure S1.
Modeling of the p.S58L mutation using the crystal structure of the motor domain of human KIF1A (PDB number 1VFV). The image was generated using PyMOL (http://www.pymol.org). The side-chain of Ser58 is involved in inferred hydrogen bonds (arrow) with other amino-acids located in the ATP-binding pocket (A). These interactions are predicted to be disrupted by the mutation (B) including the hydrogen bonds with the highly conserved Arginine 11 that interacts with ATP through a molecule of water. (JPG 499 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

{kind=link}
Cite this article
Megahed, H., Nicouleau, M., Barcia, G. et al. Utility of whole exome sequencing for the early diagnosis of pediatric-onset cerebellar atrophy associated with developmental delay in an inbred population. Orphanet J Rare Dis 11, 57 (2016). https://doi.org/10.1186/s13023-016-0436-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-016-0436-9