
Abstract
Background
Silymarin (SMN), a polyphenolic flavonoid, is involved in multiple bioactive functions including anti-inflammation. Pretreatment with SMN demonstrated chondroprotection against tumour necrosis factor-alpha (TNF-α) stimulation in a chondrocyte cell line. However, pre- and posttreatment with phytochemicals have varying effects on osteoarthritis (OA) chondrocytes, and the therapeutic potential of SMN after catabolic cytokine stimulation is not fully elucidated.
Methods
The cytotoxicity of SMN (12.5, 25, 50 and 100 μM) was evaluated in human primary chondrocytes. The chondrocytes were supplemented with SMN (25 and 50 μM) after interleukin-1beta (IL-1β) stimulation. The mRNA expression and protein production of catabolic/anabolic cytokines as well as extracellular matrix (ECM) components were evaluated.
Results
High-dose SMN (100 μM) impaired the mitochondrial activity in chondrocytes, and 50 μM SMN further caused cell death in IL-1β-stimulated cells. The addition of 25 μM SMN ameliorated cell senescence; downregulated the catabolic genes of inducible nitric oxide synthase, IL-1β, TNF-α, matrix metalloproteinase-3 (MMP-3), MMP-9 and MMP-13; upregulated the anabolic genes of tissue inhibitor of metalloproteinase-1 (TIMP-1) and collagen type II alpha 1; and restored the expression of chondrogenic phenotype genes SOX9 and sirtuin-1 (Sirt1). In addition, the production of IL-1β, MMP-3 and MMP-9 decreased with an increase in TIMP-1 secretion. However, the mRNA levels of IL-6, IL-8 and IL-10 and protein production remained high. The addition of nicotinamide, a Sirt1 inhibitor, downregulated SOX9 and attenuated the therapeutic effects of SMN on IL-1β-stimulated chondrocytes.
Conclusion
SMN regulates the chondrocyte phenotype through Sirt1 and SOX9 to improve ECM homeostasis and may serve as a complementary therapy for early-stage knee OA.
Similar content being viewed by others

Background
Disturbances in extracellular matrix (ECM) anabolism and catabolism are triggered by metabolic inflammation and oxidative stress and are known to initiate knee osteoarthritis (OA)-related pathological changes [1]. In particular, catabolic cytokines such as matrix metalloproteinases (MMPs) contribute to the degradation of the ECM components type II collagen and aggrecan [2]. Consequently, the imbalance in ECM deposition and degradation may result in cartilage destruction and eventually impair mobility in patients with OA [3]. Accordingly, interventions to modulate ECM homeostasis as well as to enhance the anti-inflammatory and antioxidant activities of articular chondrocytes are a promising therapeutic strategy against OA in the early stages [4].
Phytochemicals, involved in various bioactive functions, may be a promising choice for OA management [5]. Silymarin (SMN), a polyphenolic flavonoid, exerts anticarcinogenic, anti-inflammatory, cytoprotective and antioxidant effects, as evident in its radical scavenging activities, by regulating various cell membrane and nuclear transporters [6]. SMN reduced hepatic collagen accumulation through downregulation of procollagen α1 in rats with secondary biliary cirrhosis [7]. SMN also exerted cardioprotective effects on rats with myocardial infarction [8]. In a rat paw oedema model, SMN also exhibited anti-inflammatory activity [9]. In addition, SMN pretreatment before tumour necrosis factor alpha (TNF-α) exposure exerted chondroprotection through the modulation of interleukin 6 (IL-6), IL-8 and MMP-1 mRNA expression in human chondrocyte cells [10]. In a monoiodoacetate-induced rat OA model, SMN enhanced the anti-inflammatory activity of the nonsteroidal anti-inflammatory drug (NSAID) celecoxib [11]. Similarly, oral administration of SMN alone or in combination with an NSAID (piroxicam) reduced serum IL-1α and IL-8 levels in patients with knee OA [12]. Moreover, combination therapy with SMN reduced the adverse events of the NSAID celecoxib in an OA rat model [13].
Despite the chondroprotective potential of SMN revealed in previous studies, the benefits of pre- and posttreatment of phytochemicals for OA chondrocytes varied [14]. Furthermore, different cytokines cause glycophenotypic alterations and regulate distinctive apoptosis signalling in human articular chondrocytes [15, 16]. Therefore, this study aimed to evaluate the therapeutic effects of SMN post-treatment on human chondrocytes.
Materials and methods
Human articular chondrocyte isolation and cultivation
The experimental protocol and retrieval process of human tissues were reviewed and approved by the Research Ethics Committee of Hualien Tzu Chi Hospital, Buddhist Tzu Chi Medical Foundation (IRB106-48-A). Written informed consent was obtained from all patients. Articular cartilage samples were harvested from 12 patients with advanced knee OA (five women and five men; age range, 67–84 years; average age, 75.8 ± 6.2 years). An additional two women and one man (age range, 63–80 years; average age, 70.7 ± 8.6 years) were enrolled for a Sirt1 study. The collected cartilaginous tissues were treated with 0.1% protease (P8811, Sigma-Aldrich, Saint Louis, MO, USA) for 30 min and then digested using 0.2% type II collagenase (9001-12-1, Gibco, USA) overnight for chondrocyte isolation [17]. The released cells were maintained in Dulbecco’s modified Eagle’s medium: Nutrient Mixture F-12 (10565018, Gibco) supplemented with 10% fetal bovine serum (SH30396.03, Hyclone, USA) and 1% antibiotic (15140-122, Gibco) in an incubator at 37 °C in a humidified atmosphere with 5% CO2. Cells obtained from each donor were cultured at passages 2–6 and studied independently.
Assessment of silymarin toxicity
Human primary chondrocytes (5000 cells/well in a 96-well tissue culture plate) were maintained in regular medium overnight and subsequently cultured in media containing 0, 12.5, 25, 50 and 100 μM silymarin (SMN, S0292, Sigma-Aldrich). After 24 h of culture, cell death and mitochondrial activity of chondrocytes were evaluated using the lactate dehydrogenase (LDH) assay kit (786-210, CytoScan™, G-biosciences, USA) and the CCK-8 reagent (TEN-CCK8, TOOLS Cell Counting CCK-8 kit, Tools, Taiwan), respectively. The cell morphology of SMN-treated chondrocytes was also recorded.
Silymarin supplementation to IL-1β-stimulated chondrocytes
After treatment with 10 ng/mL IL-1β (579402, BioLegend, USA) for 24 h, the chondrocytes (40,000 cells/well in a 6-well tissue culture plate) were cultured in SMN-containing media (25 and 50 μM) for an additional 24 h. The LDH release and cell viability of the treated chondrocytes were determined again. In addition, the survival and senescence of the treated cells were evaluated using a live/dead double staining assay (R37601, LIVE/DEAD® Cell Imaging Kit, Thermo Fisher Scientific, USA) and β-galactosidase assay kit (β-gal, K320-250, Biovision, USA), respectively. Finally, glycosaminoglycan (GAG) production was detected in chondrocytes through toluidine blue staining under different treatments [18].
mRNA expression of silymarin-treated chondrocytes
The treated chondrocytes were lysed for total RNA extraction (R2052 Direct-zol™ RNA MiniPrep Kit, Zymo, USA), and the RNA was reverse transcribed into cDNA (RR037A, PrimeScript™ RT Reagent Kit, TaKaRa, Japan). The gene expression in the cells was analysed using real-time polymerase chain reaction (real-time PCR; LightCycler 96®, Roche, Germany) with SYBR Green reagents (BIO-98005, SensiFAST™ SYBR NO-ROX Kit, Bioline Meridian, UK) for inducible nitric oxide synthase (iNOS), IL-6, IL-8, IL-10, MMP-3, MMP-9, MMP-13, tissue inhibitor of metalloproteinase (TIMP)-1, aggrecan (AGCN) and sirtuin-1 (Sirt1, Table 1). Further, the Taqman system (ENZ-NUC 106-0200, AmpiGene™ qPCR Probe mix Hi-ROX, Enzo, USA) was used for the analysis of IL-1β (Hs01555410-m1), TNF-α (Hs01113624-g1), collagen type II (COL2A1, Hs00264051_m1) and SOX9 (Hs01001343-g1) with a reference of the housekeeping gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as a reference.
Quantifications of anabolic and catabolic cytokines
The anabolic and catabolic cytokines in the culture supernatants of the treated chondrocytes were determined using a relevant enzyme-linked immunosorbent assay (ELISA). The levels of IL-1β (437004, ELISA MAX™ Deluxe Set Human IL-1β, BioLegend), IL-6 (88-7066, IL-6 Human Uncoated ELISA Kit, Invitrogen, Thermo Fisher Scientific), IL-8 (BMS204/3, IL-8 Human ELISA Kit, Invitrogen, Thermo Fisher Scientific), IL-10 (88-7106, Invitrogen, Thermo Fisher Scientific), TNF-α (88-7346, TNF alpha Human Uncoated ELISA Kit, Invitrogen, Thermo Fisher Scientific), MMP-3 (444807, LEGEND MAX™ Human Total MMP-3 ELISA Kit, BioLegend), MMP-9 (440707, LEGEND MAX™ Human MMP-9 ELISA Kit with precoated plates, BioLegend), TIMP-1 (DY970, Human TIMP-1 DuoSet ELISA, R&D, USA) and TIMP-2 (DY971, Human TIMP-2 DuoSet ELISA, R&D) were quantified.
Nicotinamide treatment of SMN-treated chondrocytes
To verify the role of Sirt1 in ECM homeostasis in IL-1β-stimulated chondrocytes, 10 nM nicotinamide (NAM, 72340, Sigma-Aldrich), a Sirt1 inhibitor, was added to the culture medium with 25 μM SMN [19]. After cultivation for 24 h, the mRNA levels of AGCN, COL2A1, COL10, MMP-13, Sirt1 and SOX9 were determined again through qPCR in the treated cells.
Statistical analysis
The mean and standard error of the obtained data were calculated and analysed using analysis of variance and Tukey’s post hoc test for multiple group comparisons. At least six patients were included in one experiment, except the three patients included in the NAM study. A p value of < 0.05 was considered statistically significant.
Results
SMN in high doses impaired cell viability of articular chondrocytes
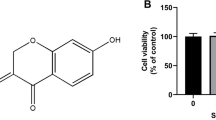
The LDH level and mitochondrial activity of the treated chondrocytes were determined. SMN supplementation (12.5–100 μM) had no effect on the LDH release (Fig. 1a) but significantly reduced the mitochondrial activity of the treated cells (p < 0.01 for the 12.5, 25 and 50 μM SMN groups and p < 0.001 for the 100 μM SMN group; Fig. 1b). In addition, chondrocytes treated with 100 μM SMN exhibited significantly lower cell viability than the other three SMN groups (p < 0.01). The treated chondrocytes did not exhibit a typical cobble-stone morphology (0 μM group), and SMN treatment caused a slightly cytoplasmic extension to chondrocytes (Fig. 1c). However, no hypertrophic differentiation or proliferation was found in the SMN-treated cells.

Cytotoxicity of SMN to articular chondrocytes. Human chondrocytes were incubated with the indicated concentrations of SMN for 24 h and then analysed. a SMN supplementation did not increase LDH release. b SMN additions reduced the mitochondrial activity in chondrocytes, and high-dose SMN (100 μM) further impaired cell viability. c No evidence of hypertrophic transformation or proliferation in SMN-treated chondrocytes was obtained
Cell death, viability, GAG production, survival, and senescence after IL-1β stimulation and SMN supplementation
IL-1β stimulation significantly increased cell death (LDH release, p < 0.01, Fig. 2a) and impaired mitochondrial activity (p < 0.01, Fig. 2b). Treatment of the IL-1β-stimulated chondrocytes with 25 μM SMN had no effect on cell death, whereas 50 μM SMN increased LDH release (p < 0.05). By contrast, no beneficial effects were observed on mitochondrial activity after treatment with 50 μM SMN, but treatment with 25 μM SMN improved cell viability during injury. IL-1β stimulation also impaired GAG production (toluidine blue staining, blue colour), reduced the number of surviving cells (green fluorescence) and resulted in cell senescence (β-gal staining, blue colour). Moreover, supplementation with 25 μM SMN ameliorated GAG production, cell survival and β-gal expression in IL-1β-injured chondrocytes (Fig. 2c).

Treatment of IL-1β-stimulated chondrocytes with SMN. a Addition of 50 μM SMN further increased cell death (LDH release) of injured cells. b Addition of 25 μM SMN partially restored the viability of stimulated chondrocytes. c IL-1β stimulation impaired GAG production (toluidine blue staining), reduced the number of surviving cells (green fluorescence), and resulted in cell senescence (β-gal staining). Supplementation with 50 μM SMN had no substantial effects, whereas the addition of 25 μM SMN ameliorated GAG production, cell survival and β-gal expression in IL-1β-injured chondrocytes
SMN modulated the mRNA expression of catabolic and anabolic cytokines
The results of real-time PCR showed that supplementation with 25 μM SMN significantly downregulated the IL-1β-stimulated expression of catabolic iNOS (p < 0.05), IL-1β (p < 0.05), TNF-α (p < 0.05), MMP-3 (p < 0.05), MMP-9 (p < 0.01), and MMP-13 (p < 0.05) and upregulated anabolic IL-10 (p < 0.05), TIMP-1 (p < 0.05), and COL2A1 (p < 0.01) in the chondrocytes. Moreover, the chondrocyte phenotype genes SOX9 (p < 0.01) and Sirt1 (p < 0.05) were restored. However, SMN did not cause substantial changes in the mRNA levels of IL-6, IL-8, and AGCN in the stimulated cells (Fig. 3).

The mRNA expression profile of SMN-treated chondrocytes. Supplementation with 25 μM SMN significantly downregulated catabolic iNOS, IL-1β, TNF-α, MMP-3, MMP-9 and MMP-13 and upregulated anabolic IL-10, TIMP-1 and COL2A1 mRNA expression in IL-1β-stimulated chondrocytes. The chondrogenic phenotype genes SOX9 and Sirt1 were also restored. However, SMN did not cause substantial changes in the mRNA levels of IL-6, IL-8 and AGCN in the injured cells
SMN modulated anabolic and catabolic cytokines
SMN supplementation decreased IL-1β (p < 0.01), TNF-α (p < 0.05), MMP-3 (p < 0.01), MMP-9 (p < 0.01), but increased upregulated TIMP-1 (p < 0.05) productions in IL-1β-stimulated chondrocytes (Fig. 4). By contrast, the secretion of IL-6, IL-8, IL-10 and TIMP-2 was not altered upon SMN treatment.

Modulation of anabolic and catabolic cytokines by SMN. ELISA assays revealed that treatment with 25 μM SMN reduced the production of catabolic cytokine-stimulated IL-1β, TNF-α, MMP-3 and MMP-9 and increased the production of TIMP-1 in the injured chondrocytes. However, the levels of IL-6, IL-8, IL-10 and TIMP-2 proteins remained unaltered upon SMN addition
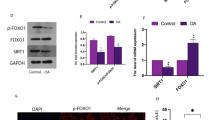
NAM downregulated Sirt1 and SOX9 in SMN-treated chondrocytes
NAM additions downregulated Sirt1 expression (p < 0.05) in IL-1β-stimulated chondrocytes even after SMN supplementation (Fig. 5). In spite of SOX9 level was decreased, there was no significant difference between IL-1β + SMN and IL-1β + SMA + NAM groups. Similarly, the cartilaginous ECM genes AGCN (p < 0.05) and COL2A1 (p < 0.05) were downregulated, whereas the hypertrophic genes IL-10 and MMP-13 were not changed in IL-1β + SMA + NAM group.

NAM attenuated the therapeutic effects of SMN, as evidenced by the downregulation of Sirt-1, AGCN and COL2A1
Discussion
OA is one the most common degenerative joint diseases, and the application of phytochemicals is recommended to adjust ECM homeostasis during arthritis progression [20]. Because dietary nutraceuticals and antioxidants are generally not sufficiently effective for protection against OA development, direct administration of bioactive compounds into the knee joint capsule is a promising option [21, 22]. Pretreatment with SMN has been shown to alleviate TNF-α-stimulated inflammatory responses in chondrocytes, and SMN has also been used in combination with NASIDs to treat OA [10]. However, the therapeutic potential of SMN against catabolic cytokine stimulations is not fully elucidated. In particular, SMN pre- and posttreatment may exert different effects on OA chondrocytes. Therefore, this study demonstrated the effects of SMN posttreatment on IL-1β-stimulated human chondrocytes.
Although phytochemicals possess multiple bioactive functions, high doses have deteriorating effects on cells, especially the primary human OA chondrocytes [17, 23]. Dvořák et al. reported that SMN (10–100 μM) did not exert cytotoxicity (LDH release) toward primary human hepatocytes [24]. Similarly, Gharagozloo and Amirghofran demonstrated that SMN (50 and 100 μM) increased viability in Jurkat cells [25]. However, we found that SMN did not cause cell death (Fig. 1a), but rather reduced the mitochondrial activity (Fig. 1b) in primary chondrocytes. In addition, the responses of OA chondrocytes to external stimulation differ from those of healthy cells [23]. Our study demonstrated that 50 μM SMN supplementation did not cause cell death in unstimulated chondrocytes (Fig. 1a); however, cells with IL-1β-induced injury were more sensitive to SMN toxicity, and 50 μM SMN treatment resulted in cell death (Fig. 2a). Our findings revealed that the cytotoxic effects of SMN may be cell type-dependent. Although SMN is reported be cytoprotective, we found that LDH release and mitochondrial activity (Fig. 2b) were not fully restored in IL-1β-treated chondrocytes upon treatment with 25 μM SMN. Similarly, the GAG production, survival and senescence in chondrocytes were partially ameliorated (Fig. 2c).
Overexpression of catabolic cytokines such as IL-1β can trigger an inflammatory cascade, and the proinflammatory mediator-driven positive feedback loop contributes to the OA by promoting ECM destruction [26, 27]. Zheng et al. demonstrated that pretreatment with silibinin, the main active component of SMN, attenuated OA through inhibition of the expression of iNOS, TNF-α, NO, COX2 and prostaglandin E2 and cartilage ECM degradation in vitro [28]. Similarly, pretreatment with SMN regulated the mRNA expression of IL-6, IL-8 and MMP-1 which ameliorated the TNF-α-stimulated responses in chondrocytes [10]. Despite the downregulation of iNOS, IL-1β and TNF-α after SMN posttreatment, our results indicated that IL-6 and IL-8 expression was not restored in the stimulated chondrocytes (Fig. 3). Moreover, SMN posttreatment downregulated catabolic MMP-3, MMP-9 and MMP-13; upregulated anabolic IL-10 and TIMP-1; and enhanced the expression of ECM gene COL2A1. Similarly, the levels of chondrogenic phenotype genes SOX9 and Sirt1 were also restored. Because IL-6 is known to downregulate COL2A1 and AGCN and IL-8 was found to induce hypertrophic transformation in chondrocytes [29, 30], our data suggest that SMN posttreatment may not affect ECM balance through the regulation of IL-6 and IL-8. In addition, SOX9 and Sirt1 were upregulated in our study, which is consistent with a previous study that demonstrated that the combination of SOX9 and SOX5/6 enhanced COL2A1 transcription [31]. A similar study demonstrated that COL2A1 expression is positively regulated by Sirt1 through the chromatin-binding regions of the COL2A1 promoter and enhancer, which implied that the disruption of Sirt1 could accelerate OA progression during ageing [32, 33]. Taken together, SMN posttreatment may modulate ECM homeostasis through the regulation of SOX9 and Sirt1.
Although SMN posttreatment upregulated IL-10 mRNA level, IL-10 protein production was not further increased (Fig. 4). Ortved et al. found that IL-10 expression was increased in IL-1β-stimulated chondrocytes, and overexpression of IL-10 reduced the IL-1β levels but did not rescue GAG synthesis [34]. In addition, another study reported that IL-10 overexpression antagonised TNF-α-stimulated downregulation of AGCN, while the suppression of COL2A1 was barely affected [35]. Although the SMN-treated chondrocytes exhibited upregulated IL-10 levels and increased IL-10 secretion, the AGCN levels remained unchanged. Therefore, the correlation between SMN addition and IL-10 expression to ECM component production requires further investigation.
Regarding the role of Sirt1 in cartilage homeostasis, several cartilage-specific genes such as AGCN and COL2A1 are known to be modulated through Sirt1-mediated deacetylation of SOX9 [36]. The Sirt1 inhibitor NAM attenuated the therapeutic effects of SMN, as evidenced by the downregulation of anabolic genes such as AGCN and COL2A1, which revealed the correlations among Sirt1, SOX9 and ECM components.
In conclusion, high-dose SMN (100 μM) impaired the mitochondrial activity in unstimulated chondrocytes, and 50 μM SMN supplementation increased cell death in IL-1β-stimulated cells, suggesting that injured chondrocytes are more sensitive to SMN toxicity. Treatment with 25 μM SMN ameliorated cell senescence, downregulated catabolic mRNA expression and upregulated anabolic mRNA expression in IL-1β-stimulated chondrocytes. Furthermore, levels of IL-1β, MMP-3 and MMP-9 proteins were decreased, whereas that of TIMP-1 was increased. Although the IL-6, IL-8 and IL-10 mRNA levels as well as protein production were still high, the upregulation in COL2A1 found in the current study revealed that SMN may maintain the functional phenotype of chondrocytes through the regulation of Sirt1 and SOX9 (Fig. 6). Therefore, SMN posttreatment may modulate catabolic cytokines to restore ECM homeostasis in OA chondrocytes.

Regulation of SMN on anabolism and catabolism in inflammatory chondrocytes by SMN
Availability of data and materials
The necessary data were provided to support the assumption of this study (data will be made available on demand).
Abbreviations
- AGCN:
-
Aggrecan
- β-gal:
-
β-galactosidase
- COL2A1:
-
Collagen type II alpha 1
- ECM:
-
Extracellular matrix
- ELISA:
-
Enzyme-linked immunosorbent assay
- GAPDH:
-
Glyceraldehyde 3-phosphate dehydrogenase
- IL-1β:
-
Interleukin-1 beta
- IL-6:
-
Interleukin-6
- IL-8:
-
Interleukin-8
- IL-10:
-
Interleukin-10
- iNOS:
-
Inducible nitric oxide synthase
- LDH:
-
Lactate dehydrogenase
- MMP-3:
-
Matrix metalloproteinase-3
- MMP-9:
-
Matrix metalloproteinase-9
- MMP-13:
-
Matrix metalloproteinase-13
- NAM:
-
Nicotinamide
- NSAID:
-
Nonsteroidal anti-inflammatory drug
- OA:
-
Osteoarthritis
- PCR:
-
Polymerase chain reaction
- SMN:
-
Silymarin
- Sirt1:
-
Sirtuin-1
- TIMP-1:
-
Tissue inhibitor of metalloproteinase-1
- TIMP-2:
-
Tissue inhibitor of metalloproteinase-2
- TNF-α:
-
Tumour necrosis factor-alpha
References
Wang X, Hunter D, Xu J, Ding C. Metabolic triggered inflammation in osteoarthritis. Osteoarthritis Cartilage. 2015;23:22–30.
Nam J, Perera P, Liu J, Rath B, Deschner J, Gassner R, et al. Sequential alterations in catabolic and anabolic gene expression parallel pathological changes during progression of monoiodoacetate-induced arthritis. Plos One. 2011;6:e24320.
Mueller MB, Tuan RS. Anabolic/Catabolic balance in pathogenesis of osteoarthritis: identifying molecular targets. PM R. 2011;3:S3–11.
Guilak F, Nims RJ, Dicks A, Wu CL, Meulenbelt I. Osteoarthritis as a disease of the cartilage pericellular matrix. Matrix Biol. 2018;71-72:40–50.
Mobasheri A. The future of osteoarthritis therapeutics: targeted pharmacological therapy. Curr Rheumatol Rep. 2013;15(10):364.
Karimi G, Vahabzadeh M, Lari P, Rashedinia M, Moshiri M. “Silymarin”, a promising pharmacological agent for treatment of diseases. Iran J Basic Med Sci. 2011;14:308–17.
Jia JD, Bauer M, Cho JJ, Ruehl M, Milani S, Boigk G, et al. Antifibrotic effect of silymarin in rat secondary biliary fibrosis is mediated by downregulation of procollagen alpha1(I) and TIMP-1. J Hepatol. 2001;35:392–8.
Rao PR, Viswanath RK. Cardioprotective activity of silymarin in ischemia-reperfusion-induced myocardial infarction in albino rats. Exp Clin Cardiol. 2007;12:179–87.
Juma'a KM, Ahmed ZA, Numan IT, Hussain SAR. Dose-dependent anti-inflammatory effect of silymarin in experimental animal model of chronic inflammation. Afr J Pharm Pharmacol. 2009;3:242–7.
Can V, Abouelnour A, Annie IL, Getting BS. The anti-inflammatory effect of silymarin in chondrocytes. Biochem Pharmacol. 2017;139:137.
Ashkavand Z, Malekinejad H, Amniattalab A, Rezaei-Golmisheh A, Vishwanath BS. Silymarin potentiates the anti-inflammatory effects of Celecoxib on chemically induced osteoarthritis in rats. Phytomedicine. 2012;19:1200–5.
Hussain SA, Jassim NA, Numan IT, Al-Khalifa II, Abdullah TA. Anti-inflammatory activity of silymarin in patients with knee osteoarthritis. A comparative study with piroxicam and meloxicam. Saudi Med J. 2009;30:98–103.
Ashkavand Z, Malekinejad H, Aghazadeh AM, Attari JA, Vishwanath BS. Potentiality and safety assessment of combination therapy with silymarin and celecoxib in osteoarthritis of rat model. Biomed Prev Nutr. 2013;3:209–12.
Wu CC, Chen YR, Lu DH, Hsu LH, Yang KC, Sumi S. Evaluation of the post-treatment anti-inflammatory capacity of osteoarthritic chondrocytes: An in vitro study using baicalein. Regen Ther. 2020;14:177–83.
Pabst M, Wu SQ, Grass J, Kolb A, Chiari C, Viernstein H, et al. IL-1beta and TNF-alpha alter the glycophenotype of primary human chondrocytes in vitro. Carbohydr Res. 2010;345:1389–93.
Caramés B, López-Armada MJ, Cillero-Pastor B, Lires-Dean M, Vaamonde C, Galdo F, et al. Differential effects of tumor necrosis factor-alpha and interleukin-1beta on cell death in human articular chondrocytes. Osteoarthritis Cartilage. 2008;16:715–22.
Chen YR, Yang KC, Lu DH, Wu WT, Wang CC, Tsai MH. The chondroprotective effect of diosmin on human articular chondrocytes under oxidative stress. Phytother Res. 2019;33:2378–86.
Yang KC, Chen IH, Yang YT, Hsiao JK, Wang CC. Effects of scaffold geometry on chondrogenic differentiation of adipose-derived stem cells. Mater Sci Eng C Mater Biol Appl. 2020;110:110733.
Yan J, Chen X, Pu C, Zhao Y, Liu X, Liu T, et al. Synovium stem cell-derived matrix enhances anti-inflammatory properties of rabbit articular chondrocytes via the SIRT1 pathway. Mater Sci Eng C Mater Biol Appl. 2020;106:110286.
Alamgeer HUH, Uttra AM, Qasim S, Ikram J, Saleem M, et al. Phytochemicals targeting matrix metalloproteinases regulating tissue degradation in inflammation and rheumatoid arthritis. Phytomedicine. 2020;66:153134.
D'Adamo S, Cetrullo S, Panichi V, Mariani E, Flamigni F, Borzì RM. Nutraceutical activity in osteoarthritis biology: a focus on the nutrigenomic role. Cells. 2020;9:1232.
Veen L, Hantikainen E, Bellocco R, Ye W, Serafini M, Ponzano M, et al. Dietary antioxidants, non-enzymatic antioxidant capacity and the risk of osteoarthritis in the Swedish National March Cohort. Eur J Nutr. 2020. https://doi.org/10.1007/s00394-020-02239-8.
Huang TL, Yang SH, Chen YR, Liao JY, Tang Y, Yang KC. The therapeutic effect of aucubin-supplemented hyaluronic acid on interleukin-1beta-stimulated human articular chondrocytes. Phytomedicine. 2019;53:1–8.
Dvorák Z, Kosina P, Walterová D, Simánek V, Bachleda P, Ulrichová J. Primary cultures of human hepatocytes as a tool in cytotoxicity studies: cell protection against model toxins by flavonolignans obtained from Silybum marianum. Toxicol Lett. 2003;137:201–12.
Gharagozloo M, Amirghofran Z. Effects of silymarin on the spontaneous proliferation and cell cycle of human peripheral blood leukemia T cells. J Cancer Res Clin Oncol. 2007;133:525–32.
Ma Z, Wang Y, Piao T, Liu J. Echinocystic acid inhibits IL-1β-induced COX-2 and iNOS expression in human osteoarthritis chondrocytes. Inflammation. 2016;39:543–9.
Zheng W, Zhang H, Jin Y, Wang Q, Chen L, Feng Z, et al. Butein inhibits IL-1β-induced inflammatory response in human osteoarthritis chondrocytes and slows the progression of osteoarthritis in mice. Int Immunopharmacol. 2017;42:1–10.
Zheng W, Feng Z, Lou Y, Chen C, Zhang C, Tao Z, et al. Silibinin protects against osteoarthritis through inhibiting the inflammatory response and cartilage matrix degradation in vitro and in vivo. Oncotarget. 2017;8:99649–65.
Legendre F, Dudhia J, Pujol JP, Bogdanowicz P. JAK/STAT but not ERK1/ERK2 pathway mediates interleukin (IL)-6/soluble IL-6R down-regulation of Type II collagen, aggrecan core, and link protein transcription in articular chondrocytes. Association with a down-regulation of SOX9 expression. J Biol Chem. 2003;278:2903–12.
Merz D, Liu R, Johnson K, Terkeltaub R. IL-8/CXCL8 and growth-related oncogene alpha/CXCL1 induce chondrocyte hypertrophic differentiation. J Immunol. 2003;171:4406–15.
Yasuda H, Oh CD, Chen D, de Crombrugghe B, Kim JH. A novel regulatory mechanism of type II collagen expression via a SOX9-dependent enhancer in intron 6. J Biol Chem. 2017;292:528–38.
Oppenheimer H, Kumar A, Meir H, Schwartz I, Zini A, Haze A, et al. Set7/9 impacts COL2A1 expression through binding and repression of SirT1 histone deacetylation. J Bone Miner Res. 2014;29:348–60.
Matsuzaki T, Matsushita T, Takayama K, Matsumoto T, Nishida K, Kuroda R, et al. Disruption of Sirt1 in chondrocytes causes accelerated progression of osteoarthritis under mechanical stress and during ageing in mice. Ann Rheum Dis. 2014;73:1397–404.
Ortved KF, Begum L, Stefanovski D, Nixon AJ. AAV-mediated overexpression of IL-10 mitigates the inflammatory cascade in stimulated equine chondrocyte pellets. Curr Gene Ther. 2018;18:171–9.
Müller RD, John T, Kohl B, Oberholzer A, Gust T, Hostmann A, et al. IL-10 overexpression differentially affects cartilage matrix gene expression in response to TNF-alpha in human articular chondrocytes in vitro. Cytokine. 2008;44:377–85.
Feng D, Kang X, Wang R, Chen H, Zhang K, Feng W, et al. Progranulin modulates cartilage-specific gene expression via sirtuin 1-mediated deacetylation of the transcription factors SOX9 and P65. J Biol Chem. 2020;295:13640–50.
Acknowledgements
Not applicable.
Funding
This study was supported by the Tzu Chi Medical Mission Project, Buddhist Tzu Chi Medical Foundation (grant no. TCMMP 106-05-03), provided to Dr. Wen-Tien Wu.
Author information
Authors and Affiliations
Contributions
YRC participated in conducting the experiments and data analysis. WTW, CCW, and KCY contributed to the research design, data analysis, writing of the manuscript, and grant application. DHL and Fedor SS contributed to the writing of the manuscript. All authors have read and approved the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Research Ethics Committee of Hualien Tzu Chi Hospital, Buddhist Tzu Chi Medical Foundation (IRB106-48-A). Written informed consent was provided by all the patients.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wu, WT., Chen, YR., Lu, DH. et al. Silymarin modulates catabolic cytokine expression through Sirt1 and SOX9 in human articular chondrocytes. J Orthop Surg Res 16, 147 (2021). https://doi.org/10.1186/s13018-021-02305-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13018-021-02305-9