
Abstract
Background
Steroid-induced osteonecrosis of the femoral head (ONFH) is a common hip joint disease and is difficult to be diagnosed early. At present, the pathogenesis of steroid-induced ONFH remains unclear, and recognized and effective diagnostic biomarkers are deficient. The present study aimed to identify potentially important genes and signaling pathways involved in steroid-induced ONFH and investigate their molecular mechanisms.
Methods
Microarray data sets GSE123568 (peripheral blood) and GSE74089 (cartilage) were obtained from the Gene Expression Omnibus database, including 34 ONFH samples and 14 control samples. Morpheus software and Venn diagram were used to identify DEGs and co-expressed DEGs, respectively. Besides, we conducted Kyoto Encyclopedia of Genome (KEGG) and gene ontology (GO) pathway enrichment analysis. We construct a protein-protein interaction (PPI) network through GEO2R and used cytoHubba to divide the PPI network into multiple sub-networks. Additionally, quantitative real-time polymerase chain reaction (qRT-PCR) was performed to verify the bioinformatics analysis results.
Results
A total of 118 intersecting DEGs were obtained between the peripheral blood and cartilage samples, including 40 upregulated genes and 78 downregulated genes. Then, GO and KEGG pathway enrichment analysis revealed that upregulated DEGs focused on the signaling pathways related to staphylococcus aureus infection, leishmaniasis, antigen processing, and presentation, as well as asthma and graft-versus-host disease. Downregulated genes were concentrated in the FoxO signaling pathway, AMPK signaling pathway, signaling pathway regulating stem cell pluripotency, and mTOR signaling pathway. Some hub genes with high interactions such as CXCR1, FPR1, MAPK1, FOXO3, FPR2, CXCR2, and TYROBP were identified in the PPI network. The results of qRT-PCR demonstrated that CXCR1, FPR1, and TYROBP were upregulated while MAPK1 was downregulated in peripheral blood of steroid-induced ONFH patients. This was consistent with the bioinformatics analysis.
Conclusions
The present study would provide novel insight into the genes and associated pathways involved in steroid-induced ONFH. CXCR1, FPR1, TYROBP, and MAPK1 may be used as potential drug targets and biomarkers for the diagnosis and prognosis of steroid-induced ONFH.
Similar content being viewed by others

Introduction
Osteonecrosis of the femoral head (ONFH) is a common disease of the hip with a higher incidence in elderly patients, and the most common clinical symptom is severe pain [1]. There are about 150,000–200,000 new cases in China each year [2], and about 20,000 new cases in the USA each year [3]. The pathological features of steroid-induced ONFH include decreased blood supply to the hip joint, the collapse of the femoral head, and accumulation of microfractures without continuous remodeling [4, 5]. Besides, ONFH can cause rapid destruction and dysfunction of the hip joint, and approximately 65–70% of patients with advanced ONFH require total hip replacement [6]. The pathogenesis and molecular mechanism of steroid-induced ONFH remain unclear, and there are few effective prevention and early treatment methods.
Studies have indicated that successful results can be obtained when joint-preserving surgery is used in the pre-collapse stage of steroid-induced ONFH [7]. After the femoral head necrosis collapses, cartilage wrinkles and the hip joint space gradually narrows. Meanwhile, the failure rate of joint-preserving treatment is high. Therefore, searching for new biomarkers in the blood is of great clinical significance for the diagnosis of steroid-induced ONFH, as well as the further early diagnosis and treatment of ONFH. Some genetic factors are considered key factors in the progress of steroid-induced ONFH, such as peroxisome proliferator-activated receptor γ, matrix metallopeptidase 9, and SMAD family member 3 and gremlin 1 [8,9,10]. Currently, microarray analysis using a high-throughput platform is promising and efficient for exploring the molecular mechanisms of diseases and identifying useful biomarkers for diagnosis and prognosis of diseases [11, 12]. However, there is little comprehensive research on steroid-induced ONFH with high-throughput platforms. During the development of steroid-induced ONFH, the cartilage on the surface of the femoral head and the labrum of the acetabulum are significantly damaged. Degeneration of cartilage on the surface of the femoral head and tearing of the labrum enhance the instability of the hip joint and accelerate the development of steroid-induced ONFH [13]. Prevention and early treatment of hip cartilage damage may slow the development of steroid-induced ONFH and relieve hip dysfunction. However, there are few studies on the molecular mechanism of steroid-induced ONFH hip cartilage damage. Therefore, it is hypothesized in this paper that differentially expressed genes co-exist in the peripheral serum and cartilage of ONFH patients; this can be used for early diagnosis and observation of disease progression.
In this study, hub genes are screened out from the differences expressed genes (degrees) using bioinformatics methods based on the National Center for Biotechnology Information (NCBI http://www.ncbi.nlm.nih.gov/geography/) from the Gene Expression Dataset GSE123568 (peripheral serum) and GSE74089 (hip articular cartilage) Comprehensive (GEO) database. Quantitative real-time PCR (qRT-PCR) was conducted to validate the gene expression profiling results based on an independent sera sample of eight patients with ONFH and twelve healthy controls. The present study aimed to identify potentially important genes and signaling pathways involved in ONFH and investigate their molecular mechanisms.
Materials and methods
Microarray data source
The gene chip data related to femoral head necrosis (GSE123568, GSE74089) is downloaded in the GEO (gene expression omnibus) database (https://www.ncbi.nlm.nih.gov/geo/). GSE123568 chip data contains 10 normal patient serum samples and 30 steroid-induced femoral head necrosis serum samples. Besides, the GSE74089 chip data is composed of hip cartilage specimens from 4 NFH patients and 4 healthy controls. All the above data sets are based on GPL15207 and GPL13497 platform GEO database downloaded, and the information of the above two data sets are detailed in Table 1.
Differentially expressed genes analysis
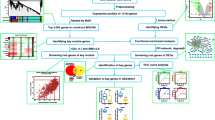
GEO 2R (http://www.ncbi.nlm.nih.gov/geo/geo2r/) is an online analysis program based on the R language provided by the GEO database (R 3.2.3 version) [14]. The Bioconductor R package in GEO2R is used to perform background correction, normalization, and expression value calculation on the GSE123568 and GSE74089 data; the limma 3.26.8 package is adopted to calculate the differential genes between the two groups and derive the GEO2R processed data. Set P< 0.01, adjust P value < 0.01, and the criteria for screening differential genes is that the expression change range is greater than or equal to twice (|log2 FC|≥1.0). Besides, log2FC≥1.0 indicates upregulation of gene expression; log2FC ≤− 1.0 suggests downregulation of gene expression. Finally, the differentially expressed genes of the femoral head necrosis group and the healthy control group, namely the differentially expressed genes (DEGs) of femoral head necrosis, were obtained. Using the online analysis website ClustVis (https://biit.cs.ut.ee/clustvis/) [15], the heat map and cluster analysis of the selected differential genes are completed, and the -log10 conversion is performed for the adjust P value in the data processed by GEO2R. Then, according to log2 FC, -log10 (adjust P value) was divided into groups of upregulated genome, downregulated genome, and non-statistically different genome. Next, the processed data is imported into GraphPad Prism 7 to draw a heat map. Finally, the Venn diagrams of the common upregulated and downregulated DEGs in the two data sets is illustrated using the online Venn diagram production website bioinfogp (https://bioinfogp.cnb.csic.es/tools/venny/index.html) [16].
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis
The online database DAVID (DAVID Bioinformatics Resources 6.8) was used to analyze the function and pathway enrichment of the upregulated and downregulated common differentially expressed genes obtained from the intersection of the two chip data sets to further explore the potential pathogenesis of the disease. The upregulated and downregulated differentially expressed genes are imported according to the set differential gene screening criteria from the data after GEO2R analysis and processing into the online enrichment analysis website DAVID (https://david.ncifcrf.gov/tools.jsp) for Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) signal pathway analysis [17, 18]. The GO analysis mainly includes the cellular components (CC), molecular functions (MF), and biological processes (BP) of differential genes.
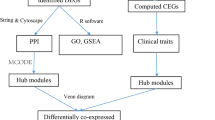
Protein-protein interaction (PPI) network analysis
The online analysis website STRING was used to construct a protein-protein interaction network (protein-protein interaction, PPI). The screened upregulated and downregulated common differentially expressed genes were uploaded to the STRING online analysis website (https://string-db.org/); “highest confidence > 0.700” was set in the lowest interaction score, and the protein-protein interaction result data were exported. Furthermore, the protein-protein interaction network (PPI network) of differentially expressed genes was obtained using Cytoscape 3.7.2 software [19], and the cytoHubba plug-in was adopted to screen Hub genes [20]. Meanwhile, three different algorithms (MNC, Degree, and MCC) were used to calculate the top 10 upregulated differentially expressed genes and the top 10 downregulated differentially expressed genes in their respective scores. Besides, the key genes are obtained by the intersection of the gene sets obtained by three different algorithms with the online analysis platform Draw Venn Diagram (http://bioinformatics. psb.ugent.be/webtools/Venn/).
Peripheral serum collection
From August 2020 to December 2020, peripheral serum samples from 12 steroid-induced ONFH patients (5 males and 7 females; mean age 33.5 ± 9.16) and 8 healthy adults (3 males and 5 females; mean age:32.5 ± 5.9) in the First Affiliated Hospital of Guangzhou University of Chinese Medicine were collected to determine mRNA levels. Two experienced orthopedic surgeons diagnosed the patient with ONFH. To avoid bias, the process of patient selection and matching has been determined prior to the start of the study. The study protocol was reviewed and approved by the Committees of Clinical Ethics of the First Affiliated Hospital of Guangzhou University of Chinese Medicine. Each participant signed the informed consent documents.
Quantitative real-time PCR analysis
Total RNA from peripheral serum samples was isolated with Trizol LS. The protocol for cDNA synthesis was reverse transcription at 37 °C for 15 min and at 85 °C for 5 s. The qPCR protocol was 95 °C for 30 s, 40 cycles at 95 °C for 5 s, and 60 °C for 30 s. The relative expression level was determined as targeting genes divided by β-Actin (ACTB). Relative miRNA expression was generated with the 2−△△Cq method. Primers used in this study are presented in Table 2. All experiments were performed independently in triplicate.
Statistical analysis
GraphPad 8.0 was used to perform statistical analysis. The results are presented as mean ± standard deviation (mean ± SD). Data conforming to normal distribution were compared using Student t test, while those with non-normally distributed were tested using Mann-Whitney U test. P < 0.05 indicates statistical significance. All experiments were performed in triplicate.
Results
Identification of DEGs
This study screened out two chip data sets (GSE123568 and GSE74089). Among them, 30 serum samples of patients with steroid hormone-induced osteonecrosis of the femoral head in the chip data set GSE123568 (including 13 males and 17 females, aged 23.07 ± 3.01 years, ARCO stage I-II patients accounted for 10 patients, ARCO III patients accounted for 10 cases, ARCO IV patients accounted for 10 cases) and 10 normal serum samples (including 7 males and 3 females, aged 25.02 ± 2.87 years). Besides, there are 4 hip cartilage samples from patients with femoral head necrosis (including 3 males and 1 female) and 4 healthy adult hip cartilage specimens (including 3 males and 1 female) in the chip dataset GSE74089. The baseline characteristics of the patients are provided in Table 3. According to the adjust P value < 0.01, the expression change range is greater than or equal to twice (|log2 FC|≥1.0) as the criteria for screening differential genes. A total of 750 differentially expressed genes were screened in the GSE123568 data set, including 454 upregulated genes and 296 downregulated genes. A total of 5211 differentially expressed genes were screened in the GSE74089 data set, including 1594 upregulated genes and 3617 downregulated genes. The DEGs in GSE123568 and GSE74089 were selected based on the adjust P value to screen the top 25 most significant upregulated and downregulated differentially expressed genes, so as to draw heat maps (Fig. 1). The -log10 conversion is performed for the adjust P value in the data processed by GEO2R. Then, according to log2 FC, -log10 (adjust P value) was divided into groups of upregulated genome, downregulated genome, and non-statistically different genome. The processed data was imported into GraphPad Prism 7 to draw a volcano map (Fig. 2). Furthermore, a total of 118 intersecting DEGs were obtained between the peripheral blood and cartilage samples, including 40 upregulated genes and 78 downregulated genes (Fig. 3).

Heat map of top 50 DEGs in ONFH. a Heat map of top 50 DEGs in GSE123568. b Heat map of top 50 DEGs in GSE74089 (tissue samples are presented as columns; individual genes are presented as rows. Red represents upregulated genes; green indicates downregulated genes in patients with ONFH. The patients in the top rows in pink indicate the control columns; the patients in the top rows in blue indicate columns were patients with NFH. DEGs, differentially expressed genes; NFH, necrosis of the femoral head)

The volcano plot was constructed using fold-change (Log2FC) values and adjust P values. a The volcano plot of a total of 750 DEGs in GSE123568. b The volcano plot of total 5211 DEGs in GSE74089 (the vertical blue lines correspond to upregulated and downregulated Log2FC; the horizontal Orange line represents an adjust P value of 0.01. The red point represents the differentially upregulated genes with statistical significance; the green point suggests the differentially downregulated genes with statistical significance)

Venn diagrams showing the overlap of DEGs between peripheral serum samples and hip articular cartilage samples. a Upregulated DEGs. b Downregulated DEGs (Venn diagram was drawn using the online tool (https://bioinfogp.cnb.csic.es/tools/venny/index.html). Numbers represented the number of DEGs. The percentage was represented by the ratio of current DEGs to the total)
Enrichment analysis of KEGG pathway and GO terms
In this study, the DAVID database was used to perform GO and KEGG pathway enrichment analysis on the upregulated and downregulated genes. GO analysis of upregulated DEGs demonstrates that the biological process (BP) is concentrated in inflammation, immune response, and signal transduction. Cellular component (CC) focuses on plasma membrane, endoplasmic reticulum membrane, and lysosome. Molecular functions (MF) are manifested in immune regulation process, IL-8 receptor activity, superoxide-producing NADPH oxidase activity, and MAP kinase activity (Table 4, Fig. 4). Besides, GO analysis of downregulated genes revealed that BP is rich in cell cycle regulation, apoptosis process, translation process regulation, autophagy, and cell response to hypoxia. CC is mainly condensed in cytoplasm, nucleus, mitochondria, autophagosomes, and exosomes. MF is manifested in protein binding, transcription factor binding, and DNA binding. The GO analysis of the top 10 downregulated genes with the smallest adjust P value is illustrated in Table 5 and Fig. 5. Enrichment analysis of the upregulated gene KEGG pathway revealed that it focuses on the signaling pathways related to staphylococcus aureus infection, leishmaniasis, antigen processing and presentation, asthma, and graft-versus-host disease. Downregulated genes are concentrated in the FoxO signaling pathway, AMPK signaling pathway, signaling pathway regulating stem cell pluripotency, and mTOR signaling pathway (Table 6, Fig. 6).

The significant terms identified by GO enrichment analysis for the upregulated DEGs in necrosis of the femoral head samples (p<0.05)

The significant terms identified by GO enrichment analysis for the downregulated DEGs in necrosis of the femoral head samples (p<0.05)

Results of KEGG pathway enrichment analysis of differentially expressed genes
Construction of PPI network and identification of hub genes
Three different algorithms (MNC, MCC, and Degree) in the Cytoscape software’s cytoHubba plug-in were used to calculate top 10 upregulated key genes and top 10 downregulated key genes (Table 7 and Table 8, Fig. 7 and Fig. 8) and obtain the intersection. Hub genes for bone necrosis are as follows: (1) upregulated genes include HLA-DRB5, HLA-DRB1, CXCR1, LPAR2, FPR1, FPR2, CXCR2, TYROBP, and MNDA and (2) downregulated genes include MKRN1, FBXL4, GABARAPL2, MAP1LC3B, SIAH2, UBE2H, AKT2, MAPK1, and FOXO3.

Module identified from the PPI network of the top 10 hub proteins of upregulated DEGs based on MNC centrality, MCC centrality, and degree centrality. a The PPI network of the top 10 hub proteins of upregulated DEGs based on MNC centrality; b the PPI network of the top 10 hub proteins of upregulated DEGs based on MCC centrality; c the PPI network of the top 10 hub proteins of upregulated DEGs based on degree centrality; d Venn diagrams of the overlap of upregulated DEGs among MNC centrality, MCC centrality, and degree centrality

Module identified from the PPI network of the top 10 hub proteins of downregulated DEGs based on MNC centrality, MCC centrality, and degree centrality. a The PPI network of the top 10 hub proteins of downregulated DEGs based on MNC centrality; b the PPI network of the top 10 hub proteins of downregulated DEGs based on MCC centrality; c the PPI network of the top 10 hub proteins of downregulated DEGs based on degree centrality; d Venn diagrams of the overlap of downregulated DEGs among MNC centrality, MCC centrality, and degree centrality
Validation of the hub genes in ONFH patients
Among the identified DEGs, CXCR1, FPR1, MAPK1, FOXO3, FPR2, CXCR2, and TYROBP were selected to verify the integrated result. Peripheral serum samples from 12 steroid-induced ONFH patients and 8 healthy adults were collected to determine mRNA levels. The result revealed that CXCR1, FPR1, and TYROBP were upregulated, and MAPK1 was downregulated in peripheral blood of steroid-induced ONFH patients (P < 0.05). There was no significant difference in the expression of FOXO3, FPR2, and CXCR2 in peripheral serum samples between the two groups (P > 0.05) (Fig. 9). The expression of CXCR1, FPR1, MAPK1, and TYROBP were consistent with bioinformatics analysis except for FOXO3, FPR2, and CXCR2. Moreover, CXCR1, FPR1, and TYROBP were upregulated, and MAPK1 was downregulated in the peripheral blood of steroid-induced ONFH patients.

Expression of hub genes in ONFH patients. a–c CXCR1, FPR1, and TYROBP were upregulated in peripheral blood of steroid-induced ONFH patients(P < 0.05). d MAPK1 was downregulated in peripheral blood of steroid-induced ONFH patients (P < 0.05); e–g there was no significant difference in the expression of CXCR2, FPR2, and FOXO3 in peripheral serum samples between the two groups (P > 0.05)
Discussion
Steroid-induced ONFH is a devastating and difficult joint disease and primarily caused by the interruption of blood supply and the dysfunction of the coagulation system, resulting in the death of bone cells, the decrease of the elastic modulus of the femoral head, and the collapse of the femoral head [21]. Various molecular and genetic studies have explored the etiology and pathogenesis [22, 23]. However, the exact pathogenesis remains unclear. In this paper, the genome-wide method was used to study the differential expression of genes in the peripheral blood serum and hip joint cartilage specimens of steroid-induced ONFH patients and healthy controls. Through bioinformatics analysis of GSE123568 and GSE74089, potentially important genes in ONFH were identified. In addition, the role of the identified genes in ONFH and their interaction were analyzed. A total of 118 intersecting DEGs were obtained in peripheral blood and cartilage samples, of which 40 were upregulated genes and 78 were downregulated genes. These DEGs may be important biomarkers related to the pathogenesis and progression of ONFH. Finally, qRT-PCR verified the expression patterns of several genes such as CXCR1, FPR1, MAPK1, FOXO3, FPR2, CXCR2, and TYROBP in vitro. However, due to different inclusion criteria and a small number of patients in the validation set, some results are inconsistent with microarray analysis.
The DEGs screened in this study contribute to the identification of new molecules or new pathways related to steroid-induced ONFH. These new molecules or pathways may be essential targets for disease diagnosis and treatment. The upregulated DEGs were generally involved in inflammation, immune response, and signal transduction. Molecular functions were manifested in immune regulation process, IL-8 receptor activity, superoxide-producing NADPH oxidase activity, and MAP kinase activity. The upregulated DEGs were enriched in pathways engaged in staphylococcus aureus infection, leishmaniasis, antigen processing and presentation, asthma, and graft-versus-host disease. Particularly, the background of this femoral head necrosis can be understood using corticosteroids to treat related acute or chronic graft-versus-host disease (aGVHD, cGVHD) [24]. Socie et al. [25] retrospectively analyzed the risk factors of osteonecrosis after allogeneic hematopoietic stem cell transplantation. The study revealed that the odds ratios of aGVHD and cGVHD were 3.73 and 3.52, respectively. The severity of graft-versus-host disease was associated with a higher frequency of osteonecrosis. Additionally, GO analysis of downregulated genes demonstrated that BP focused on cell cycle regulation, apoptosis process, translation process regulation, autophagy, and cell response to hypoxia. MF was manifested in protein binding, transcription factor binding, and DNA binding. The downregulated genes were concentrated in the FoxO signaling pathway, AMPK signaling pathway, signaling pathway regulating stem cell pluripotency, and mTOR signaling pathway. Moreover, AMPK was an essential energy state sensor responding to energy supply and demand through coordinated metabolic pathways [26]. Studies have indicated that AMPK can promote cell survival under various stress conditions [27]. The forced activation of AMPK is a good strategy to protect osteoblasts from Dex [28, 29]. The mTOR pathway may be a vital mediator of bone homeostasis [30]. Furthermore, the impaired differentiation of the osteogenic/adipogenic lineage of bone marrow mesenchymal stem cells (BMSCs) was caused by the activation of the mTOR signaling pathway [31]. Meanwhile, blocking the mTOR pathway can prevent the development of phenotypes related to aging and enhance the proliferation ability of BMSCs [32]. Therefore, regulating the mTOR signaling pathway may be one of the crucial methods to treat steroid-induced ONFH.
The construction of the PPI network of DEGs was used to study the relationship between GO enrichment and important proteins identified by pathway analysis. The upregulated DEGs included TYROBP, CXCR1, FPR1, LPAR2, FPR2, HLA-DRB5, HLA-DRB1, CXCR2, and MNDA. TYROBP is a protein, which is involved in osteoclast differentiation and function, such as the generation of actin cytoskeleton, which is very important for bone resorption [33]. In this study, both biosynthesis analysis and specimen verification found that the expression of TYROBP was upregulated, indicating that TYROBP may play an important role in regulating osteoclast differentiation in steroid-induced ONFH. Specifically, CXCR1 (IL 8 receptor), a chemokine, is recognized to be abundant in the synovial fluid of bone marrow mesenchymal stem cells and has been verified to be a potent inducer of bone marrow mesenchymal stem cell migration [33]. Chemokines can induce the migration response of bone marrow mesenchymal stem cells to cartilage defect areas [34]. Hence, CXCR1 may play an important role in the repair of steroid-induced ONFH cartilage. Besides, FPR is a chemotactic receptor, and its expression is often identified in phagocytes (monocytes and neutrophils, etc.). FPR may play a role in promoting stem cell reproliferation because MSCs functionally express FPR and are positive for n-formyl peptides [35]. Matsushita et al. [36] found an important role of fMLP or FRP1 in MSC osteogenic and adipogenic differentiation. Moreover, the important role of fMLP is to promote the osteoblast differentiation of MSCs, and the knockdown of FPR1 inhibits this osteogenic differentiation. Downregulated DEGs identified in the present study included MAPK1, AKT2, MKRN1, FBXL4, GABARAPL2, MAP1LC3B, SIAH2, UBE2H, and FOXO3. MAPK was a type of serine/threonine protein kinase in cells that can regulate cell growth and differentiation. Experiments have demonstrated that activating the MAPK signaling pathway can promote the mineralization of osteoblasts and the expression of ALP and BMP-2, contributing to bone formation. ERK, a downstream factor of MAPK, can inhibit osteoclast formation when it is inactivated [35]. We discovered that MAPK1 was downregulated in steroid-induced ONFH blood samples compared with normal controls, providing another pathogenic role in bone disease. Moreover, AKT2 is a pro-survival protein in the AKT family and is activated through the PI3K pathway. Zhang et al. [37] revealed that AKT2 plays a crucial role in the development of zebrafish bone. Furthermore, inhibiting the PI3k/Akt signaling pathway may be one of the vital mechanisms of glucocorticoid-induced osteoblast apoptosis [38]. In the present study, the decreased expression of AKT2 was observed in steroid-induced ONFH, suggesting a vital role in bone formation and metabolism.
There are limitations to our study. First, the sample size in the qRT-PCR data set was small, and larger numbers of blood samples of ONFH patients are needed for further research. Second, the genes investigated and their pathways were not confirmed through in vitro studies or other functional studies. Therefore, the potential role of DEGs and PPIs identified in ONFH patients requires to be further investigated in vivo and in vitro. For example, the effects of downregulated or upregulated DEGs in cellular or in vivo models could be determined.
In conclusion, the present study would provide novel insight into the genes and associated pathways involved in steroid-induced ONFH. CXCR1, FPR1, TYROBP, and MAPK1 could serve as potential drug targets and biomarkers for the diagnosis and prognosis of steroid-induced ONFH.
Availability of data and materials
All the data will be available upon motivated request to the corresponding author of the present paper.
Abbreviations
- ONFH:
-
Osteonecrosis of the femoral head
- KEGG:
-
Kyoto Encyclopedia of Genome
- GO:
-
Gene Ontology
- PPI:
-
Protein-protein interaction
- NCBI:
-
National Center for Biotechnology Information
References
Zhang QY, Li ZR, Gao FQ, Sun W. Pericollapse stage of osteonecrosis of the femoral head: a last chance for joint preservation. Chin Med J (Engl). 2018;131(21):2589–98. https://doi.org/10.4103/0366-6999.244111.
Song Y, Du Z, Ren M, et al. Association of gene variants of transcription factors PPARγ, RUNX2, Osterix genes and COL2A1, IGFBP3 genes with the development of osteonecrosis of the femoral head in Chinese population. Bone. 2017;101:104–12. https://doi.org/10.1016/j.bone.2017.05.002.
Seamon J, Keller T, Saleh J, Cui Q. The pathogenesis of nontraumatic osteonecrosis. Arthritis. 2012;2012:601763.
Hauzeur JP, Malaise M, de Maertelaer V. A prospective cohort study of the clinical presentation of non-traumatic osteonecrosis of the femoral head: spine and knee symptoms as clinical presentation of hip osteonecrosis. Int Orthop. 2016;40(7):1347–51. https://doi.org/10.1007/s00264-015-3079-x.
Moya-Angeler J, Gianakos AL, Villa JC, Ni A, Lane JM. Current concepts on osteonecrosis of the femoral head. World J Orthop. 2015;6(8):590–601. https://doi.org/10.5312/wjo.v6.i8.590.
Johnson AJ, Mont MA, Tsao AK, Jones LC. Treatment of femoral head osteonecrosis in the United States: 16-year analysis of the Nationwide Inpatient Sample. Clin Orthop Relat Res. 2014;472(2):617–23. https://doi.org/10.1007/s11999-013-3220-3.
Mont MA, Cherian JJ, Sierra RJ, Jones LC, Lieberman JR. Nontraumatic osteonecrosis of the femoral head: where do we stand today? A ten-year update. J Bone Joint Surg Am. 2015;97(19):1604–27. https://doi.org/10.2106/JBJS.O.00071.
Du J, Liu W, Jin T, et al. A single-nucleotide polymorphism in MMP9 is associated with decreased risk of steroid-induced osteonecrosis of the femoral head. Oncotarget. 2016;7(42):68434–41. https://doi.org/10.18632/oncotarget.12034.
Gu C, Xu Y, Zhang S, Guan H, Song S, Wang X, et al. miR-27a attenuates adipogenesis and promotes osteogenesis in steroid-induced rat BMSCs by targeting PPARγ and GREM1. Sci Rep. 2016;6(1):38491. https://doi.org/10.1038/srep38491.
Hao C, Yang S, Xu W, Shen JK, Ye S, Liu X, et al. MiR-708 promotes steroid-induced osteonecrosis of femoral head, suppresses osteogenic differentiation by targeting SMAD3. Sci Rep. 2016;6(1):22599. https://doi.org/10.1038/srep22599.
Franco D, Bonet F, Hernandez-Torres F, Lozano-Velasco E, Esteban FJ, Aranega AE. Analysis of microRNA microarrays in cardiogenesis. Methods Mol Biol. 2016;1375:207–21. https://doi.org/10.1007/7651_2015_247.
Kang S, Song J. Robust gene selection methods using weighting schemes for microarray data analysis. BMC Bioinformatics. 2017;18(1):389. https://doi.org/10.1186/s12859-017-1810-x.
Magnussen RA, Guilak F, Vail TP. Articular cartilage degeneration in post-collapse osteonecrosis of the femoral head. Radiographic staging, macroscopic grading, and histologic changes. J Bone Joint Surg Am. 2005;87(6):1272–7. https://doi.org/10.2106/JBJS.D.01936.
Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, et al. NCBI GEO: archive for functional genomics data sets--update. Nucleic Acids Res. 2013;41(Database issue):D991–5. https://doi.org/10.1093/nar/gks1193.
Metsalu T, Vilo J. ClustVis: a web tool for visualizing clustering of multivariate data using principal component analysis and heatmap. Nucleic Acids Res. 2015;43(W1):W566–70. https://doi.org/10.1093/nar/gkv468.
Oliveros JC. Venny. An interactive tool for comparing lists with Venn's diagrams. 2015. https://bioinfogp.cnb.csic.es/tools/venny/index.html.
Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37(1):1–13. https://doi.org/10.1093/nar/gkn923.
Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. https://doi.org/10.1038/nprot.2008.211.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504. https://doi.org/10.1101/gr.1239303.
Chin CH, Chen SH, Wu HH, et al. cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst Biol. 2014;8(Suppl 4):S11.
Cohen-Rosenblum A, Cui Q. Osteonecrosis of the femoral head. Orthop Clin North Am. 2019;50(2):139–49. https://doi.org/10.1016/j.ocl.2018.10.001.
Wei B, Wei W. Identification of aberrantly expressed of serum microRNAs in patients with hormone-induced non-traumatic osteonecrosis of the femoral head. Biomed Pharmacother. 2015;75:191–5. https://doi.org/10.1016/j.biopha.2015.07.016.
Song Y, Du ZW, Yang QW, et al. Erratum: Association of genes variants in RANKL/RANK/OPG signaling pathway with the development of osteonecrosis of the femoral head in Chinese population: Erratum. Int J Med Sci. 2018;15(12):1355. https://doi.org/10.7150/ijms.29056.
Jones KB, Seshadri T, Krantz R, Keating A, Ferguson PC. Cell-based therapies for osteonecrosis of the femoral head. Biol Blood Marrow Transplant. 2008;14(10):1081–7. https://doi.org/10.1016/j.bbmt.2008.06.017.
Socié G, Cahn JY, Carmelo J, Vernant JP, Jouet JP, Ifrah N, et al. Avascular necrosis of bone after allogeneic bone marrow transplantation: analysis of risk factors for 4388 patients by the Société Française de Greffe de Moëlle (SFGM). Br J Haematol. 1997;97(4):865–70. https://doi.org/10.1046/j.1365-2141.1997.1262940.x.
Carling D, Thornton C, Woods A, Sanders MJ. AMP-activated protein kinase: new regulation, new roles? Biochem J. 2012;445(1):11–27. https://doi.org/10.1042/BJ20120546.
Wang S, Song P, Zou MH. AMP-activated protein kinase, stress responses and cardiovascular diseases. Clin Sci (Lond). 2012;122(12):555–73. https://doi.org/10.1042/CS20110625.
Guo S, Chen C, Ji F, Mao L, Xie Y. PP2A catalytic subunit silence by microRNA-429 activates AMPK and protects osteoblastic cells from dexamethasone. Biochem Biophys Res Commun. 2017;487(3):660–5. https://doi.org/10.1016/j.bbrc.2017.04.111.
Liu W, Mao L, Ji F, Chen F, Hao Y, Liu G. Targeted activation of AMPK by GSK621 ameliorates H2O2-induced damages in osteoblasts. Oncotarget. 2017;8(6):10543–52. https://doi.org/10.18632/oncotarget.14454.
Chen C, Akiyama K, Wang D, Xu X, Li B, Moshaverinia A, et al. mTOR inhibition rescues osteopenia in mice with systemic sclerosis. J Exp Med. 2015;212(1):73–91. https://doi.org/10.1084/jem.20140643.
Liu Y, Kou X, Chen C, Yu W, Su Y, Kim Y, et al. Chronic high dose alcohol induces osteopenia via activation of mTOR signaling in bone marrow mesenchymal stem cells. Stem Cells. 2016;34(8):2157–68. https://doi.org/10.1002/stem.2392.
Gharibi B, Farzadi S, Ghuman M, Hughes FJ. Inhibition of Akt/mTOR attenuates age-related changes in mesenchymal stem cells. Stem Cells. 2014;32(8):2256–66. https://doi.org/10.1002/stem.1709.
Park MS, Kim YH, Jung Y, Kim SH, Park JC, Yoon DS, et al. In situ recruitment of human bone marrow-derived mesenchymal stem cells using chemokines for articular cartilage regeneration. Cell Transplant. 2015;24(6):1067–83. https://doi.org/10.3727/096368914X681018.
Koelling S, Kruegel J, Irmer M, Path JR, Sadowski B, Miro X, et al. Migratory chondrogenic progenitor cells from repair tissue during the later stages of human osteoarthritis. Cell Stem Cell. 2009;4(4):324–35. https://doi.org/10.1016/j.stem.2009.01.015.
Matsushita T, Chan YY, Kawanami A, Balmes G, Landreth GE, Murakami S. Extracellular signal-regulated kinase 1 (ERK1) and ERK2 play essential roles in osteoblast differentiation and in supporting osteoclastogenesis. Mol Cell Biol. 2009;29(21):5843–57. https://doi.org/10.1128/MCB.01549-08.
Shin MK, Jang YH, Yoo HJ, Kang DW, Park MH, Kim MK, et al. N-formyl-methionyl-leucyl-phenylalanine (fMLP) promotes osteoblast differentiation via the N-formyl peptide receptor 1-mediated signaling pathway in human mesenchymal stem cells from bone marrow. J Biol Chem. 2011;286(19):17133–43. https://doi.org/10.1074/jbc.M110.197772.
Zhang D, Wang J, Zhou C, Xiao W. Zebrafish akt2 is essential for survival, growth, bone development, and glucose homeostasis. Mech Dev. 2017;143:42–52. https://doi.org/10.1016/j.mod.2017.01.004.
Piovan E, Yu J, Tosello V, Herranz D, Ambesi-Impiombato A, da Silva AC, et al. Direct reversal of glucocorticoid resistance by AKT inhibition in acute lymphoblastic leukemia. Cancer Cell. 2013;24(6):766–76. https://doi.org/10.1016/j.ccr.2013.10.022.
Acknowledgements
Grateful acknowledgement is made to my supervisor Professor He Wei who gave me considerable help by means of suggestion, comments, and criticism. In addition, I deeply appreciate the contribution to this thesis made in various ways by my friends and classmates.
Funding
Publication charges for this article were funded by the Natural Science Foundation of China (81873327, 81904226), Excellent Doctoral Dissertation Incubation Grant of First Clinical School of Guangzhou University of Chinese Medicine.
Author information
Authors and Affiliations
Contributions
L.T.Y was responsible for the design and implementation of the study presented. C.W.J, Y.P, W.Q.S, W.H.B, and L.D conducted the experiments and were responsible for the acquisition of the data. L.T.Y prepared the initial draft of the manuscript. H.W and Z.Q.W gave critical feedback during the study and critically revised the submitted manuscript for important intellectual content. All authors have read and approved the final manuscript to be submitted.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was conducted in agreement with the Declaration of Helsinki and its later amendments or comparable ethical standards and had been approved by the ethics board of The First Affiliated Hospital of Guangzhou University of Chinese Medicine (No: Y《2019》118).
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lin, T., Chen, W., Yang, P. et al. Bioinformatics analysis and identification of genes and molecular pathways in steroid-induced osteonecrosis of the femoral head. J Orthop Surg Res 16, 327 (2021). https://doi.org/10.1186/s13018-021-02464-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13018-021-02464-9