
Abstract
Background
Centipeda minima (Ebushicao) has been used for the treatment of various diseases, such as nasal allergies, rhinitis and sinusitis, nasopharyngeal carcinoma, cough, and headache. This study aims to investigate the anticancer activities of Centipeda minima ethanol extracts (CME) against nasopharyngeal carcinoma cell CNE-1 and their underlying mechanism.
Methods
CNE-1 cells were treated with different concentrations (15–50 μg/mL) of CME for different time intervals (24, 48, and 72 h). Cytotoxicity of CME was determined by MTT assay. Cell morphological changes were observed by fluorescence microscopy after HO 33258 staining. Cell cycle status was evaluated by flow cytometry following propidium iodide staining. Apoptosis was detected by flow cytometry following annexin V-FITC/PI staining. The levels of apoptosis-associated and PI3K-Akt-mTOR signaling related proteins were measured by western blotting analysis.
Results
CME (15–50 μg/mL) significantly inhibited the proliferation of CNE-1 in a dose- and time-dependent manner (P = 0.026 for 15 μg/mL, P < 0.001 for 25, 30, 40, and 50 μg/mL, respectively); the IC50 values (μg/mL) were 41.57 ± 0.17, 30.34 ± 0.06 and 24.98 ± 0.08 for 24, 48 and 72 h treatments, respectively. Significant morphological changes of CNE-1 cells displaying apoptosis were observed after CME treatment. CME showed low cytotoxicity toward normal LO2 cells. CNE-1 cells were arrested in the G2/M phase while treated with 15, 25, 40 μg/mL of CME, respectively (P = 0.032, P = 0.0053, P < 0.001). CME (15, 25, 40 μg/mL) down-regulated Bcl-2 expression (P = 0.032, P = 0.0074, P < 0.001), and up-regulated Bax (P = 0.026, P = 0.0056, P < 0.001) with activation of caspase-3, caspase-8, caspase-9, and PARP observed in CNE-1 cells (P = 0.015, P = 0.0067, P < 0.001 for caspase 3; P = 0.210, 0.028, < 0.001 for caspase 8; P = 0.152, 0.082, 0.0080 for caspase 9; P = 0.265, 0.0072, < 0.001 for PARP). CME suppressed the activation of the PI3K-AKT-mTOR pathway (P = 0.03, 0.0007, 0.004, 0.006, 0.022 for p-PI3K, p-Akt-Ser473, p-Akt-Thr308, p-mTOR-Ser2448, p-mTOR-Ser2481, respectively after 40 μg/mL of CME treated for 24 h).
Conclusion
CME inhibited the proliferation of CNE-1 cells and activation of the PI3K-AKT-mTOR signaling pathway.
Similar content being viewed by others

Background
Nasopharyngeal carcinoma (NPC), a malignant epithelial tumor derived from the nasopharyngeal surface epithelium, has a low incidence in Western countries but a high incidence in Southeast Asia, southern China and the Mediterranean basin [1]. Epstein-Barr virus infection is a critical factor in NPC pathogenesis [2, 3]. Additional etiological factors for NPC include genetic susceptibility, environmental contamination, and dietary habits such as excessive consumption of salt-preserved fish during childhood [1, 4]. Radiotherapy is the standard primary treatment for NPC [5], and a combination of radiotherapy and chemotherapy has been shown to be an effective therapeutic strategy for treating advanced NPC [6]. The most prevalent chemotherapeutic drugs used to treat NPC are cisplatin-based drugs, which are highly toxic and prone to drug resistance [7, 8]. Chinese medicine (CM) in combination with conventional cancer therapies has been applied to NPC [9]. Controlled clinic trials demonstrated that CM therapies achieve prolonged survival and improved quality of life in NPC patients [10, 11].
Some CMs exhibit antitumor activities toward NPC cells via growth inhibition, apoptotic induction and cell cycle arrest [12, 13]. Centipeda minima (L.) A. et Aschers. (Ebushicao) (Asteraceae) has been used to treat nasal allergies, rhinitis, sinusitis, cough, headache [14, 15] and NPC [16]. Recent researches have focused on its anti-NPC activities, leading to the isolation of a few sesquiterpene lactones against NPC cells [17, 18]. Volatile oils from C. minima extracted by steam distillation and supercritical fluid extraction induced CNE cell death via induction of intrinsic apoptosis by regulating the expression of the Bcl-2 family of proteins [16]. Ethyl acetate extracts of C. minima exhibited anti-proliferative activity against CNE-2 cells [19]. However, the anticancer activities of C. minima ethanol extracts (CME) against the NPC cell line CNE-1, and their underlying mechanism, remain unclear.
This study aims to investigate the anticancer activities of CME against the nasopharyngeal carcinoma cell line CNE-1 and determine their underlying mechanism.
Methods
Plant materials and sample preparation
Centipeda minima was collected in August 2010 in Xiangfan, Hubei Province, China (latitude, 32°04′ N; longitude, 112°05′ E), and authenticated by Si-bao Chen based on morphological features. A voucher specimen (EBSC-016-09) was deposited at the herbarium of the State Key Laboratory of Chinese Medicine and Molecular Pharmacology, Department of Applied Biology and Chemical Technology, The Hong Kong Polytechnic University. The herb was air-dried and ground to coarse powder. The powder (30 g) was macerated with 0.5 L of 95 % ethanol at room temperature for 72 h. The extraction was repeated twice. After extraction, the ethanol extracts were combined, filtered through Whatman filter paper (Whatman, Maidstone, UK), and evaporated to dryness using a rotary evaporator (Precision MLG3, Heidolph, Germany) on a water bath at 40 °C, and further lyophilized to dried powder (3.80 g). CME samples with a yield of 12.65 % were stored in the refrigerator at 4 °C until further use. CME was dissolved in DMSO and filtered to 0.22 μm to obtain the stock concentration of 20 mg/mL. Finally, a serial dilution to concentrations of 15, 25, 30, 40, and 50 μg/mL CME was performed. DMSO-treated cells were employed as a vehicle control in all experiments.
Chemicals
3-(4,5-Dimethylthiazole-2-yl)-2,5-diphenyltetrazolium bromide (MTT), dimethyl sulfoxide (DMSO), Hoechst 33258 and cisplatin were obtained from Sigma (St. Louis, MO, USA). The annexin V-FITC apoptosis detection kit and cell cycle analysis kit were purchased from Beyotime (Invitrogen, Carlsbad, CA, USA). Precision Plus Protein Standards (Dual Color) and Immun-Star™ WesternC™ Chemiluminescent Kit were purchased from Bio-Rad (Hercules, CA, USA). Antibodies against procaspase 8, cleaved caspase 9, cleaved PARP, PI3K p110α, Akt, p-Akt (Thr308 and Ser473), mTOR and p-mTOR (Ser2448 and Ser2481) were obtained from Cell Signaling Technology (Beverly, MA, USA). All other primary antibodies, as well as anti-rabbit and anti-mouse secondary horseradish peroxidase antibodies, were purchased from Abcam (Cambridge, MA, USA). All other common chemicals were reagent grade.
Cell culture
CNE-1 human nasopharyngeal carcinoma cells (depository no. CBP60002) were purchased from the Cancer Institute and Hospital, Chinese Academy of Medical Sciences (Beijing, China). LO2 human normal liver cells (depository no. GNHu 6) were purchased from the Cell Bank of Chinese Academy of Sciences (Shanghai, China). Cells were cultured in RPMI-1640 medium (Invitrogen, USA) supplemented with 10 % fetal bovine serum (Gibco, Life Technologies, Grand Island, NY, USA), 100 IU/mL penicillin, 100 μg/mL streptomycin (Thermo Fisher Scientific, Madison, WI, USA) at 37 °C in a humidified incubator with a 5 % CO2 atmosphere. The medium was renewed three times a week. Cells in logarithmic growth phase were used for all experiments.
Cell viability assay
The effects of CME on the viability of CNE-1 and LO2 cells were evaluated by MTT assay [20]. Cells (3 × 104 cells/mL) were seeded into the wells of 96-well culture plates (Thermo Fisher Scientific) in 100 μL of medium per well and then allowed to adhere for 24 h at 37 °C in a 5 % CO2 atmosphere. After incubation, the cells were treated with the target concentrations of CME (15–50 μg/mL), 0.1 % DMSO as the vehicle control, and cisplatin (4 μg/mL) as a positive control, for 24 h. After incubation for 24, 48, and 72 h at 37 °C in a CO2 incubator, 10 μL of MTT solution [5 mg/mL in phosphate-buffered saline (PBS)] was added to each well and incubated for a further 4 h. Then, excess medium was removed and 150 μL of DMSO was added to each well to dissolve the formazan crystals. The optical density in each well was measured using a microplate spectrophotometer (BMG POLARstar Galaxy, Offenburg, Germany) at 490 nm. Triplicate experiments were performed for treatment with each concentration. The percentage cell viability (%) was calculated by comparison with a sample’s corresponding control. IC50 values were calculated to evaluate the cytotoxic effects of treatments on CNE-1 cells.
Morphological assessment
The cells (1 × 106 cells/mL) were seeded into 6-well plates, and then treated with different concentrations (15, 25, 30, 40, or 50 μg/mL) of CME for 24 h. Similarly, cells were treated with 25 μg/mL CME for 24, 48, and 72 h, respectively. DMSO (0.1 %)-treated cells were employed as controls. Subsequently, the cells were washed twice with cold PBS, fixed with 4 % paraformaldehyde solution for 10 min, stained with Hoechst 33258 (10 μg/mL) for 15 min in the dark, and then observed under an inverted fluorescence microscope (Leica DMRB, Weitzl, Germany) at the excitation wavelength of 352 nm and emission wavelength of 461 nm.
Apoptosis analysis by annexin V-FITC/PI staining
The numbers of apoptotic cells were assessed using an annexin V-FITC apoptosis detection kit (Invitrogen, USA) according to the manufacturer’s protocol. Cells (1 × 106 cells/mL) were treated with various concentrations of CME for 24, 48, and 72 h. Cells were then harvested, and re-suspended in binding buffer. Subsequently, cells were stained with 5 mL of annexin V-FITC and 5 mL of propidium iodide (PI) for 15 min at room temperature in the dark. The apoptotic cell percentages were then immediately detected using a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA, USA).
Cell cycle analysis by PI staining
Cells (1 × 106 cells/well) were treated with various concentrations of CME (15, 25, and 40 μg/mL) for 24, 48, and 72 h. Cells were then collected and washed twice with 1 mL of ice-cold PBS, suspended in 1 mL of ice-cold 70 % ethanol (containing 1 × 105 cells) and kept overnight at 4 °C. The next day, cells were washed with PBS and incubated with 500 μL of PI staining solution (containing PI 40 μg/mL, RNase A 100 μg/mL and PBS) for 30 min in the dark at 37 °C. Then, samples were filtered using 70-μm filters into test tubes and detected by flow cytometry (FACSCalibur, Bio-Rad). Analysis of cell cycle status was performed using ModFit LT 3.0™ software (BD Biosciences).
Western blot analysis
CNE-1 cells (1 × 106 cells/well) were seeded into 100 mL culture dishes for 24 h and then treated with various concentrations of CME (15, 25, and 40 μg/mL). After incubation for 24 h, the cells were harvested and prepared in NP-40 lysis buffer (2 mM Tris–Cl pH 7.5, 150 mM NaCl, 10 % glycerol and 0.2 % NP-40 plus a protease inhibitor cocktail) for 30 min on ice. Cell lysates were ultra-centrifuged (Allegra™ 21R Centrifuge, Beckman Coulter, Krefeld, Germany) at 14,000×g for 10 min at 4 °C, and then the supernatant was collected as the total cellular proteins. Protein concentrations were determined using a BCA protein assay kit (Beijing ComWin Biotech Co.,Ltd., Beijing, China). Proteins (10 μg for β-actin and Bax, 20 μg for PARP and cleaved PARP, 30 μg for caspase-3, -9, and -8, PI3K p110α, Akt, and p-Akt, 50 μg for Bcl-2, mTOR, and p-mTOR) were resolved on 12–15 % SDS-PAGE gels, then transferred to PVDF (polyvinylidene difluoride) membranes (Roche Applied Science, Basel, Switzerland) and blocked with 5 % non-fat dry milk in Tris-buffered saline containing 0.1 % Tween-20 (T-TBS) overnight at 4 °C. After washing with T-TBS, the membranes were incubated with primary antibodies against procaspase-3, -8, and -9; caspase-3, -8, and -9; Bcl-2, Bax, PARP, cleaved PARP; PI3K p110α, Akt, p-Akt (Thr308 and Ser473), mTOR, p-mTOR (Ser2448 and Ser2481), and β-actin overnight at 4 °C. Subsequently, the membranes was incubated with goat anti-rabbit or horseradish peroxidase-conjugated goat anti-rabbit secondary antibodies (Merck Millipore) at room temperature for 2 h at dilutions of 1:5000 and 1:10,000, respectively. Antibody-bound proteins were detected, normalized to β-actin and quantified in a ChemiDoc™ XRS system (Universal Hood II, Bio-Rad) using an Immun-Star™ HRP Chemiluminescence Kit (Bio-Rad).
Statistical analysis
All data are expressed as the mean ± standard deviation (SD) of at least three independent experiments. Analyses of dose-, concentration- and time-dependent effects were performed by sigmoidal non-linear regression using GraphPad Prism 5.02 software (GraphPad Software, Inc., La Jolla, CA, USA). The percentage cell viability (%) after CME treatment with different concentrations and time intervals was used for the fitting. The two-tailed Student’s t test was used for comparisons of two groups, and multi-group differences were analyzed by one-way analysis of variance (ANOVA) followed by Dunnett’s multiple comparison test. P values < 0.05, 0.01, or 0.001 were considered statistically significant.
Results
Cytotoxic effects of CME on CNE-1 cells
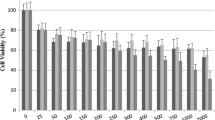
CNE-1 cells were treated with various concentrations (15–50 μg/mL) of CME for 24, 48, and 72 h, and the cell viability was determined by MTT assay. CME decreased CNE-1 cell viability in a dose- and time-dependent manner (Fig. 1; Table 1). After treatment with CME (15, 25, 30, 40 and 50 μg/mL) for 24 h, the percentage of live cells were 95.41, 81.27, 60.10, 50.0 and 42.41 %, (P = 0.026 for 15 μg/mL, P < 0.001 for 25, 30, 40, and 50 μg/mL), respectively. Meanwhile, those were 51.14, 34.0, and 25.05 % (P < 0.001 for each test), while CNE-1 cells were treated with CME (40 μg/mL) for 24, 48, and 72 h, respectively. The IC50 values were 41.57 ± 0.17, 30.34 ± 0.06 and 24.98 ± 0.08 μg/mL for 24, 48, and 72 h incubations, respectively. Despite its potency in CNE-1 cells, CME exhibited no significant cytotoxic effect on normal LO2 cells (data not shown due to no significant variation versus vehicle control).

Dose- and time-dependent cytotoxic effects of CME on CNE-1 cells. CNE-1 cells were treated with 0.1 % DMSO (vehicle control), 4 μg/mL cisplatin (positive control) and various concentrations (15–50 μg/mL) of CME for 24, 48, and 72 h, respectively, and then cell viability was determined by MTT assay. Values are the mean ± SD of at least three independent experiments. *P < 0.05, **P < 0.001 versus control
Morphological changes in CNE-1 cells after CME treatment
Morphological changes and cell death in CNE-1 cells were observed under an inverted fluorescence microscope (DFC 420 C, Leica, Germany) after HO 33258 staining. Vehicle-treated (control) cells showed normal cell architecture with clear cytoskeletons (Fig. 2). Typical morphological changes associated with apoptosis, including chromatin condensation, apoptotic body formation and nuclear degradation, were observed in CME-treated cells. These morphological changes were enhanced with increasing concentrations of CME (Fig. 2a), and with increasing treatment duration (Fig. 2b).

Cell morphological changes induced by CME. a Cells treated with various concentrations of CME (15, 25, 30, 40, and 50 μg/mL) for 24 h. b Cells treated with 25 μg/mL for 24, 48, and 72 h. The cells were fixed after CME treatment, stained with HO 33258, and their nuclear morphology was photographed under fluorescence using a blue filter (×400). The arrow points to an apoptotic body
Apoptosis analysis by flow cytometry
The timing of induction of apoptosis was determined by flow cytometry following annexin V-FITC/PI double staining. After exposure to CME (15, 25, or 40 μg/mL) for 48 h, the proportion of apoptotic cells dramatically increased in a dose-dependent manner; the percentages of apoptotic cells were 39.53, 56.43, and 76.42 %, respectively (P = 0.0086, P < 0.001, P < 0.001) (Fig. 3a). Similarly, CME induced CNE-1 cell apoptosis in a time-dependent manner. The percentages of apoptotic cells were 43.30, 76.42, and 91.29 % after treatment with 40 μg/mL CME for 24, 48, and 72 h, respectively (P < 0.001 for each test) (Fig. 3b).

Flow cytometry analysis of the induction of apoptosis in CNE-1 cells by CME. Results are shown in logarithmic fluorescence intensity with the x-axis (annexin V-FITC) versus y axis (PI). Four quadrants represent necrotic cells (Q1: AV−/PI+), late apoptotic cells (Q2: AV+/PI+), early apoptotic cells (Q3: AV+/PI−) and viable cells (Q4: AV−/PI−). Numbers indicate the percentages of cells in each quadrant and a minimum of 5000 events were read (n = 3). a CNE-1 cells were treated with 15, 25, and 40 μg/mL CME for 48 h; b CNE-1 cells were treated with 40 μg/mL CME for 24, 48, and 72 h
Flow cytometric analysis of the cell cycle status of CNE-1 cells
Cell cycle progression was analyzed by flow cytometry to determine whether the cytotoxic effect of CME toward CNE-1 cells was associated with the induction of cell cycle arrest. After treatment with 15, 25, and 40 μg/mL CME for 48 h, the percentages of cells in the G2/M phase were 16.00, 24.71, and 60.85 %, respectively (P = 0.032, P = 0.0052, P < 0.001); while the percentages of those in the G0/G1 phase were 50.74, 47.67, and 21.94 %, respectively (P = 0.102, 0.067, P < 0.001) (Fig. 4a). Meanwhile, after treatment with CME (40 μg/mL) for 24, 48, and 72 h, the percentages of cells in the G2/M phase were 29.38, 35.21, and 41.13 %, respectively (P < 0.001 in each test); while the percentages of those in the G0/G1 phase were 42.69, 30.20, and 25.64 %, respectively (P = 0.0072, P = 0.0047, P < 0.001) (Fig. 4b). No significant variations in the percentages of cells in S phase were observed after CME treatment. These results indicated that the inhibition of CNE-1 cell proliferation induced by CME could be attributed to cell cycle arrest in the G2/M phase.

Cell cycle distribution of CNE-1 cells in the flow cytometric analysis after CME treatment. a CNE-1 cells were treated with 0.1 % DMSO (control) and 15, 25, and 40 μg/mL CME for 48 h; b CNE-1 cells were treated with 40 μg/mL CME for 0 (control), 24, 48, and 72 h
Expression of CME-regulated apoptosis-related proteins
The expression levels of apoptosis-related proteins were analyzed by western blotting to study CME-induced apoptosis of CNE-1 cells in more detail. After treatment with CME (15, 25, 40 μg/mL) for 24 h, the levels of cleaved caspase 3, 8, 9 and cleaved PARP increased in a dose-dependent manner (P = 0.020, P = 0.0060, P < 0.001 for caspase 3; P = 0.210, 0.028, < 0.001 for caspase 8; P = 0.152, 0.082, 0.0082 for caspase 9; P = 0.265, P = 0.0060, P < 0.001 for PARP), while the levels of pro-caspases 3, 8, 9 and PARP decreased (P = 0.0045, P < 0.001, P < 0.001 for pro-caspase 3; P = 0.147, 0.015, 0.036 for pro-caspase 8; P = 0.250, 0.031, 0.0085 for pro-caspase 9; P = 0.092, < 0.001, 0.001 for pro-PARP) (Fig. 5a). Meanwhile, the anti-apoptotic protein Bcl-2 was down-regulated (P = 0.035, P = 0.0055, P < 0.001), while the pro-apoptotic protein Bax was up-regulated (P = 0.020, P = 0.0035, P < 0.001) (Fig. 5b), and the Bax/Bcl-2 ratio was significantly increased as the concentrations of CME increasing (P = 0.072, 0.034, 0.0005) (Fig. 5c).

Effects of CME on the expression levels of apoptosis-related proteins in CNE-1 cells after 24 h of treatment. a Expression levels of procaspase-3, -8, and -9; caspase-3, -8, and -9; PARP and cleaved-PARP. b Expression level of Bcl-2 and Bax. c Bax/Bcl-2 ratios; each value is based on three independent experiments. *P = 0.034, **P = 0.0005 versus control
Inhibition of PI3K-Akt-mTOR signaling in CME-treated CNE-1 cells
We investigated whether CME treatment could also down-regulate the expression levels of related proteins in the PI3K-Akt-mTOR pathway. After treatment with CME (15, 25, 40 μg/mL) for 24 h, the levels of PI3Kinase P110α, the catalytic subunit of PI3K (P = 0.042, 0.040, 0.030), and the amount of phosphorylation of Akt at Ser473 (P = 0.047, 0.0008, 0.0007) and Thr308 (P = 0.021, 0.005, 0.004) were remarkably decreased in a concentration-dependent manner, while the amount of total Akt remained constant following CME treatment (Fig. 6). Consistently, phosphorylation of the downstream effector mTOR at Ser2448 (P = 0.037, 0.025, 0.006) and Ser2481 (P = 0.046, 0.039, 0.022) also showed a dose-dependent decrease, and the amount of total mTOR was slightly down-regulated upon CME treatment of CNE-1 cells.

Inhibition of the PI3K-Akt-mTOR signaling pathway in CNE-1 cells by CME. After treatment with the indicated concentrations of CME for 24 h, cell lysates were subjected to western blot analysis using the indicated antibodies against PI3K p110α, Akt and p-Akt (Thr308 and Ser473), mTOR and p-mTOR (Ser2448 and Ser2481). β-actin served as a loading control
Discussion
In the present study, the anti-cancer effects of CME toward human nasopharyngeal carcinoma CNE-1 cells in terms of cytotoxicity, apoptosis induction and cell cycle arrest were investigated. CME possessed significant cytotoxic activities against CNE-1 cells, in a time- and dose-dependent manner, but very low cytotoxicity toward normal LO2 cells. These results suggest that CME has potential selective anti-cancer effects on CNE-1 cells.
Apoptotic induction in cancer cells is a crucial therapeutic strategy for cancer treatment. CME induced morphological changes including cell shrinkage, the appearance of apoptotic vacuoles, and chromatin condensation, detected by fluorescence microscopy after CME treatment followed by Ho 35288 staining. This apoptotic induction was confirmed by flow cytometry using an annexin V-FITC/PI double-staining approach.
In this study, changes in cell cycle progression were analyzed by flow cytometry. CME induced obvious G2/M phase arrest in CNE-1 cells in a dose- and time-dependent manner. G2/M, the most important checkpoint for DNA damage, is critical to cell cycle progression, and strictly regulated by specific genes [21]. If phase arrest cannot be repaired, cells may undergo apoptosis.
Changes in the expression levels of apoptosis-related proteins in CNE-1 cells after CME treatment were evaluated by western blotting to explore the mechanism underlying the induction of apoptosis by CME in CNE-1 cells. The activation of caspase 9 through apoptotic protease activating factor-1 (APAF-1) and cytochrome c release [22] initiates activation of the caspase cascade, cleaves executioner caspase-3, and eventually results in apoptosis [23]. Cleaved caspase 8 activates downstream caspases such as caspase-3, -6, and -7 [24], which cleave specific intracellular substrates, such as polyADP-ribose polymerase (PARP), lamins and inhibitor of caspase activated DNase (ICAD), finally resulting in programmed cell death [25]. This study revealed that CME could significantly induce CNE-1 cell apoptosis via the activation of caspase-8, -9, -3, as well as PARP, through both extrinsic and intrinsic pathways.
The B cell lymphoma 2 (Bcl-2) family proteins, including pro-apoptotic members such as Bax, Bak and anti-apoptotic members such as Bcl-2, Bcl-xL, play important roles in the regulation of mitochondrial-mediated apoptosis [26, 27]. In this study, CME treatments downregulated the expression of Bcl-2, and upregulated the expression of Bax, resulting in CNE-1 cell apoptosis. Furthermore, elevated intracellular ratios of Bax/Bcl-2 were observed during a period of increased apoptotic cell death. These experimental findings suggested that CME induced CNE-1 cell apoptosis via a mitochondrial pathway.
The PI3K-AKT-mTOR pathway is a major signaling pathway in cancer [28]. This pathway is frequently mutated and overactivated in human cancers [29]. The PI3K-AKT-mTOR pathway promotes survival through inhibition of proapoptotic factors and activation of anti-apoptotic factors [30]. Through phosphorylation of pathway components, this pathway inhibits the activity of proapoptotic members while activating anti-apoptotic members [31]. Therefore, blockade of the PI3K-AKT-mTOR pathway has become a cancer therapy strategy. Our present results indicate for the first time that CME downregulates the PI3K-AKT-mTOR-mediated signaling in CNE-1 cells (Fig. 7). Additionally, 14-thienyl methylene matrine (YYJ18), a derivative of matrine, has been reported to induce apoptosis in NPC cells by targeting PI3K-Akt [32], which also provides evidence for the involvement of this pathway in NPC carcinogenesis.

Schematic diagram showing the hypothetical effects of CME on the PI3K-Akt-mTOR/signaling pathway and apoptosis in CNE-1 cells
Conclusion
CME inhibited the proliferation of CNE-1 cells and activation of the PI3K-AKT-mTOR signaling pathway.
Abbreviations
- CME:
-
Centipeda minma ethanol extracts
- CM:
-
Chinese medicine
- CNE-1:
-
high differentiated human nasopharyngeal carcinoma cells
- CNE-2:
-
low differentiated human nasopharyngeal carcinoma cells
- LO2:
-
human hepatocytes
- NPC:
-
nasopharyngeal carcinoma
- MTT:
-
3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyltetrazolium bromide
- DMSO:
-
dimethyl sulfoxide
- FBS:
-
fetal bovine serum
- RPMI1640:
-
Roswell Park Memorial Institute1640 medium
- PBS:
-
phosphate buffered saline
- PI:
-
peromide iodine
- FITC:
-
fluorescein-isothiocyanate
- SDS:
-
sodium dodeeyl sulphate
- PAGE:
-
polyacrylamidegel electrophoresis
- T-TBS:
-
tween-20
- PVDF:
-
polyrinylidene difluoride
- RNase A:
-
ribonuclease A
- ICAD:
-
lamins and inhibitor of caspase activated DNase
- PARP:
-
polyADP-ribose polymerase
- PI3K:
-
phosphatidylinositol 3-kinase
- Akt:
-
protein kinase B
- mTOR:
-
the mammalian target of rapamycin
- Bcl-2:
-
B cell lymphoma/lewkmia-2
- Bax:
-
Bcl-2 assaciated X protein
- YYJ18:
-
14-thienyl methylene matrine (YYJ18)
- Caspase:
-
cysteinyl aspartate specific proteinase
References
Yu MC, Yuan JM. Epidemiology of nasopharyngeal carcinoma. Semin Cancer Biol. 2002;12:421–9.
Raab-Traub N. Epstein—Barr virus in the pathogenesis o f NPC. Semin Cancer Biol. 2002;12:431–41.
Tao Q, Young LS, Woodman CB, Murray PG. Epstein-Barr virus (EBV) and its associated human cancers-genetics, epigenetics, pathobiology and novel therapeutics. Front Biosci. 2006;11:2672–713.
Lo Angela KF, Dawson CW, Jin DY, Lo KW. The pathological roles of BART miRNAs in nasopharngeal carcinoma. J Pathol. 2012;227:392–403.
Lee N, Xia P, Quivey JM, Sultanem K, Poon I, Akazawa C, Akazawa P, Weinberg V, Fu KK. Intensity-modulated radiotherapy in the treatment of nasopharyngeal carcinoma: an update of the UCSF experience. Int J Radiat Oncol Biol Phys. 2002;53:12–22.
Chan AT, Teo PM, Ngan RK, Leung TW, Lau WH, Zee B, Leung SF, Cheung FY, Yeo W, Yiu HH, Yu KH, Chiu KW, Chan DT, Mok T, Yuen KT, Mo F, Lai M, Kwan WH, Choi P, Johnson PJ. Concurrent chemotherapy-radio-therapy compared with radiotherapy alone in loco regionally advanced nasopharyngeal carcinoma: progression-free survival analysis of a phase III randomized trial. J Clin Oncol. 2002;20:2038–44.
Cheng SH, Jian JJ, Tsai SY, Yen KL, Chu NM, Chan KY, Tan TD, Cheng JC, Leu SY, Hsieh CY, Huang AT. Long-term survival of nasopharyngeal carcinoma following concomitant radiotherapy and chemotherapy. Int J Radiat Oncol. 2000;48:1323–30.
Xie SM, Fang WY, Liu Z, Wang SX, Li X, Liu TF, Xie WB, Yao KT. Lentivirus-mediated RNAi silencing targeting ABCC2 increasing the sensitivity of a human nasopharyngeal carcinoma cell line against cisplatin. J Transl Med. 2008;6:55–63.
Cho WC, Chen HY. Clinical efficacy of traditional Chinese medicine as a concomitant therapy for nasopharyngeal carcinoma: a systematic review and meta-analysis. Cancer Invest. 2009;27:334–44.
Li X, Yang GY, Li XX, Zhang Y, Yang JL, Chang J, Sun XX, Zhou XY, Guo Y, Xu Y, Liu J, Bensoussan A. Traditional Chinese medicine in cancer care: a review of controlled clinical studies published in Chinese. PLoS One. 2013;8:e60338.
Wei BH. Progression on treatment of nasopharyngeal carcinoma by combined therapy of radiation and Chinese herbal medicine. Zhongguo Zhong Xi Yi Jie He Za Zhi. 2001;21:477–9.
Liu HH, Peng H, Ji ZH, Zhao SW, Zhang YF, Wu J, Fan JP, Liao JC. Reactive oxygen species-mediated mitochondrial dysfunction is involved in apoptosis in human nasopharyngeal carcinoma CNE cells induced by Selaginella doederleinii extract. J Ethnopharmacol. 2011;138:184–91.
Su MX, Wu P, Li YL, Chung HY. Antiproliferative effects of volatile oils from Centipeda minima on human nasopharyngeal cancer CNE cells. Nat Prod Commun. 2010;5:151–6.
Shi Z, Fu GX. Flora of China, vol. 76. Beijing: Science Publishing House; 1983. p. 132.
China Pharmacopoeia Committee. Pharmacopoeia of the People’s Republic of China, 1st division of 2010 edition. Beijing: Chinese Medicine And Technology Publishing House; 2010. p. 326.
Su MX, Li YL, Chung HY, Ye WC. 2β-(Isobutyryloxy) florilenalin, a sesquiterpene lactone isolated from the medicinal plant Centipeda minima, induces apoptosis in human nasopharyngeal carcinoma CNE cells. Molecules. 2009;14:2135–46.
Wu P, Li XG, Liang N, Wang GC, Ye WC, Zhou GX, Li YL. Two new sesquiterpene lactones from the supercritical fluid extract of Centipeda minima. J Asian Nat Prod Res. 2012;14:515–20.
Wu P, Su MX, Wang Y, Wang GC, Ye WC, Chung HY, Li J, Jiang RW, Li YL. Supercritical fluid extraction assisted isolation of sesquiterpene lactones with antiproliferative effects from Centipeda minima. Phytochemistry. 2012;76:133–40.
Su MX, Chung HY, Li YL. Deoxyelephantopin from Elephantopus scaber L. induces cell-cycle arrest and apoptosis in the human nasopharyngeal cancer CNE cells. Biochem Biophys Res Commun. 2011;2011(411):342–7.
Van Meerloo J, Kaspers GJ, Cloos J. Cell sensitivity assays: the MTT assay. Method Mol Biol. 2011;731:237–45.
Weinert TA, Hartwell LH. Cell cycle arrest of cdc mutants and specificity of the RAD9 checkpoint. Genetics. 1993;134:63–80.
Zou H, Li Y, Liu X, Wang X. An APAF-1.cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J Biol Chem. 1999;274:11549–56.
Scaffidi C, Fulda S, Srinivasan A, Friesen C, Li F, Tomaselli KJ, Debatin KM, Krammer PH, Peter ME. Two CD95 (APO-1/Fas) signaling pathways. EMBO J. 1998;17:1675–87.
Wilson MR. Apoptotic signal transduction: emerging pathways. Biochem Cell Biol. 1998;76:573–82.
Korsmeyer SJ, Wei MC, Saito M, Weiler S, Oh J, Schlesinger PH. Pro-apoptotic cascade activates BID, which oligomerized BAK or BAX into pores that result in the release of cytochrome C. Cell Death Differ. 2000;7:1166–73.
Wolter KG, Hsu YT, Smith CL, Nechushtan A, Xi XG, Youle RJ. Movement of Bax from the cytosol to mitochondria during apoptosis. J Cell Biol. 1997;139:1281–92.
Bruce-Keller AJ, Begley JG, Fu W, Butterfield DA, Bredesen DE, Hutchins JB, Hensley K, Mattson MP. Bcl-2 protects isolated plasma and mitochondrial membranes against lipid peroxidation induced by hydrogen peroxide and amyloid beta-peptide. J Neurochem. 1998;70:31–9.
Hay N. The Akt-mTOR tango and its relevance to cancer. Cancer Cell. 2005;8:179–83.
Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–19.
Ocana A, Vera-Badillo F, Al-Mubarak M, Templeton AJ, Corrales-Sanchez V, Diez-Gonzalez L, Cuenca-Lopez MD, Seruga B, Pandiella A, Amir E. Activation of the PI3K/mTOR/AKT pathway and survival in solid tumors: systematic review and meta-analysis. PLoS One. 2014;9:e95219.
Choe G, Horvath S, Cloughesy TF, Crosby K, Seligson D, Palotie A, Inge L, Smith BL, Sawyers CL, Mischel PS. Analysis of the phosphatidylinositol 3′-kinase signaling pathway in glioblastoma patients in vivo. Cancer Res. 2003;63:2742–6.
Xie M, Yi X, Wang R, Wang L, He G, Zhu M, Qi C, Liu Y, Ye Y, Tan S, Tang A. 14-thienyl methylene matrine (YYJ18), the derivative from matrine, induces apoptosis of human nasopharyngeal carcinoma cells by targeting MAPK and PI3K/Akt pathways in vitro. Cell Physiol Biochem. 2014;33:1475–83.
Authors’ contributions
SBC and ZLY conceived and designed the study. YQG, HYS, BBL and JHW performed the experiments. YQG, HYS and SWC performed the statistical analysis. YQG and HYS wrote the manuscript. COC, DKWM and AKWT revised the manuscript. All the authors read and approved the final version of the manuscript.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 81303203; 81373953) and Shenzhen Basic Research Program (No. JCYJ20120618173411244), China. It was part of the work of the Hong Kong Chinese Material Medica Standard Project.
Compliance with ethical guidelines
Competing interests The authors declare that they have no conflict of interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Guo, Yq., Sun, Hy., Chan, Co. et al. Centipeda minima (Ebushicao) extract inhibits PI3K-Akt-mTOR signaling in nasopharyngeal carcinoma CNE-1 cells. Chin Med 10, 26 (2015). https://doi.org/10.1186/s13020-015-0058-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13020-015-0058-5