
Abstract
Background
Rapid-onset Obesity with Hypothalamic Dysfunction, Hypoventilation, and Autonomic Dysregulation (ROHHAD) is thought to be a genetic disease caused by de novo mutations, though causative mutations have yet to be identified. We searched for de novo coding mutations among a carefully-diagnosed and clinically homogeneous cohort of 35 ROHHAD patients.
Methods
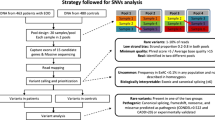
We sequenced the exomes of seven ROHHAD trios, plus tumours from four of these patients and the unaffected monozygotic (MZ) twin of one (discovery cohort), to identify constitutional and somatic de novo sequence variants. We further analyzed this exome data to search for candidate genes under autosomal dominant and recessive models, and to identify structural variations. Candidate genes were tested by exome or Sanger sequencing in a replication cohort of 28 ROHHAD singletons.
Results
The analysis of the trio-based exomes found 13 de novo variants. However, no two patients had de novo variants in the same gene, and additional patient exomes and mutation analysis in the replication cohort did not provide strong genetic evidence to implicate any of these sequence variants in ROHHAD. Somatic comparisons revealed no coding differences between any blood and tumour samples, or between the two discordant MZ twins. Neither autosomal dominant nor recessive analysis yielded candidate genes for ROHHAD, and we did not identify any potentially causative structural variations.
Conclusions
Clinical exome sequencing is highly unlikely to be a useful diagnostic test in patients with true ROHHAD. As ROHHAD has a high risk for fatality if not properly managed, it remains imperative to expand the search for non-exomic genetic risk factors, as well as to investigate other possible mechanisms of disease. In so doing, we will be able to confirm objectively the ROHHAD diagnosis and to contribute to our understanding of obesity, respiratory control, hypothalamic function, and autonomic regulation.
Similar content being viewed by others


Background
Rapid-onset Obesity with Hypothalamic Dysfunction, Hypoventilation, and Autonomic Dysregulation (ROHHAD) is a complex and devastating disease whose etiology is poorly understood, despite its initial description 50 years ago [1]. The condition, previously termed “late-onset central hypoventilation syndrome with hypothalamic dysfunction” is extremely rare, with fewer than 100 cases reported in the literature, and occurs sporadically, with no clear-cut family history for inheritance of the phenotype [1–7]. In addition to the hypothalamic, respiratory and autonomic manifestations that are hallmarks of the disease, about 40 % of ROHHAD patients develop benign tumours of neural crest origin [2–4]. Because of the heralding feature of the rapid-onset obesity (20–30 lb over a 3–6 month period in, typically, a 2–7 year old, otherwise healthy, child), affected children should come to the attention of their pediatricians early in the clinical course. However, due to the variable timing and onset of other features, coupled with the presumption of exogenous obesity, a ROHHAD diagnosis is often delayed or missed, potentially leading to fatal central hypoventilation, cardiorespiratory arrest, and impaired neurocognitive development. Conversely, many children with marked and potentially rapid weight gain, who do not meet the additional clinical criteria for ROHHAD, are inappropriately labeled as having ROHHAD. While diagnostic criteria and knowledge of the disease course have improved since the introduction of the ROHHAD acronym by Ize-Ludlow and coworkers in 2007 [2], these issues remain a challenge. Thus, unambiguous diagnosis of ROHHAD is difficult but essential – not only for scientific inquiry but also for appropriate patient care. The identification of a diagnostic marker (genetic or otherwise) has the potential to decrease morbidity and mortality of patients with ROHHAD, and to guide future intervention and research on this disease.
As many ROHHAD features are reminiscent of other neurocristopathies of genetic origin, such as Congenital Central Hypoventilation Syndrome (CCHS) [7], a genetic basis to ROHHAD has been hypothesized. The sporadic appearance of a syndrome with tumour predisposition and an apparently primary disturbance of body growth is consistent with somatic mosaicism or constitutional inheritance of a de novo dominant mutation (as seen in Sotos syndrome [8], Proteus syndrome [9], CLOVES syndrome [10], and Weaver syndrome [11]), though epigenetic mechanisms could also be postulated, as have been shown to occur in Beckwith-Wiedemann syndrome [12]. However, previous candidate gene studies in ROHHAD using Sanger sequencing have not revealed a clear candidate gene that can account for the full ROHHAD phenotype [2, 4, 13].
To date, most known disease-causing mutations have been identified within the coding portion of the genome. Whole exome sequencing (WES) among small, carefully-phenotyped cohorts has proven to be a very effective means of identifying the cause of rare pediatric diseases, a fact well demonstrated in a large national exome sequencing effort (FORGE Canada Consortium) that found disease-causing variants for 146 rare pediatric diseases, including variants in 67 genes not previously linked to any human disease [14]. Thus, WES is a highly cost-effective and efficient method for solving the genetic basis of rare diseases. To search for both constitutional and somatic de novo sequence variants that could be the cause of this severe pediatric disease, we used WES to analyze seven ROHHAD trios, the unaffected monozygotic (MZ) twin of one of these seven patients, and tumours isolated from four of these seven patients as a discovery cohort. We also assembled a replication cohort of 28 singleton ROHHAD cases in which to analyze candidate genes identified through WES.
Methods
Criteria for preliminary diagnosis
The basic criteria for consideration of the diagnosis of ROHHAD were published in Ize-Ludlow et al. (2007) [2]. Briefly, features included: 1) onset of rapid and extreme weight gain after age 1.5 years (typically 2–7 years) in a previously non-obese and seemingly normal child, 2) evidence of hypothalamic dysfunction, 3) alveolar hypoventilation, and 4) features of autonomic dysregulation.
Patient selection
The Center for Autonomic Medicine in Pediatrics (CAMP) at Ann & Robert H. Lurie Children’s Hospital of Chicago and the Stanley Manne Children’s Research Institute is a Center of Excellence for the study of ROHHAD. Medical records for each proband referred to CAMP were reviewed to confirm ROHHAD characteristics. Cases were then tested and confirmed negative for any CCHS-related PHOX2B gene mutations. All of the patients selected for exome sequencing were evaluated clinically in the CAMP laboratory for serial characterization of the ROHHAD phenotype (control of breathing, hypothalamic dysfunction, and autonomic dysregulation). Patients who met strict diagnostic criteria for ROHHAD were offered inclusion into both the International ROHHAD REDCap Registry and the ROHHAD Genetic Inquiry project (both IRB-approved). In this cohort, all patients demonstrated rapid-onset obesity, hypothalamic dysfunction, hypoventilation requiring artificial ventilation, and autonomic dysregulation. All (35) patients who consented to the Genetic Inquiry project, provided a peripheral blood sample and, where appropriate, provided a frozen neural crest tumour tissue sample, were included in this study. All (7) patients whose parents also consented to the Genetic Inquiry project and provided a peripheral blood sample were included in the discovery cohort for trio analysis. The other 28 participants formed the replication cohort, of which exome sequence data were obtained for 9 and the remaining 19 were analyzed by Sanger sequencing, only in genes of interest.
Ethics, consent and permissions
This study was approved by the Ann & Robert H. Lurie Children’s Hospital of Chicago IRB (study ID: 2009–13904) and the University of Calgary Conjoint Health Research Ethics Board (study ID: REB13-0164_REN2). All participants provided informed consent to participate, including consent to publish the data herein.
Sample collection and DNA extraction
Genomic DNA was isolated from blood and tumour samples, using a Puregene reagent kit (Qiagen). Tumours included both ganglioneuroma and ganglioneuroblastoma samples (Table 1).
Whole exome sequencing and analysis
Exome captures were completed using the Agilent SureSelect V5 + UTRs capture kit. Massively parallel sequencing was performed on a SOLiD platform, at the Alberta Children’s Hospital Research Institute (ACHRI), and sequences were aligned to the human reference genome (GRCh37) using Lifescope Genomic Analysis Software 2.5 (Life Technologies), (see Table 2). Variants were called using DeNovoGear [15] for the trio, twin, and tumour analyses; and the Genome Analysis Toolkit (GATK) Haplotype Caller (Version 3.3) [16] for the recessive and dominant analyses; and annotated for filtration and prioritization using ANNOVAR [17]. For the trio analysis, candidate variants were identified as novel or rare (MAF < 0.005, according to 1000 Genomes Project [18], the Exome Variant Server [EVS; [19]] and the Exome Aggregation Consortium [ExAC; [20]]) exonic, UTR, or splice site (within 2 bp of an exon) variants not within a segmental duplication. Candidate variants were further filtered to exclude UTR variants for the recessive analysis, and both UTR and synonymous variants for the dominant analysis. Candidate variants were assessed using Combined Annotation Dependent Depletion (CADD) [21] and PolyPhen-2 [22] (for non-synonymous variants) to predict deleteriousness. CADD scores are phred-scaled, so a score of 10 indicates the pathogenicity score of a variant is in the top 10 % of the scores for all possible human variants; a score of 20 indicates it is in the top 1 % etc. PolyPhen-2 scores represent the probability that the variant is deleterious.
Structural variation analysis
Bellerophon v1.03 [23] was used with the default settings to search for chromosomal translocation and inter-chromosomal insertions located inside exons in all 16 ROHHAD proband exomes (the 7 discovery cohort trio probands and the 9 replication cohort exomes) and 4 tumour samples. FishingCNV v2.1 [24] was used to search for genomic copy number variants (CNVs) in the seven trio ROHHAD probands within the discovery cohort. Exomes from 22 healthy individuals, sequenced with the same enrichment kit (Agilent SureSelect capture kit V5 + UTRs), were used as controls. We used Benjamini-Hochberg multiple testing correction to detect CNVs with a p-value < 0.05.
Variant validation and mutation analysis by sanger sequencing
The NCBI Primer Blast tool [25] was used to design site-specific primers to amplify genomic regions of interest. The amplicons were purified using the E.Z.N.A. Cycle Pure spin purification protocol (Omega Biotek) and then sequenced using the fluorescent dideoxy terminator method (Sanger method). Amplicon sequences were compared to the reference genome (GRCh37) using Mutation Surveyor (SoftGenetics) in order to identify variant positions within the exons or UTRs. All de novo candidate variants identified by DeNovoGear [15] with a posterior probability of 0.9 or greater were validated in this way, as were select candidate variants identified in the singleton (autosomal dominant and autosomal recessive) analyses. Selected candidate genes identified in the discovery cohort of 7 ROHHAD probands underwent mutation analysis by Sanger sequencing in 19 additional ROHHAD patients.
Mutation analysis by exome sequencing
WES was obtained for nine members of the ROHHAD replication cohort, and was used to search for candidate variants in the genes identified as potential candidates through the analysis of the trio exomes (i.e., genes carrying de novo or compound heterozygous variants in one of the seven trios; or heterozygous, rare, protein-altering variants in three of the seven trio probands). Exome captures were completed using the Agilent SureSelect capture kit, 38 MB or V4, or the Illumina All Exon 65 MB kit. Massively parallel sequencing was performed on either a SOLiD or Illumina platform, at one of three institutions, and sequences were aligned to the human reference genome (GRCh37) using BWA 0.5.9 [26], Lifescope Genomic Analysis Software 2.5 (Life Technologies), NovoAlign 2.07.13 (Novocroft), or Bowtie 0.12.7 [27] (see Table 2). Variants were called using the GATK Haplotype Caller (Version 3.3) [16] and annotated for filtration and prioritization using ANNOVAR [17]. The coverage of these nine exomes was not sufficient for a robust whole exome analysis (Table 2), but provided a reasonable data set in which to perform an exploratory mutation analysis (Additional file 1 shows the low-coverage proportions for each candidate gene that underwent mutation analysis in these replication exomes).
Results
Cohort characteristics
A total of 35 ROHHAD patients in whom the clinical diagnosis was confirmed were included in this cohort for genetic investigation (Table 1). Specifically, 7 of these patients were included in the initial, trio-based, exome sequencing analysis (discovery cohort), while 28 other ROHHAD patients were included in the secondary exome and Sanger sequencing analysis (replication cohort). In addition, neural crest tumours from four ROHHAD patients (all of whom were trio probands) were analyzed. Blood DNA from 14 parents of the probands (7 trios) and from the monozygotic twin of one ROHHAD proband (discordant for the ROHHAD phenotype) was also included in our analysis. Our cohort represents a highly homogeneous group of ROHHAD patients, and all of the 35 patients included in the study showed rapid-onset obesity, hypothalamic dysfunction, hypoventilation, and autonomic dysregulation; all required artificial ventilation; and 15 (43 %) developed tumours of neural crest origin. Copy number variation (CNV) analysis by array comparative genomic hybridization (CGH), using the Nimblegen 720 k platform, in 26 of the 35 ROHHAD patients did not identify any ROHHAD-specific CNVs. Table 1 reveals a slight gender bias within our cohort (21 females, 14 males), however in our extensive experience studying ROHHAD we have not identified any specific gender prevalence, and careful review of ROHHAD referrals subsequent to this study identified equal distribution of females and males in our broader ROHHAD cohort.
Exome sequencing
WES was completed on a total of 31 individuals including 16 ROHHAD patients (7 as part of trios in the discovery cohort and 9 in the replication cohort), 15 unaffected relatives (parents and one monozygotic twin), and tumour samples from four of the ROHHAD probands (Table 2).
De novo inheritance model - trio analysis
To identify de novo variants, we sequenced the exomes of seven ROHHAD trios. In this set of exomes, we achieved 130-fold mean coverage (after mapping and removal of PCR duplicate reads) with an average of 91.8 % +/− 0.81 % (SD) of bases covered at least 20-fold (Table 2). Using DeNovoGear [15] to analyze the trios, 13 candidate de novo variants were identified and validated by Sanger analysis (Table 3). Each patient carried between zero and three exonic or UTR de novo variants (average 1.86 per exome; 0.71 amino acid altering per exome), a value that is consistent with previous findings [28] and the expected mutation rate [29]. However, no two of these seven ROHHAD patients had de novo variants in the same gene.
In a second iteration of the genetic analysis, the 7 ROHHAD trio probands plus 9 additional ROHHAD exomes were then searched for other candidate variants (not necessarily de novo) in the 13 genes that had been flagged as containing de novo variants. Four genes were identified (C17ORF53, PDE11A, WDFY4, and FAM199X) that contained de novo variants in one patient each, and one or more additional candidate variants (novel or rare [MAF < 0.005] exonic, UTR, or splice site [within 2 bp of an exon] variants not within a segmental duplication) in other exomes (Table 4). From these four genes, C17ORF53 was selected for mutation analysis in the replication cohort. It was selected because both of the candidate variants identified among the 16 patient exomes were amino acid altering, and neither was known to be inherited (one is known to be de novo; there was no inheritance information for the other). Although four different rare variants were identified among five patients in PDE11A, it was not selected for mutation analysis because in two cases, the variant was inherited from clinically unaffected parents, while another variant resulted in a synonymous change. WDFY4 and FAM199X were each mutated in two individuals, but they were not considered for mutation analysis as, in both cases, one variant was shown to be inherited while the other variant resulted in a synonymous change.
Somatic mutation model (tumour and MZ twin analysis)
It is possible that de novo mutations giving rise to ROHHAD may be so severe that they are normally incompatible with life. Such a mechanism has been hypothesized for mutations in AKT1, which cause the Proteus syndrome, but have only been found in a mosaic state and never in all somatic cells [9]. Similarly, surviving patients with ROHHAD may be mosaic for the disease-causing mutation, carried in only a subset of their cells and tissues. If this model were applicable, the subset of ROHHAD patients with neural crest tumours would be the most likely to have an identifiable mutation, and the tumours themselves would be the most likely tissue to carry the causative mutation. To test this somatic mutation hypothesis, we sequenced the exomes of tumours from four of the seven patients that were included in the trio analysis.
We assumed that the tumour samples would contain a variable amount of normal non-tumourous tissue. Because this mosaic state would reduce our ability to detect rare mutations, we increased the sequencing coverage to approximately double that obtained from the trios. This achieved a mean coverage of 260-fold (after mapping and removal of PCR duplicate reads) with an average of 95.1 % +/− 0.54 % (SD) of targeted bases covered at least 20-fold (Table 2). Since each analyzed tumour sample came from a ROHHAD case in a trio, DeNovoGear [15] was used to compare the tumour samples to the parental samples. Any de novo variants identified in a tumour were then compared to the de novo variants identified in that patient’s genomic DNA to single out any de novo variants unique to the tumour. All 13 of the de novo variants identified in the trio analysis were also present in the corresponding tumours; however, no additional tumour-only variants were detected.
Our cohort also contains a pair of monozygotic (MZ) twins discordant for the ROHHAD phenotype [30]. To evaluate the possibility of somatic mosaicism for a causative mutation in the affected twin, we used DeNovoGear to compare the variant profile between the twins, but did not identify any sequence variants in the affected twin’s exome that were not also present in the unaffected twin.
Autosomal recessive model
To identify potential recessive candidates, the trios were used to identify genes containing compound heterozygous inherited rare coding variants (or homozygous, as long as each parent was carrying the variant). We identified between one and four compound heterozygous candidate genes in each patient. These are genes that contained two rare variants inherited in trans (i.e., one from each parent) (Table 5). No individual compound heterozygous candidate was seen in more than one individual, none of the nine replication exomes were found to contain two sequence variants in any of these genes, and none of these candidates have a known function that would strongly suggest an association with ROHHAD. Thus, we have not pursued any of them further at this time. We acknowledge that, in light of the fact that ROHHAD may be highly genetically heterogeneous, one or more of these genes may indeed be disease-causing in an individual proband – but further studies would be required in order to gain additional evidence in support of any particular gene.
Autosomal dominant model
Though a familial dominant inheritance model is not consistent with the sporadic occurrence of this disease, we did search the 7 ROHHAD patient exomes for any genes that contained rare protein-altering (i.e., missense, nonsense, splice-site and indel) variants in all or a large subset of the patients after censoring the de novo or inherited status. After further filtering by visual inspection in the Integrative Genomics Viewer (IGV; [31, 32]), multiple variant calls were removed as probable false positives (e.g. eliminating rare variants on reads that hosted multiple other rare variants – we assumed that these reads were poorly mapped). At this stage, we identified no genes with candidate variants in the same gene among 4 or more of the 7 patients. We did identify four genes (FRAS1, RELN, RIF1, POLE) that each had different rare, protein-altering variants among 3 of the 7 patients (Table 6). We searched the 9 exomes from the replication cohort for rare protein-altering variants in these four genes and identified only one additional patient with a candidate variant in RIF1 (Table 6). All four of the RIF1 variants were validated by Sanger sequencing. All four RIF1 variants as well as all variants in the three genes in which three of 16 probands have candidate variants (FRAS1, RELN, POLE), were inherited from unaffected parents, with both maternal and paternal transmission (Table 6).
Additionally, 187 genes were identified with potential variants in 2 of the 7 discovery cohort patients (Additional file 2), but these have not been inspected in IGV, and a portion of them are likely false positive calls. While it remains possible that sequence variants in one or more of these genes are causative of ROHHAD, with such a large number of candidate genes, and without inheritance information to support any particular one, it presently remains difficult to obtain enough genetic evidence to identify one as a causative gene. In one attempt to narrow down this list, we looked only at loss-of-function (LOF) variants, but did not identify any gene with LOF variants in more than one proband.
Structural variation analysis
In addition to identifying SNVs and small indels (as described in the previous sections), we also analyzed the ROHHAD exome data (including the tumour samples) for larger variations such as chromosomal translocations and inter chromosomal insertions and CNVs. No CNVs and no exonic translocations or insertions were detected in any of the samples. However, with exome data alone, we cannot rule out the possibility of fusion events in intronic or intergenic regions.
Mutation analysis
We performed mutation analysis (by Sanger sequencing) of two candidate genes (C17ORF53 and MAPKAPK5) in 19 additional ROHHAD patients.
C17ORF53
In total, we identified three novel or rare (MAF < 0.005) variants in a search of the exons and UTRs of the C17ORF53 gene among 35 ROHHAD patients (16 by exome sequencing and 19 by Sanger sequencing) (Table 4). Two were non-synonymous variants, and one was in the 3’UTR. We do not have genetic samples from the parents of two of the patients carrying sequence variants in this gene, and so are unable to determine whether or not they are de novo variants. Since very little is known about the protein encoded by this open reading frame, we do not have information to model the effect these sequence variants would have. Further investigation into the protein (when and where it is expressed, and what its function is) and the effect of these variants would be required to further implicate them in ROHHAD.
MAPKAPK5
Improvement in a subset of phenotypic features in Patient 27 was observed shortly after a regimen of caffeine treatment (100 mg BID) was initiated [33]. A study found that transcript levels of MAPKAPK5 (in which Patient 27 is carrying a de novo missense variant; Table 3) were transiently increased in response to forskolin treatment (which, like caffeine, elevates cAMP) [34], leading us to hypothesize that MAPKAPK5 might be involved in this patient’s ROHHAD phenotype. However, mutation analysis in our ROHHAD cohort did not identify any additional novel or rare exonic, splice site, or UTR sequence variants in MAPKAPK5.
Discussion
Our hypothesis that ROHHAD might be caused by germline de novo mutations or by mosaicism for somatic mutation(s) in one or more genes was tested using seven trios, including neural crest tumours of four of these patients and the unaffected MZ twin of one (Table 2), as well as an additional 28 singleton ROHHAD patients used for mutation analysis of candidate genes. Despite the homogeneity of this ROHHAD cohort, and the depth of coverage established in our exome sequencing, we did not identify any two patients who had de novo variants in the same gene, any de novo coding variants unique to the tumours, or any variants discordant between the affected and unaffected MZ twins. We also did not identify any structural variations in the exomes. While the sporadic occurrence of ROHHAD suggests a de novo inheritance model, we were also able to use the exome sequencing data to assess autosomal recessive and autosomal dominant inheritance models. Neither model revealed a major gene that would explain ROHHAD in our cohort. One or more of the genes containing a) de novo variants (Table 3, Table 4), b) inherited compound heterozygous variants (Table 5), or c) inherited heterozygous variants in 3 or 4 patients (Table 6), may be causative of ROHHAD in some cases, but further genetic and/or functional evidence is required before these variants could be considered truly pathogenic for the ROHHAD phenotype. In particular, it is difficult to determine the significance of the RIF1 finding (that 4 of 16 patients have inherited a rare protein-altering variant in this gene). The gene is large (41 exons encoding 2472 amino acids), and harbours a substantial number of variants (2492 variants in the ExAC database [20], 937 of which are rare protein-altering variants, and 20 of which are loss-of-function (LOF) variants [i.e., nonsense, frameshift, or canonical splice site mutations]). Thus, additional evidence - genetic (i.e., identifying additional ROHHAD patients with RIF1 mutations) or functional (i.e., discovering a RIF1 function that could explain the ROHHHAD phenotype) – would be required to prove that this is more than a random chance occurrence. An in silico search using Ingenuity Pathway Analysis (Qiagen) for functional connections among these 31 genes did not identify any potential links between different candidate genes from more than two patients.
Previous studies postulated eight genes (NDN, ASCL1, PHOX2B, NTRK2, BDNF, HTR1A, OTP, and ADCYAP1) as candidate ROHHAD genes, based on their roles in the development or function of the systems (hypothalamic, autonomic, and neuroendocrine) known to be deficient in ROHHAD, but failed to identify any disease-associated variants in these genes among their cohorts [2, 4, 13]. Likewise, we did not identify any candidate variants in any of these eight genes in our 7 ROHHAD patient exomes (for each gene, at least 96 % of the coding bases were covered at least 20-fold with the exception of OTP, for which one of three exons was not effectively captured), confirming that none of these eight genes is a major ROHHAD gene. A recent study described a novel nonsense mutation in the Smith-Magenis gene, RAI1, in a patient previously diagnosed with ROHHAD (though the authors note that deeper evaluation revealed his phenotype was not consistent with ROHHAD or Smith-Magenis syndrome) [35]. We did not identify any candidate variants in RAI1 in our 7 ROHHAD patient exomes, despite the fact that >96 % of coding bases were covered at least 20-fold, suggesting that mutations in this gene are not characteristic of ROHHAD.
Although the present analysis did not uncover a causative gene for ROHHAD, it does not completely rule out the possibility that ROHHAD is caused by de novo coding mutations. There are several aspects of the disease that may lead our analysis, as currently designed, to miss a de novo coding answer, even if one exists. For one, the possibility that somatic mosaicism represents the major mechanism for ROHHAD complicates our analysis in that we may not have sampled the major tissue(s) carrying the ROHHAD-causing mutations, such as the brainstem or hypothalamic nuclei. Our use of the patients’ neuroendocrine tumours represents one approach to addressing this challenge, because these tissues would be expected to contain the causative ROHHAD genetic lesion if one exists. The fact that we did not identify any coding mutations distinct to the tumours suggests that the genetic lesion(s) we are looking for may be rare and highly specific (such as regulatory mutations or fusion genes) or are located outside the exonic region. Secondly, there is the possibility that ROHHAD is characterized by a high level of genetic heterogeneity, whereby a discovery set of seven probands is not sufficiently large to contain more than one patient with a mutation in any single ROHHAD gene. However, we think that this is unlikely, as we did extend our exome analysis to include a total of 16 ROHHAD patients, and our entire study cohort included a total of 35 ROHHAD patients, suggesting that exome sequencing of additional trios or individuals is not likely to lead to an answer for the underlying genetics of ROHHAD. Finally, some limitations remain as to what can be discovered, even within the coding region, by WES. The mutations causing ROHHAD may be of a nature that is difficult to detect by WES, such as triplet repeat expansion. Alternatively, causative mutations might have occurred in parts of genes poorly enriched by our capture kit, and thus not covered by our sequencing. A list of those genes reveals several genes with known functions that could potentially be linked to ROHHAD; those will require a more extensive follow up sequencing project.
Given the many recent successes of WES in identifying disease-causing mutations, our study represents a major effort toward discovering the cause of ROHHAD. Though no coding pathogenic mutations for this disease were identified, our experimental design was sufficiently robust for us to conclude that exome sequencing of trios and tumours is not likely to be an efficient means of identifying the cause of ROHHAD. A reasonable next step would be whole genome sequencing to look for de novo mutations beyond the coding exome, that may disrupt promoter regions, transcription factor binding sites, microRNA genes or binding sites, or other important non-coding regions.
It is also important to consider alternative hypotheses wherein ROHHAD may not have a genetic etiology. Older literature has suggested that ROHHAD may be a paraneoplastic condition, resulting from pathophysiology triggered by a specific subset of neuroendocrine tumours [6, 36, 37]. Nevertheless, the fact that many patients with ROHHAD do not have neuroendocrine tumours suggests that the paraneoplastic hypothesis cannot be the single unifying model. Similarly, the fact that patients whose tumours were removed do not experience remission of other symptoms, and the ROHHAD phenotype continues to evolve with advanced hypoventilation and autonomic dysregulation, makes this model less attractive as an explanation. An autoimmune etiology for ROHHAD has also been postulated [6, 38, 39] though it is not clear that all the study subjects actually had ROHHAD, or that the intervention modified the phenotype. No technology yet available can rule out an autoimmune etiology for a particular disease, though autoimmune-mediated inflammation cannot explain a predisposition to neoplasia.
Conclusion
Using a comprehensive, well-designed and carefully executed exome sequencing study in an expertly-phenotyped cohort of 35 ROHHAD patients, we were not able to identify de novo pathogenic mutations causing ROHHAD. Our findings suggest that clinical exome sequencing is unlikely to be a useful diagnostic test in patients with true ROHHAD. However, because ROHHAD is a devastating disease that is characterized by a high incidence of cardiorespiratory arrest in childhood and by the need for artificial ventilation for life-support, it remains imperative to refine the search for genetic risk factors for ROHHAD, as well as to investigate other possible mechanisms of disease.
References
Fishman LS, Samson JH, Sperling DR. Primary alveolar hypoventilation syndrome (Ondine’s Curse). Am J Dis Child. 1965;110:155–61.
Ize-Ludlow D et al. Rapid-onset obesity with hypothalamic dysfunction, hypoventilation, and autonomic dysregulation presenting in childhood. Pediatrics. 2007;120(1):E179–88.
Bougneres P et al. Endocrine manifestations of the rapid-onset obesity with hypoventilation, hypothalamic, autonomic dysregulation, and neural tumor syndrome in childhood. J Clin Endocrinol Metab. 2008;93(10):3971–80.
De Pontual L et al. Delineation of late onset hypoventilation associated with hypothalamic dysfunction syndrome. Pediatr Res. 2008;64(6):689–94.
Onal H, Ersen A. A case of late-onset central hypoventilation syndrome with hypothalamic dysfunction: through a new phenotype. Turk J Pediatr. 2010;52(2):198–202.
Paz-Priel I, Cooke DW, Chen AR. Cyclophosphamide for rapid-onset obesity, hypothalamic dysfunction, hypoventilation, and autonomic dysregulation syndrome. J Pediatr. 2011;158(2):337–9.
Weese-Mayer DE et al. An official Ats clinical policy statement: congenital central hypoventilation syndrome: genetic basis, diagnosis, and management. Am J Respir Crit Care Med. 2010;181(6):626–44.
Douglas J et al. Nsd1 mutations are the major cause of sotos syndrome and occur in some cases of weaver syndrome but are rare in other overgrowth phenotypes. Am J Hum Genet. 2003;72(1):132–43.
Lindhurst MJ et al. A mosaic activating mutation in Akt1 associated with the Proteus syndrome. N Engl J Med. 2011;365(7):611–9.
Kurek KC et al. Somatic mosaic activating mutations in Pik3ca Cause Cloves syndrome. Am J Hum Genet. 2012;90(6):1108–15.
Gibson WT et al. Mutations in Ezh2 cause Weaver syndrome. Am J Hum Genet. 2012;90(1):110–8.
Weksberg R et al. Beckwith-Wiedemann syndrome demonstrates a role for epigenetic control of normal development. Hum Mol Genet. 2003;12 Spec No 1:R61–8.
Rand CM et al. Rapid-onset obesity with hypothalamic dysfunction, hypoventilation, and autonomic dysregulation: analysis of hypothalamic and autonomic candidate genes. Pediatr Res. 2011;70(4):375–8.
Beaulieu CL et al. Forge Canada consortium: outcomes of A 2-year national rare-disease gene-discovery project. Am J Hum Genet. 2014;94(6):809–17.
Ramu A et al. Denovogear: De Novo Indel and point mutation discovery and phasing. Nat Methods. 2013;10(10):985–7.
Van Der Auwera, GA, et al. From Fastq Data To High Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Current Protocols In Bioinformatics/Editoral Board, Andreas D. Baxevanis et al. 2013. 11(1110): 11 10 1–11 10 33.
Wang K, Li M, Hakonarson H. Annovar: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):E164.
Abecasis GR et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491(7422):56–65.
Exome Variant Server, Nhlbi Go Exome Sequencing Project (Esp), Seattle, Wa. December, 2014. Available From: Http://Evs.Gs.Washington.Edu/Evs/.
Exome Aggregation Consortium (Exac), Cambridge, Ma. Available From: http://exac.broadinstitute.org.
Kircher M et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310–5.
Adzhubei IA et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–9.
Hayes M, Li J. Bellerophon: a hybrid method for detecting interchromosomal rearrangements at base pair resolution using next-generation sequencing data. BMC Bioinformatics. 2013;14 Suppl 5:S6.
Shi Y, Majewski J. Fishingcnv: a graphical software package for detecting rare copy number variations in exome-sequencing data. Bioinformatics. 2013;29(11):1461–2.
Ye J et al. Primer-blast: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics. 2012;13:134.
Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics. 2009;25(14):1754–60.
Langmead B et al. Ultrafast and memory-efficient alignment of short Dna sequences to the human genome. Genome Biol. 2009;10(3):R25.
O’roak BJ et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet. 2011;43(6):585–9.
Lynch M. Rate, molecular spectrum, and consequences of human mutation. Proc Natl Acad Sci U S A. 2010;107(3):961–8.
Patwari PP et al. Monozygotic twins discordant for Rohhad phenotype. Pediatrics. 2011;128(3):E711–5.
Robinson JT et al. Integrative genomics viewer. Nat Biotechnol. 2011;29(1):24–6.
Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (Igv): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14(2):178–92.
Gordon, SC, et al. The evolving phenotype in a patient with Rapid-Onset Obesity With Hypothalamic Dysfunction, Hypoventilation, And Autonomic Dysregulation (Rohhad) and response to caffeine treatment. Am J Respir Crit Care. 2015. Med(191): P. A5923, 2015 - Presented At American Thoracic Society International Conference, Denver, Co, May 2015.
Gerits N et al. The transcriptional regulation and cell-specific expression of the Mapk-activated protein kinase Mk5. Cell Mol Biol Lett. 2009;14(4):548–74.
Thaker VV et al. Whole exome sequencing identifies rai1 mutation in a morbidly obese child diagnosed with rohhad syndrome. J Clin Endocrinol Metab. 2015;100(5):1723–30.
Ouvrier R et al. Idiopathic hypothalamic dysfunction: a paraneoplastic syndrome? Lancet. 1995;346(8985):1298.
Sirvent N et al. Hypothalamic dysfunction associated with neuroblastoma: evidence for a new paraneoplastic syndrome? Med Pediatr Oncol. 2003;40(5):326–8.
Sartori S et al. Intrathecal synthesis of oligoclonal bands in rapid-onset obesity with hypothalamic dysfunction, hypoventilation, and autonomic dysregulation syndrome: new evidence supporting immunological pathogenesis. J Child Neurol. 2014;29(3):421–5.
Chow C et al. Rapid-Onset Obesity with Hypothalamic Dysfunction, Hypoventilation, and Autonomic Dysregulation (Rohhad) syndrome may have a hypothalamus-periaqueductal gray localization. Pediatr Neurol. 2015;52(5):521–5.
Acknowledgments
Funding for the exome and Sanger sequencing at the ACHRI Genomic and Bioinformatic Platform and UCDNA Services, University of Calgary was generously provided by ROHHAD Fight, Inc. SFB, NTBH, RJAW, CMR and DEW-M were also funded, in part, by ROHHAD Fight, Inc. SFB is also funded by the ACHRI-CIHR Training Program. NTBH is also funded by CIHR. PAG is funded by NIH HL089742. RJAW is funded by AIHS and CIHR. WTG is funded by the CFRI and the CIHR. Sincere thanks to Richard Pon for his assistance in designing and planning the exome sequencing experiments. The authors would like to thank the Exome Aggregation Consortium and the groups that provided exome variant data for comparison. A full list of contributing groups can be found at http://exac.broadinstitute.org/about.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SFB designed the study, planned and carried out the bioinformatic data analysis, designed and performed variant validations, designed mutation analysis, led data interpretation, and drafted and revised the paper. CMR designed the study, assisted with case designation, collected and extracted DNA from tissue samples, assisted with data interpretation, and drafted and revised the paper. LAB and LN designed and performed variant validations and mutation analyses, and revised the draft paper. PAG conceived the initial study, and contributed to the design of the overall study, assisted with data interpretation, and revised the draft paper. RJAW designed the study, assisted with data interpretation, and revised the draft paper. WTG designed the study, assisted with clinical assessments/case designation, assisted with data interpretation, and revised the draft paper. PMKG performed exome sequence alignments, assisted with planning bioinformatics analyses, designed and performed structural variation analyses, and revised the draft paper. ZA wrote scripts for bioinformatic data analysis and revised the draft paper. EMB-K and DI-L assisted with clinical assessments/case designation, assisted with data interpretation, and revised the draft paper. DEW-M initiated the international collaboration and continues to serve as its leader, conceived and designed the study, clinically assessed all patients, led the interpretation of clinical data/case designation, assisted with genetics data interpretation, and drafted and revised the paper. NTB-H designed and planned the study, led and supervised genetic analyses, assisted with data interpretation and drafted and revised the paper. All authors read and approved the final manuscript.
Debra E. Weese-Mayer and N. Torben Bech-Hansen contributed equally to this work.
Additional files
Additional file 1:
Low-coverage proportions of candidate genes in replication exomes. (XLSX 43 kb)
Additional file 2:
Genes carrying putative candidate mutations in 2 of 7 ROHHAD probands in the discovery cohort. (XLSX 44 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Barclay, S.F., Rand, C.M., Borch, L.A. et al. Rapid-Onset Obesity with Hypothalamic Dysfunction, Hypoventilation, and Autonomic Dysregulation (ROHHAD): exome sequencing of trios, monozygotic twins and tumours. Orphanet J Rare Dis 10, 103 (2015). https://doi.org/10.1186/s13023-015-0314-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-015-0314-x