
Abstract
Background
Siegesbeckia pubescens Makino (SP) is one of the important plant origins for the anti-inflammatory Chinese herbal medicine of Siegesbeckiae Herba. The current investigations indicated that the anti-inflammatory effects of SP were associated with the toll-like receptors (TLRs)-mediated nuclear factor-κB (NF-κB) and the mitogen-activated protein kinase (MAPK) signaling pathways.
Methods
Raw 264.7 macrophages were pretreated with the 50% ethanol extract of SP (SPE, 50–200 µg/mL) and then co-treated with Pam3CSK4 (200 ng/mL) for another 12 h. The inhibitory effect of SPE on Pam3CSK4-stimulated NO release and post-inflammatory cytokines secretions were determined using Griess reagent and Elisa kits, respectively. The influence of SPE on NF-κB and MAPKs signaling relevant proteins was measured by Western blotting analysis, while the intracellular nitric oxide (NO) generation and NF-κB/p65 nuclear translocation were determined using Leica TCS SP8 laser scanning confocal microscope. Moreover, the effect of SPE on luciferase reporter gene in NF-κB-luc DNA transfected raw 264.7 cells was determined using the Dual-Glo luciferase assay system kit.
Results
SPE dose-dependently (50–200 µg/mL) attenuated Pam3CSK4-induced NO release, post-inflammatory cytokines (IL-6, TNF-α and MCP-1) secretions and intracellular NO generation in raw 264.7 cells. Biologically, SPE suppressed Pam3CSK4-induced expressions of cyclooxygenase-2 (COX-2), inducible nitric oxide synthase (iNOS), phosphorylation of NF-κB/p65 and IκBα, but did not significantly show effect on the proteins involved in MAPKs signaling (p38, ERK and JNK). The results were further confirmed by NF-κB-luc reporter gene assay and p65 nuclear translocation assay.
Conclusions
In conclusion, SPE ameliorated Pam3CSK4-induced inflammation in raw 264.7 cells through suppressing TLR 1/2-mediated NF-κB activation.
Similar content being viewed by others
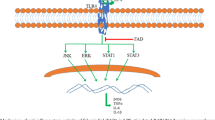


Background
Inflammation is an innate (non-specific) immune response and plays an important role in the physiological defense in response to various trauma or infection to the body [1]. An appropriate inflammatory response is necessary for the organism’s healing potential and facilitates tissue repair. However, an excessive or prolonged response might cause continuously damage to the body and induce many chronic diseases, organ dysfunction or organ failure [2, 3]. Therefore, an effective means of modulating systemic inflammation is beneficial for patients with chronic inflammatory autoimmune diseases, such as rheumatoid arthritis and diabetic nephropathy.
In the past decades, numerous studies indicated that transcription factors NF-κB target genes were involved in the occurrence and progress of various inflammations [4,5,6,7,8]. Activation of NF-κB stimulated macrophage recruitment and maturation, as well as the further production of pro-inflammatory cytokines and chemokines, such as tumor necrosis factor (TNF)-α, interleukin (IL)-1β, IL-6, monocyte chemoattractant protein (MCP)-1, and so on [9, 10]. Subsequently, the secreted inflammatory mediators further accelerated the degree of inflammation and the development of diseases [11]. On the other hand, being a family of transmembrane receptors closely related to the innate immune response [12], toll-like receptors (TLRs) (TLR1–TLR10 for human TLRs) present different functions on regulating inflammatory signaling and mediators based on their capacity to recognize the host derived agonists mostly released from the damaged cells or tissues during the progression of the diseases [13,14,15,16]. In triacyl lipoprotein-induced inflammations, the activation of NF-κB signaling pathways and production of various pro-inflammatory cytokines through TLR1/TLR2 (a heterodimer of TLR1 and TLR2) activation have been investigated and reported [17,18,19,20]. Therefore, targeting TLR1/TLR2 heterodimer-induced inflammation might be the potential therapeutic approach for such inflammatory diseases.
Siegesbeckia pubescens Makino (SP) is one of the plant origins of the traditional Chinese herbal medicine of Siegesbeckiae Herba, which has been widely used for various inflammatory diseases in China from the Tang dynasty. Currently, the chemical analysis indicated that SP mainly contained diterpenoids [21], sesquiterpenoids [22], flavonoids [23], glycosides [24] and some other constituents [25]. Moreover, the SP extracts or derived components were investigated to present various pharmacological activities such as anti-inflammatory [22, 26, 27], anti-allergic [28], and anti-cancer effects [29, 30]. The anti-inflammatory activity of SP was demonstrated to be related to its suppression on lipopolysaccharide (LPS)-induced nitric oxide (NO) [26] and inflammatory mediators [31] productions via NF-κB inactivation [32]. However, in our preliminary studies, the 50% ethanol extract of SP has been observed to have better activity against Pam3CSK4-(a specific TLR1/TLR2 agonist) than LPS-induced NO production in RAW 264.7 macrophages. In this study, the potential mechanisms of SP on Pam3CSK4-induced inflammation were further investigated and reported.
Methods
The Minimum Standards of Reporting Checklist contains details of the experimental design, and statistics, and resources used in this study (Additional file 1).
Chemical and reagents
Rutin, kirenol and darutoside (the purities of all standards were higher than 98% by HPLC analysis) were purchased from Chengdu Pufei De Biotech Co., Ltd. (Chengdu, China). Hoechst 33342, 3-(4, 5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and Griess reagent were purchased from Sigma Chemicals Ltd. (St. Louis, MO, USA). Milli-Q water was prepared using a Milli-Q system (Millipore, MA, USA).
Dulbecco’s modified eagle medium (DMEM) and fetal bovine serum (FBS) were purchased from Gibco (Carlsbad, CA, USA). Pam3CSK4 (N-palmitoyl-S-[2,3-bis(palmitoyloxy)-(2RS)-propyl]-[R]-cysteinyl-[S]-seryl-[S]-lysyl-[S]-lysyl-[S]-lysyl-[S]-lysine·3HCl) was purchased from InvivoGen (San Diego, CA, USA). Enzyme-linked immunosorbent assay kits for IL-6, TNF-α, and MCP-1 were obtained from Neobioscience (Shenzhen, China). TurboFect transfection reagent was purchased from Thermo Fisher Scientific (Waltham, MA, USA). Antibodies were purchased from Santa Cruz Biotechnology (CA, USA) or Cell Signaling Tech (Danvers, MA, USA).
Preparation and characterization of SP extract (SPE)
The herbal material of SP was collected from Guiyang (Guizhou province, China) and authenticated by the corresponding author. The voucher specimens (No. SP-002) were deposited at the Institute of Chinese Medical Sciences, University of Macau, Macao, China.
The powdered SP (100 g) was extracted twice with 50% ethanol (1:10, w/v) for 1 h each under reflux. The combined extracts were filtered with filter paper after cooling and then concentrated under reduced pressure to remove the ethanol. The powdered SPE (yield: 27.3%) was obtained by lyophilizing the concentrated sample with a Virtis Freeze Dryer (The Virtis Company, New York, USA).
Quantification of rutin, kirenol and darutoside in SPE was performed using an Agilent HP1100 system (Hewlett Packard, Agilent, USA) coupled with an Elite Hypersil BDS C-18 analytical column (100 mm × 2.1 mm I.D., 3 μm) (Dalian, China) maintained at 25 °C. Elution was performed with a mobile phase of A (0.2% phosphoric acid in water) and B (0.2% phosphoric acid in ACN) under a gradient program by a linear increase from 10% B to 22% B in the first 30 min, and to 23% B in 10 min, then to 30% in 30 min. The flow rate was 0.35 mL/min, and the injection volume was 10 μL. The analytes were monitored at the UV wavelength of 215 nm. Prior to next injection, the column was washed with 100% B for 5 min and then equilibrated with the initial mobile phase for 10 min.
Cell culture
RAW 264.7 cells were obtained from the American Type Culture Collection (ATCC, Manassa, VA, USA). The cells were maintained in DMEM supplemented with 10% heat-inactivated FBS at 37 °C in humidified 5% CO2 atmosphere. The Minimum Standards of Reporting Checklist (Additional file 1) contains details of the experimental design, and statistics, and resources used in this study.
Cytotoxicity
The cytotoxicity of SPE on RAW 264.7 cells was detected using the MTT assay combined with lactose dehydrogenase (LDH) assay. In brief, the cells were seeded on a 96-well plate (1 × 104 cells/well) and allowed to adhere overnight. The cells were pretreated with SPE (25–200 μg/mL) for 4 h followed by co-treatment in the presence or absence of Pam3CSK4 (200 ng/mL) for 24 h. The cell proliferation was determined using the MTT assay as previous described [33]. The release of LDH in medium was determined using the LDH Cytotoxicity Detection Kit (ThermoFisher Scientific Inc., USA) according to the manufacturer’s instructions.
Nitric oxide (NO) production and inflammatory cytokines secretion assays
RAW 264.7 cells were seeded on a 24-well plate (1 × 105 cells/well) and allowed to adhere overnight. The cells were pretreatment with SPE (50, 100 and 200 μg/mL) or CU-CPT22 (4 µM, positive control) for 4 h and then co-treated with addition of Pam3CSK4 (200 ng/mL) for another 12 h. NO production was determined by measuring the accumulated nitrite in the culture medium with Griess reagent [33]. Cytokines (TNF-α, IL-6, and MCP-1) secretion in the supernatants of cultured cells was quantified using the enzyme-linked immunosorbent assay kits (Neobioscience, Shenzhen, China) following the manufacturer’s instructions.
Capture of intracellular NO generation
RAW 264.7 cells were cultured in glass bottom dish overnight and pretreated with SPE (50, 100 and 200 μg/mL) for 4 h. Subsequently, the cells were co-treated with Pam3CSK4 (200 ng/mL) for 12 h and then washed twice with ice-cold PBS. After incubated with 4-amino-5-methylamino-2′,7′-difluorofluorescein diacetate (DAF-FM, 5 μM in PBS) (ThermoFisher Scientific Inc., USA) for 30 min at room temperature, the cells were washed with PBS and then stained with 1 μg/mL Hoechst 33342 for 10 min. The images were obtained by a Leica TCS SP8 laser scanning confocal microscopy (Leica Microsystem, Wetzlar, Germany).
Western blotting analysis
RAW 264.7 cells were treated as described above. The harvested cells were washed thrice with ice-cold PBS and then extracted with RIPA buffer (Beyotime Biotechnology, Shanghai, China) containing protease inhibitor cocktails (ThermoFisher Scientific Inc., USA). The proteins (50 µg for each sample) were separated with SDS-PAGE (8%) and then transferred onto a PVDF membrane. The membranes were blocked with non-fat milk (5% in TBS containing 0.05% Tween-20, w/v) and incubated overnight at 4 °C with antibodies against iNOS, COX-2, p-IκBα, IκBα p-p65, p65 or GAPDH (1:1000). The membranes were then incubated with the corresponding secondary antibody (1:1000) at room temperature for 1 h. The signals were detected using ECL western blotting substrate (ThermoFisher Scientific Inc., USA) and ChemiDoc™ XRS+ system with Image Lab™ software (Bio-Rad Laboratories, Hercules, CA, USA).
Immunofluorescence analysis
RAW 264.7 cells were attached on confocal dish overnight. After pretreatment with SPE (200 μg/mL) for 2 h, the cells were co-treated with Pam3CSK4 (200 ng/mL) for another 4 h and then fixed with 4% Paraformaldehyde (PFA) for 10 min at room temperature. The cells were washed thrice with PBS, permeabilized with 0.05% Triton X-100 in PBS for 3 min, and followed by blocking with 3% bovine serum albumin (in PBS, w/v) for 1 h. Thereafter, the cells were incubated with antibody against p65 (1:100) overnight and reacted with Alexa Fluor 488-conjugated secondary antibody (1:1000) for 1 h. The nuclear of the cell was stained with Hoechst 33342. The images were captured with a Leica TCS SP8 laser scanning confocal microscopy.
Luciferase reporter gene assay
RAW 264.7 cells were transiently transfected with NF-κB-luc DNA for 48 h and refreshed with completed DMEM. The transfected cells were seeded in 6 plates overnight and pretreated with SPE (200 μg/mL) or CU-CPT22 (4 µM) for 2 h before Pam3CSK4 (200 ng/mL) stimulation for another 4 h. Luciferase activity was determined using a Dual-Glo luciferase assay system kit (Promega, Madison, Wisconsin, USA) following the manufacturer’s instructions.
Statistical analysis
Each experiment was performed in triplicate and was repeated for at least thrice. All results were presented as mean ± SD. Variance between two groups was evaluated by one-way ANOVA using the GraphPad Prism software (GraphPad Software, United States). The Newman–Keuls multiple comparison tests were performed for post hoc pairwise comparisons. P < 0.05 was considered statistically significant difference.
Results
Characterization of SP extract
The chromatograms of mixed standards and the SP extract were illustrated in Fig. 1. The contents of rutin, kirenol and darutoside in the extract were determined to be 0.27 ± 0.01, 1.81 ± 0.02 and 0.28 ± 0.03%, respectively.

HPLC chromatograms of (a) mixed standards (7.5 μg/mL of rutin, kirenol and darutoside) and (b) SPE (1 mg/mL). 1: rutin; 2: kirenol; 3: darutoside. The contents of rutin, kirenol and darutoside were determined to be 0.27 ± 0.01, 1.81 ± 0.02 and 0.28 ± 0.03%, respectively (n = 3)
Cytotoxicity
The cytotoxicity of SPE on RAW 264.7 cells were determined using MTT and LDH assays. As illustrated in Fig. 2, SPE did not exert any observable toxicity on RAW 264.7 cells within the concentration ranged from 25 to 200 μg/mL while incubated with or without Pam3CSK4 (200 ng/mL) in 24 h. The concentrations of 50, 100 and 200 μg/mL were selected for SPE throughout the study.

Siegesbeckia pubescens Makino (SP) or SP with Pam3CSK4-stimulated did not significantly affect the cell viability and cytotoxicity (n = 3). RAW 264.7 cells were treated with SP with different concentrations for 24 h. a The cell viability was measured by MTT assay and c cell cytotoxicity was measured by LDH assay. RAW 264.7 cells were pretreated with SP for 4 h before Pam3CSK4 (200 ng/mL) stimulation for another 12 h. b The cell viability was measured by MTT assay and d cell cytotoxicity was measured by LDH assay
SPE suppressed NO release and inflammatory cytokines secretions in Pam3CSK4-induced RAW 264.7 cells
Compared to the normal control cells, incubation with Pam3CSK4 for 12 h significantly induced the release of NO and secretion of inflammatory cytokines (IL-6, TNF-α, and MCP-1) in RAW 264.7 cells (Fig. 3). However, the stimulations were dose-dependently inhibited by SPE in the concentration ranged from 50 to 200 μg/mL. More than 50% of Pam3CSK4-stimulated NO release was observed to be decreased (> 50%) by SPE at the concentration higher than 100 μg/mL (Fig. 3a). The estimated IC50 of SPE on NO release was calculated to be 103.6 µg/mL. Moreover, SPE (200 μg/mL) significantly inhibited the Pam3CSK4-induced IL-6 (59.98%), TNF-α (42.38%), and MCP-1 (55.10%) (Fig. 3b–d). The inhibitory effects of SPE on Pam3CSK4-induced inflammation were comparable to those of the positive control of CU-CPT22 (4 µM).

Siegesbeckia pubescens Makino (SP) exhibited anti-inflammatory effects in Pam3CSK4-stimulated RAW 264.7 cells (n = 3). RAW 264.7 cells were pretreated with SP with different concentrations for 4 h before Pam3CSK4 (200 ng/mL) stimulation for another 12 h. a Nitric oxide (NO) was determined by Griess assay. TLR1/TLR2 antagonist: CU-CPT22 (CU) was selected as the positive control. We measured the levels of b IL-6, c TNF-α, and d MCP-1 by ELISA assay. *P < 0.05 vs. Pam3CSK4-induced, **P < 0.01 vs. Pam3CSK4-induced, ***P < 0.001 vs. Pam3CSK4-induced
SPE attenuated Pam3CSK4-induced intracellular NO generation
The inhibitory effect of SPE on Pam3CSK4-induced intracellular NO generation was determined by confocal microscopy. As illustrated in Fig. 4, Pam3CSK4 significantly stimulated the intracellular NO generation in RAW 264.7 cells. This stimulation was attenuated by SPE in a dose-dependent manner (50–200 μg/mL).

RAW 264.7 cells were pretreated with Siegesbeckia pubescens Makino (SP) (n = 3) for 4 h before Pam3CSK4 (200 ng/mL) stimulation for another 12 h. NO was captured by Leica TCS SP8 laser scanning confocal microscope with 5 μM DAF-FM diacetate (4-amino-5-methylamino-2′,7′-difluorofluorescein diacetate). CU-CPT22 (CU) is selected as the positive control
SPE inhibited Pam3CSK4-induced the protein expressions of iNOS and COX-2
The protein expressions of iNOS and COX-2 in SPE-treated RAW 264.7 cells were analyzed by Western blotting assay and illustrated in Fig. 5. SPE was determined to dose-dependently inhibit the Pam3CSK4-induced iNOS and COX-2 protein expressions in RAW 264.7 cells. The related amounts of iNOS and COX-2 in SPE-treated cells were determined to be decreased by 61.42 and 74.65% respectively when compared to those in the Pam3CSK4-induced cells (Fig. 5b, c). Moreover, the phosphorylation of JNK1/2 and p38, but not ERK1/2, was observed to be increased under Pam3CSK4 stimulation; whilst, SPE did not present any influence on the phosphorylation of such proteins.

Effects of Siegesbeckia pubescens Makino (SP) on relevant pathways. RAW 264.7 cells were pretreated with SP (0, 50, 100 and 200 μg/mL) for 4 h and followed with Pam3CSK4 (200 ng/mL) addition for 12 h. CU-CPT22 (CU) is selected as the positive control. a Proteins was evaluated by Western blotting assay. b, c Quantification of iNOS and COX-2 protein was detected by densitometric analysis (n = 3). **P < 0.01 vs. Pam3CSK4-induced
SPE inactivated Pam3CSK4-induced NF-κB signaling
Compared to the untreated cell group, SPE at high concentration (200 μg/mL) was observed to slightly increase the phosphorylation of IκBα but did not influence on the phosphorylation of NF-κB-p65 (Fig. 6a–c). However, under the inflammatory condition, SPE presented dose-dependent inhibition on Pam3CSK4-induced phosphorylation of IκBα and NF-κB-p65 in RAW 264.7 cells. The activated p-IκBα and p-NF-κB-p65 were decreased by 26.71 and 34.14%, respectively, while co-treated with SPE at 200 μg/mL for 12 h (Fig. 6a–c). The results suggested the involvement of NF-κB inactivation in Pam3CSK4-induced inflammation by SPE. Confirmed by luciferase reporter gene assay, SP significantly attenuated the NF-κB-driven luciferase activity in Pam3CSK4-stimulated RAW 264.7 cells (Fig. 6d). Furthermore, the Pam3CSK4-induced p65 nuclear translocation was also determined to be attenuated by SPE using the immunofluorescence staining assay (Fig. 7).

Effects of Siegesbeckia pubescens Makino (SP) on NF-κB pathways (n = 3). RAW 264.7 cells were pretreated with SP (0, 50, 100 and 200 μg/mL) for 2 h and followed with Pam3CSK4 (200 ng/mL) addition for 4 h. CU-CPT22 (CU) is selected as the positive control. a Proteins was evaluated by Western blotting assay. b, c Quantification was detected by densitometric analysis. d RAW 264.7 cells were transfected with NFκB-luc for 48 h. Cells were pretreated with SP 2 h before Pam3CSK4 (200 ng/mL) stimulation for another 4 h. Luciferase activity was determined by Dual-Glo Luciferase Assay. *P < 0.05 vs. Pam3CSK4-induced and **P < 0.01 vs. Pam3CSK4-induced

RAW 264.7 cells were pretreated with 200 μg/mL of Siegesbeckia pubescens Makino (SP) (n = 3) for 2 h before Pam3CSK4 stimulation for another 4 h. NF-κB/p65 nuclear translocation was determined by immunofluorescence assay
Discussion
In this work the anti-inflammatory effect and underlying mechanisms of SP on Pam3CSK4-induced inflammation in RAW 264.7 macrophages were investigated and reported.
TLRs have been demonstrated to play critical role in innate immune response in mammals against various infections. Up to date, 10 human TLRs (TLR1-TLR10) and 12 murine TLRs (TLR1-TLR9 and TLR11-TLR13) have been identified and demonstrated to react to various types of inflammations [13, 14]. Different to TLR4 (mainly response to LPS-induced inflammations), the TLR1–TLR2 heterodimer specifically responses to bacterial tri-acetylated lipopeptides or porins [34]. Activation of TLR1–TLR2 heterodimerization subsequently activates the NF-κB signaling [35] and MAPKs [17] pathways and up-regulates the inflammatory related proteins (such as iNOS and COX-2). Finally, the production of NO and secretion of inflammatory cytokines were increased. Using the specific TLR1–TLR2 heterodimerization stimulator of Pam3CSK4 [36], a synthetic tripalmitoylated lipopeptide with the similarity to the bacterial lipoproteins, the inflammatory responses are subsequently induced through activation on NF-κB signaling pathway.
Being a traditional anti-rheumatoid herbal medicine, SP was demonstrated to be beneficial to and has been applied for the management of various chronic inflammatory diseases [26,27,28]. The inactivation of TLR4-induced NF-κB signaling was identified to be involved in the biological mechanisms of SP on inhibiting the LPS-induced inflammations [32, 37]. However, our preliminary study, we observed that SPE presented more potency on suppressing Pam3CSK4-induced than LPS-induced NO release in RAW 264.7 macrophages. The results suggested the inhibition of SP on TLR1-TLR2 activation mediated inflammatory responses might be involved in its potential mechanisms on anti-inflammation. Further investigated in Pam3CSK4-stimulated macrophages, SPE improved the inflammatory responses of the cells by decreasing the NO release and cytokines (IL-6, TNF-α, and MCP-1) secretion into the culture medium. The biological mechanisms of such effect were identified to be associated with the suppressions of SPE on Pam3CSK4-stimulated NF-κB activation and up-regulation of the protein expressions of iNOS and COX-2. On the other hand, Pam3CSK4 stimulated inflammation by activating the MAPKs signaling, but SPE was determined to have no significant influences on the activated p38, ERK and JNK.
Previously, SP has been reported to contain multiple components. Diterpenoids, sesquiterpenoids and flavonoids have been determined to be the major components in SP [21,22,23,24]. Kirenol and darutoside, two ent-pimarane-type diterpenoids, were reported to be highly contained in SP [21, 38, 39]. Pharmacologically, the anti-inflammatory effects of SP were partially identified to be related to kirenol and darutoside [40]. Rutin, a widely distributed flavonoid in many plants, has been demonstrated to present various pharmacological activities such as anti-inflammation, anti-oxidant, anti-cancer as well as others [41]. In this study, the chemical compositions of SPE were analyzed using the HPLC method, the contents of the three representative components of kirenol (1.81 ± 0.02%), darutoside (0.28 ± 0.03%) and rutin (0.27 ± 0.01%) in SPE were quantified to be 2.36% in total. Further investigations on other chemical components in SPE as well as their relationship to TLR1-TLR2-activated inflammation are currently under progress in our research team.
Conclusions
In conclusion, the anti-inflammatory activity of SP on Pam3CSK4-stimulated RAW 264.7 macrophages was investigated and reported. The results demonstrated that the 50% ethanol extract of SP could effectively decrease Pam3CSK4-induced NO release and cytokines secretion in RAW 264.7 cells. The potential biological mechanisms of SP on anti-inflammation were associated with its inactivation on Pam3CSK4-stimulated NF-κB signaling.
Abbreviations
- SP:
-
Siegesbeckia pubescens Makino
- SPE:
-
50% ethanol extract of SP
- NF-κB:
-
nuclear factor-κB
- MAPK:
-
mitogen-activated protein kinase
- TLR:
-
toll-like receptor
- Pam3CSK4 :
-
N-palmitoyl-S-[2,3-bis(palmitoyloxy)-(2RS)-propyl]-[R]-cysteinyl-[S]-seryl-[S]-lysyl-[S]-lysyl-[S]-lysyl-[S]-lysine·3HCl
- NO:
-
nitric oxide
- COX-2:
-
cyclooxygenase-2
- iNOS:
-
inducible nitric oxide synthase
- IL-6:
-
interleukin-6
- IL-1β:
-
interleukin-1β
- TNF-α:
-
tumor necrosis factor-α
- MCP-1:
-
monocyte chemoattractant protein-1
- LPS:
-
lipopolysaccharide
- MTT:
-
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- DMEM:
-
Dulbecco’s modified eagle medium
- FBS:
-
fetal bovine serum
- LDH:
-
lactose dehydrogenase
- DAF-FM:
-
4-amino-5methylamino-2′,7′-difluorofluorescein diacetate
References
Hawiger J. Innate immunity and inflammation: a transcriptional paradigm. Immunol Res. 2001;23(2–3):99–109.
Sauaia A, Moore FA, Moore EE. Postinjury inflammation and organ dysfunction. Crit Care Clin. 2017;33(1):167–91.
Kunnumakkara AB, Sailo BL, Banik K, Harsha C, Prasad S, Gupta SC, Bharti AC, Aggarwal BB. Chronic diseases, inflammation, and spices: how are they linked? J Transl Med. 2018;16(1):14.
Tak PP, Firestein GS. NF-kappaB: a key role in inflammatory diseases. J Clin Invest. 2001;107(1):7–11.
Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol. 2009;1(6):a001651.
Shih RH, Wang CY, Yang CM. NF-kappaB signaling pathways in neurological inflammation: a mini review. Front Mol Neurosci. 2015;8:77.
Zhang H, Sun SC. NF-κB in inflammation and renal diseases. Cell Biosci. 2015;5:63.
Zhang Q, Lenardo MJ, Baltimore D. 30 years of NF-κB: a blossoming of relevance to human pathobiology. Cell. 2017;168(1–2):37–57.
Kulms D, Schwarz T. NF-kappaB and cytokines. Vitam Horm. 2006;74:283–300.
Hayden MS, Ghosh S. Regulation of NF-κB by TNF family cytokines. Semin Immunol. 2014;26(3):253–66.
Lai Y, Dong C. Therapeutic antibodies that target inflammatory cytokines in autoimmune diseases. Int Immunol. 2016;28(4):181–8.
Jiménez-Dalmaroni MJ, Gerswhin ME, Adamopoulos IE. The critical role of toll-like receptors–from microbial recognition to autoimmunity: a comprehensive review. Autoimmun Rev. 2016;15(1):1–8.
Drexler SK, Foxwell BM. The role of toll-like receptors in chronic inflammation. Int J Biochem Cell Biol. 2010;42(4):506–18.
Ospelt C, Gay S. TLRs and chronic inflammation. Int J Biochem Cell Biol. 2010;42(4):495–505.
Achek A, Yesudhas D, Choi S. Toll-like receptors: promising therapeutic targets for inflammatory diseases. Arch Pharm Res. 2016;39(8):1032–49.
Elshabrawy HA, Essani AE, Szekanecz Z, Fox DA, Shahrara S. TLRs, future potential therapeutic targets for RA. Autoimmun Rev. 2017;16(2):103–13.
Lee NY, Lee HY, Lee KH, Han SH, Park SJ. Vibrio vulnificus IlpA induces MAPK-mediated cytokine production via TLR1/2 activation in THP-1 cells, a human monocytic cell line. Mol Immunol. 2011;49(1–2):143–54.
Funderburg NT, Jadlowsky JK, Lederman MM, Feng Z, Weinberg A, Sieg SF. The toll-like receptor 1/2 agonists Pam(3) CSK(4) and human β-defensin-3 differentially induce interleukin-10 and nuclear factor-κB signalling patterns in human monocytes. Immunology. 2011;134(2):151–60.
Khan J, Sharma PK, Mukhopadhaya A. Vibrio cholerae porin OmpU mediates M1-polarization of macrophages/monocytes via TLR1/TLR2 activation. Immunobiology. 2015;220(11):1199–209.
Broz P, Monack DM. Newly described pattern recognition receptors team up against intracellular pathogens. Nat Rev Immunol. 2013;13(8):551–65.
Huo L, Jiang Z, Li H, Wang M, Ye X, Ji B, Guo X. Simultaneous determination of seven major diterpenoids in Siegesbeckia pubescens Makino by high-performance liquid chromatography coupled with evaporative light scattering detection. J Sep Sci. 2012;35(19):2585–91.
Wang R, Liu YQ, Ha W, Shi YP, Hwang TL, Huang GJ, Wu TS, Lee KH. In vitro anti-inflammatory effects of diterpenoids and sesquiterpenoids from traditional Chinese medicine Siegesbeckia pubescens. Bioorg Med Chem Lett. 2014;24(16):3944–7.
Yao Y, Liang X, Sun X, Yin L, He H, Guo X, Jiang Z. Rapid extraction and analysis method for the simultaneous determination of 21 bioflavonoids in Siegesbeckia pubescens Makino. J Sep Sci. 2015;38(7):1130–6.
Wang J, Xie K, Duan H, Wang Y, Ma H, Fu H. Isolation and characterization of diterpene glycosides from Siegesbeckia pubescens. Bioorg Med Chem Lett. 2017;27(8):1815–9.
Tao HX, Xiong W, Zhao GD, Peng Y, Zhong ZF, Xu L, Duan R, Tsim KWK, Yu H, Wang YT. Discrimination of three Siegesbeckiae Herba species using UPLC-QTOF/MS-based metabolomics approach. Food Chem Toxicol. 2018. https://doi.org/10.1016/j.fct.2017.12.068.
Lee M, Kim SH, Lee HK, Cho Y, Kang J, Sung SH. ent-kaurane and ent-pimarane diterpenes from Siegesbeckia pubescens inhibit lipopolysaccharide-induced nitric oxide production in BV2 microglia. Biol Pharm Bull. 2014;37(1):152–7.
Wang J, Cai Y, Wu Y. Antiinflammatory and analgesic activity of topical administration of Siegesbeckia pubescens. Pak J Pharm Sci. 2008;21(2):89–91.
Kim HM. Effects of Siegesbeckia pubescens on immediate hypersensitivity reaction. Am J Chin Med. 1997;25(2):163–7.
Wang J, Duan H, Wang Y, Pan B, Gao C, Gai C, Wu Q, Fu H. ent-Strobane and ent-pimarane diterpenoids from Siegesbeckia pubescens. J Nat Prod. 2017;80(1):19–29.
Lv D, Guo KW, Xu C, Huang M, Zheng SJ, Ma XH, Pan LH, Wang Q, Yang XZ. Essential oil from Siegesbeckia pubescens induces apoptosis through the mitochondrial pathway in human HepG2 cells. J Huazhong Univ Sci Technol Med Sci. 2017;37(1):87–92.
Lee SG, Kim M, Kim CE, Kang J, Yoo H, Sung SH, Lee M. Quercetin 3,7-O-dimethyl ether from Siegesbeckia pubescens suppress the production of inflammatory mediators in lipopolysaccharide-induced macrophages and colon epithelial cells. Biosci Biotechnol Biochem. 2016;80(11):2080–6.
Park HJ, Kim IT, Won JH, Jeong SH, Park EY, Nam JH, Choi J, Lee KT. Anti-inflammatory activities of ent-16alphaH,17-hydroxy-kauran-19-oic acid isolated from the roots of Siegesbeckia pubescens are due to the inhibition of iNOS and COX-2 expression in RAW 264.7 macrophages via NF-kappaB inactivation. Eur J Pharmacol. 2007;558(1–3):185–93.
Su T, Yu H, Kwan HY, Ma XQ, Cao HH, Cheng CY, Leung AK, Chan CL, Li WD, Cao H, Fong WF, Yu ZL. Comparisons of the chemical profiles, cytotoxicities and anti-inflammatory effects of raw and rice wine-processed Herba Siegesbeckiae. J Ethnopharmacol. 2014;156:365–9.
Jin MS, Kim SE, Heo JY, Lee ME, Kim HM, Paik SG, Lee H, Lee JO. Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell. 2007;130:1071–82.
Cheng K, Gao M, Godfroy JI, Brown PN, Kastelowitz N, Yin H. Specific activation of the TLR1–TLR2 heterodimer by small-molecule agonists. Sci Adv. 2015;1(3):e1400139.
Aliprantis AO, Yang RB, Mark MR, Suggett S, Devaux B, Radolf JD, Klimpel GR, Godowski P, Zychlinsky A. Cell activation and apoptosis by bacterial lipoproteins through toll-like receptor-2. Science. 1999;285(5428):736–9.
Li H, Kim JY, Hyeon J, Lee HJ, Ryu JH. In vitro antiinflammatory activity of a new sesquiterpene lactone isolated from Siegesbeckia glabrescens. Phytother Res. 2011;25(9):1323–7.
Xu FQ, Liu JQ, Wang JT, Huang JP, Zhang J. Simultaneous determination of kirenol and darutoside in Siegesbeckiae Herba by high-performance liquid chromatography. Chin Tradit Pat Med. 2011;33(8):1444–6.
Ye XX, Song JX, Ji B, Jiang Z, Guo XJ. Simultaneous determination of five major constituents in Siegesbeckia pubescens Makino by HPLC-RID. J Shenyang Pharm Univ. 2014;31(1):36–40.
Lu Y, Xiao J, Wu ZW, Wang ZM, Hu J, Fu HZ, Chen YY, Qian RQ. Kirenol exerts a potent anti-arthritic effect in collagen-induced arthritis by modifying the T cells balance. Phytomedicine. 2012;19(10):882–9.
Chua LS. A review on plant-based rutin extraction methods and its pharmacological activities. J Ethnopharmacol. 2013;150(3):805–17.
Authors’ contributions
HY and YW organized, conceived, and supervised the study. WS and ZZ designed the study and conducted the experiments. KL, WX and WSC analyzed the data. AKWT edited and revised the manuscript. All authors read and approved the final manuscript.
Acknowledgements
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Funding
This study was supported by the National Natural Science Foundation of China (NSFC, No. 81470170), the Research Committee of the University of Macau (SRG2015-00060-ICMS-QRCM, MYRG2017-00178-ICMS, and CPG2014-00012-ICMS), the Guangzhou Science and Technology Innovation funding (EF007/ICMS-YH/2018/GSTIC), the Project funded by China Postdoctoral Science Foundation (No. 2017M622811), and the opening fund of the State Key Laboratory of Quality Research in Chinese Medicine of University of Macau (No. SKL-QRCM-2014-2016).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding authors
Additional file
Additional file 1.
Minimum Standards of Reporting Checklist.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Sang, W., Zhong, Z., Linghu, K. et al. Siegesbeckia pubescens Makino inhibits Pam3CSK4-induced inflammation in RAW 264.7 macrophages through suppressing TLR1/TLR2-mediated NF-κB activation. Chin Med 13, 37 (2018). https://doi.org/10.1186/s13020-018-0193-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13020-018-0193-x