
Abstract
Radiotherapy is an effective regimen for cancer treatment alone or combined with chemotherapy or immunotherapy. The direct effect of radiotherapy involves radiation-induced DNA damage, and most studies have focused on this area to improve the efficacy of radiotherapy. Recently, the immunomodulatory effect of radiation on the tumour microenvironment has attracted much interest. Dying tumour cells can release multiple immune-related molecules, including tumour-associated antigens, chemokines, and inflammatory mediators. Then, immune cells are attracted to the irradiated site, exerting immunostimulatory or immunosuppressive effects. CC chemokines play pivotal roles in the trafficking process. The CC chemokine family includes 28 members that attract different immune subsets. Upon irradiation, tumour cells or immune cells can release different CC chemokines. Here, we mainly discuss the importance of CCL2, CCL3, CCL5, CCL8, CCL11, CCL20 and CCL22 in radiotherapy. In irradiated normal tissues, released chemokines induce epithelial to mesenchymal transition, thus promoting tissue injury. In the tumour microenvironment, released chemokines recruit cancer-associated cells, such as tumour-infiltrating lymphocytes, myeloid-derived suppressor cells and tumour-associated macrophages, to the tumour niche. Thus, CC chemokines have protumour and antitumour properties. Based on the complex roles of CC chemokines in the response to radiation, it would be promising to target specific chemokines to alleviate radiation-induced injury or promote tumour control.
Similar content being viewed by others


Background
RT is administered as a curative or palliative therapy in more than 50% of cancer patients [1]. The most frequent and lethal lesions that occur during radiation include single-strand breaks (SSBs), double-strand breaks (DSBs), DNA-DNA or DNA–protein cross-links, base release and other chemical modifications, and multiple damaged sites in DNA [2, 3]. Most research on improving the efficacy of RT has focused on tumour cells [4]. However, tumour lesions are not purely composed of cancer cells, and multiple components are present with cancer cells. These components include (1) immune cells; (2) fibroblasts and epithelial cells; (3) extracellular matrix proteins; (4) blood and lymphatic vessels; and (5) metabolites, chemokines, and cytokines [5]. RT can influence these components in different manners, thus remodelling the tumour microenvironment. In tumour tissues, radiation could induce local release of inflammatory cytokines, temporarily eradicate local radiation-sensitive immune cell lineages, and promote immune cell trafficking and immune cell activation [6]. Immune cell subtypes, including dendritic cells (DCs) or cytotoxic lymphocytes, promote antitumour immunity. However, suppressive cells could also be attracted by radiation. In these contexts, chemokines play fundamental roles.
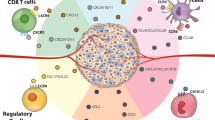
Chemokines include different subfamilies according to the domain found at the N-terminus. CC chemokines are one of these families and have an N-terminal CC domain [7, 8]. CC chemokines are represented using 28 symbols. The chemokines C–C motif chemokine ligand (CCL)9 and CCL10 are the same chemokine; thus, there are a total of 27 CC chemokines [7, 8]. These chemokines form a complex network that influences the distribution of immune cells in the tumour microenvironment [9]. Although the role of CC chemokines in cancer has been discussed elsewhere, the relationship between radiation and these chemokines has not been described. The aim of this review was to collect information about the involvement of each CC chemokine in radiation (Fig. 1).

CC chemokines in radiation responses. The figure shows the chemokines released by tumour cells or adjacent normal cells. In tumour tissues (left part, green), the released chemokines could act on tumour cells or attract different immune subsets, which could promote tumour control or metastasis. In normal tissues (right part, red), radiation promotes the production of chemokines in epithelial cells, endothelial cells or fibroblasts. The released chemokines could trigger EMT in primary cells. Multiple immune cell subsets could be recruited, including macrophages, lymphocytes, eosinophils, neutrophils and microglia. These outcomes ultimately lead to radiation-induced injury. Abbreviations: MSC, MSCs; MDSCs, myeloid-derived suppressor cells; TAMs, tumour-associated macrophages; TILs, tumour-infiltrating lymphocytes; DC, dendritic cells; Treg, regulatory T lymphocytes
CCL2
CCL2, also known as monocyte chemoattractant protein (MCP)-1, can bind to multiple receptors, including CC chemokine receptor (CCR)1 [10], CCR2 [11], CCR4 [12] and CCR5 [13]. CCL2 is also an antagonist of CCR3 [14]. The main function of CCL2 involves the recruitment of monocytes to inflammatory sites [15,16,17]. In tumours, CCL2 can be secreted by multiple cell types, including tumour cells [18,19,20,21], myeloid-derived suppressor cells (MDSCs) [22], mesenchymal stem cells (MSCs) [23], tumour-associated macrophages (TAMs) [19, 24,25,26], tumour-associated neutrophils (TANs) [27], and cancer-associated fibroblasts (CAFs) [28,29,30]. It has been reported that RT can induce the expression of CCL2 [31, 32]. In this section, we will discuss the significance of CCL2 in radiation-related events.
CCL2 is involved in the radiation-induced immune response
Promotion of macrophage infiltration
As mentioned previously, one of the most important functions of CCL2 is the recruitment of monocytes. It has been reported that radiation can promote macrophage infiltration in lung cancer. This infiltration may promote tumour progression through the induction of EMT [33] or triggering an immunosuppressive microenvironment [34]. IL-6 was shown to play a positive role in radiation-induced macrophage migration. When IL-6 signalling was blocked, radiation-induced CCL2 upregulation was inhibited. Neutralizing CCL2 significantly reduced the number of migrated macrophages, indicating that CCL2 was downstream of IL-6 signalling and mediated radiation-induced macrophage infiltration [35]. In the radioresistant pancreatic ductal adenocarcinoma (PDAC) model, tumour-derived CCL2 was significantly upregulated, resulting in increased infiltration of inflammatory monocytes and macrophages but not T cells. When CCL2 was blocked by a specific antibody, the recruitment of inflammatory monocytes was impaired, and tumour growth was delayed [36].
Recruitment of MDSCs
Radiation can induce the recruitment of MDSCs. These CCR2-expressing MDSCs contribute to radioresistance. Using CCR2-knockout mice or CCR2-depleting antibodies can reverse the inhibitory effect of MDSCs. The recruitment of MDSCs was further shown to be dependent on activation of the STING pathway. The activated STING pathway promoted the production of type I IFN, which induced CCL2, CCL7, and CCL12 expression, mobilizing monocytes to tumours [37].
Recruitment of T cells
In addition to the effect on monocytes, CCL2 is involved in the recruitment of T cells. In a sarcoma mouse model, 20 Gy radiation upregulated CCL2 expression. This outcome was associated with the activation and increased infiltration of Th1/Tc1 T cells in the tumour microenvironment [38]. In a murine model of head and neck squamous cell carcinoma, mice received 7.5 Gy radiation. Irradiation upregulated CCL2 production, promoting the infiltration of tumour necrosis factor-alpha (TNFα)-producing monocytes and CCR2+ regulatory T cells (Tregs). In this context, monocyte-derived TNFα activated Tregs, inhibiting the effect of RT [39].
The contradictory effects of CCL2 on antitumour immunity may be dependent on the experimental context, especially radiation dose. A higher dose may stimulate a stronger immune response. In most cases, CCL2 attracts immunosuppressive monocytes, which contribute to tumour progression. This finding suggests that inhibiting CCL2 with radiation may promote the efficacy of RT.
CCL2 mediates radioresistance
In a nasopharyngeal carcinoma (NPC) model, CCL2 correlated with radioresistance and metastasis. CCL2 expression was significantly elevated in HONE1-IR cells and recurrent NPC tumours. CCL2 could promote adaptive radioresistance, metastasis, and EMT in NPC cells. Inhibiting CCL2 enhanced the sensitivity of NPC cells to radiation. In clinical cohorts, high CCL2 expression could predict poorer distant metastasis-free survival [40].
CCL2 and radiation-induced toxicity in normal tissue
Although the primary tumour is the direct target of radiation, normal tissues are inevitably irradiated. Radiation-induced injury usually encompasses two phases: an early phase characterized by tissue inflammation and a late phase characterized by tissue fibrosis. Depending on the irradiated site, organs develop radiation-induced injury, including the lung, liver, intestine and neurons. This section will discuss the role of CCL2 in radiation-induced injury.
Radiation-induced lung injury
Increased expression of CCL2 is related to radiation-induced toxicity in normal tissue [41]. In a mouse model of radiation‐induced pulmonary injury, both the selective CCL2 inhibitor bindarit (BIN) and knockout of the main CCL2 receptor CCR2 protected against normal lung tissue injury in mice. RT-induced vascular dysfunction and associated adverse effects were alleviated. RT-induced expression of CCL2 was abrogated by inhibiting CCL2 signalling. Lung fibrosis was delayed by long-term CCL2 signalling inhibition [41]. EMT has been reported to be involved in radiation-induced lung toxicity. Irradiated human pulmonary alveolar epithelial cells (AECs) (HPAEpiC cells) exhibited upregulated CCL2 and CCR4 expression. In vitro treatment with CCL2 induced EMT in HPAEpiC cells. This effect was weakened by the bioactive component of Radix Ophiopogonin B, CCR4 knockdown, or U0126 (a MEK/ERK inhibitor) [42]. However, not all radiation doses or modes promoted CCL2 secretion. Release of CCL2 from human umbilical vein endothelial cells (HUVECs) depended on the dose rate, and 4.1 mGy/h was optimal [43]. CCR2, the receptor for CCL2, is a marker of inflammatory monocytes. Mice deficient for CCR2 exhibited no pulmonary fibrosis after being irradiated and showed decreased numbers of infiltrating and interstitial macrophages [44]. Reactive oxygen species (ROS) have been implicated in radiation-induced pulmonary injury and fibrosis [45]. A study revealed that inducible nitric oxide synthase (iNOS) promoted the production of inflammatory cytokines such as CCL2 [46]. This finding suggests that radiation-induced injury was not due to a single factor but to the interaction between multiple factors.
Radiation-induced brain and liver injury
CCR2 has been shown to mediate cognitive impairments induced by irradiation. CCR2 knockout (-/-) mice received 10 Gy cranial irradiation. Compared with wild-type mice, CCR2 deficiency prevented hippocampus-dependent spatial learning and memory impairments induced by cranial irradiation. Moreover, CCR2 deficiency normalized the fraction of pyramidal neurons [47]. Irradiation to treat brain tumours could create a chronic neuroinflammatory state resulting in progressive cognitive decline. This effect was associated with the upregulation of CCL2, and inhibiting CCL2 signalling could attenuate this effect [48]. CCL2-deficient mice exhibited attenuated chronic microglial activation, which allowed for the recovery of neurogenesis. In radiation-induced liver damage, multiple chemokines, including CCL2, are released after irradiation. (Myo)fibroblasts in portal vessels may be one of the major sources of these chemokines [49].
Interestingly, not all radiation induces normal tissue injury. Compared with conventional RT (CRT), synchrotron microbeam radiation therapy (MRT) does not induce CCL2 expression in normal tissue [50]. This may explain why synchrotron MRT achieved equivalent tumour control as CRT but showed reduced normal tissue damage. This finding highlights the importance of radiation technology in radiation-induced injury.
CCL2 as a therapeutic target
Based on these findings, CCL2 may be a therapeutic target or marker of normal tissue injury during irradiation. Numerous studies have investigated drugs that could prevent the release of radiation-induced inflammatory cytokines. Pravastatin is used to lower cholesterol, and it was found that pravastatin could limit the increase in blood CCL2 induced by radiation [51]. Fibroblast growth factor 2 peptide (FGF-P) is a peptide derived from the receptor-binding domain of fibroblast growth factor 2. In acute gastrointestinal syndrome (AGS) after radiation, FGF-P could reduce several proinflammatory cytokines, including CCL2 [52]. Inhibiting uPA and uPAR abrogated CCL2 expression in meningioma cell lines that were treated with radiation [53]. This outcome was accompanied by reduced ERK activation, nuclear translocation of Rel-A, and NF-κB-DNA binding activity. CCL2 is a marker of late rectal toxicity in prostate cancer patients undergoing RT. In patients with grade 2 late rectal toxicity, CCL2 levels were significantly increased [54]. Evaluating CCL2 levels at an earlier phase and targeting CCL2 may help prevent the development of rectal toxicity.
CCL3
CCL3, also known as macrophage inflammatory protein-1α (MIP-1α), was first purified from the supernatant of endotoxin-induced murine macrophages [55]. Multiple cell types can produce CCL3, including immune cell subtypes, fibroblasts, and epithelial cells, as well as platelets, basophils, and eosinophils [56, 57]. CCL3 binds to CCR1, CCR4, and CCR5, exerting various effects on multiple immune cell subtypes. Due to the diverse functions of CCL3, it is involved in diverse biological processes.
CCL3 is involved in the radiation-induced immune response
In a hepatocellular carcinoma model, mice treated with MIP-1α and irradiation exhibited significantly enhanced antitumour effects. Immunological markers, including CD8A and CD107A, were also increased [58]. In a phase I clinical trial, patients with breast cancer were treated with intratumoural H2O2 and RT. Blood marker analysis highlighted significant associations between MIP-1α and the tumour response [59]. This finding suggests that blood CCL3 may serve as a predictive marker for treatment response in specific cancer types.
CCL3 and radiation-induced injury
CCL3 was reported to contribute to the development of radiation-induced injury. Both in vitro and in vivo experiments showed that CCL3 was increased by radiation. Mice deficient in CCL3 exhibited reduced inflammation, no significant septal thickening, and small collagen deposition foci in lung tissues after radiation. This effect was mediated by CCR1 but not CCR5. Both CCR1−/− mice and the CCR1 inhibitor BX-471 ameliorated radiation-induced injury. CCR1 has been reported to promote Th2 cell recruitment. In CCR1−/− mice, Th2 cytokines were not increased after irradiation. These results indicated that Th2 cytokines and cells are involved in the development of fibrosis [60]. Samples from radiated human conduit arteries exhibited sustained expression of inflammatory genes, including CCL3, and this radiation-induced CCL3 upregulation was due to NF-κB activation [61]. However, a study showed that in patients with prostate cancer who received three-dimensional conformal radiation therapy, MIP-1α levels were not sensitive to irradiation [62]. Based on these results, CCL3 could be a predictive biomarker for treatment response. This discrepancy in the effects of CCL3 may be attributed to the different responses in specific irradiated sites.
CCL3 as a therapeutic target
Cryptotanshinone (CTS) is a major lipophilic extract from Salvia miltiorrhiza Bunge (Danshen). CTS can attenuate collagen deposition and pulmonary fibrosis in radiation-induced lung injury (RILI) rats by mitigating radiation-induced activation of CCL3 and its receptor CCR1 [63]. The reduced secretion of CCL3 alleviates the radiation-induced inflammatory microenvironment, subsequently attenuating the initiation of fibrosis. Since mitotic cells are highly sensitive to radiation, reducing the number of cells undergoing mitosis may be an ideal approach to protect normal tissues. MIP-1α induces a transient 50% reduction in the number of mitotic cells in small intestinal crypts [64]. BB-10010 is an analogue of MIP-1, and pretreatment with BB-10010 before irradiation significantly increased the number of surviving crypts (10%), which could prevent the side effects associated with RT [65]. This finding may support the use of BB-10010 in patients undergoing abdominal or pelvic treatments. ECI301 is a human MIP-1α variant. Daily administration of 2 µg of ECI301 for 3–5 consecutive days after local irradiation prolonged survival and eradicated tumour growth in CT26 and LLC models. The abscopal effect was observed, and this antitumour effect depended on CD8+ and CD4+ lymphocytes and NK1.1 cells [66].
CCL5
CCL5 (also known as regulated on activation, normally T cell expressed and secreted (RANTES)) belongs to the C–C motif chemokine family. CCL5 binds to CCR5 with high affinity, but it can also bind to CCR1, CCR3 and CCR4 [67]. CCL5 can be found in cancer cells, CAFs, MSCs, MDSCs, TAMs, and anticancer tumour-infiltrating lymphocytes (TILs) [68]. The role of CCL5 in cancer is complicated; it has both protumour and antitumour properties. CCL5 is elevated in multiple tumours and indicates a poor prognosis [69]. However, studies have shown that CCL5 promotes antitumour immunity by recruiting antitumour T cells and DCs to the tumour microenvironment [67]. Thus, CCL5 is a double-edged sword in cancer.
CCL5 promotes tumour progression
Tumour cell-derived CCL5
Macrophages are predominantly found in the tumour microenvironment. Although RT is effective at reducing primary tumours, it may enhance macrophage infiltration to tumour sites, accelerating tumour progression [70, 71]. In oesophageal squamous cell carcinoma, RT enhanced the expression levels of 12-LOX in tumour cells. Subsequently, CCL5 expression was upregulated through the AKT/NF-κB pathway. Elevated CCL5 can recruit macrophages to tumour tissues and promote their polarization to the immunosuppressive M2 subtype, thereby facilitating metastasis [72]. Similarly, radiation promotes macrophage migration in non-small-cell lung cancer models. CCL5 is responsible for this effect, as blocking CCL5 significantly reduces the number of migrated macrophages. Further analysis showed that CCL5 was upregulated by IL-6, and IL-6 inhibition impaired macrophage migration [35].
MSC-derived CCL5
MSCs play important roles in cancer metastasis and are involved in radiation-induced cancer lung metastasis. In the 4T1 breast cancer model, irradiated MSCs promote lung metastasis. The cGAS–STING signalling pathway is activated in irradiated MSCs. Knockdown of cGAS–STING signalling in MSCs abolished their prometastatic effect. Further analysis identified CCL5 as a downstream molecule. cGAS–STING pathway knockdown impaired CCL5 expression. Knockout of CCL5 in MSCs inhibited lung metastasis and CCR5+ macrophage infiltration. This finding highlights the role of the cGAS–STING-CCL5 pathway in metastasis [73].
Based on these results, CCL5 may indicate a worse prognosis in a specific context. A randomized phase 2 trial compared the efficacy of gemcitabine to capecitabine-based chemoradiotherapy in locally advanced pancreatic cancer. Baseline plasma samples were collected to identify circulating biomarkers. It was found that higher baseline circulating levels of CCL5 indicated worse overall survival [74].
CCL5 promotes antitumour immunity
CD8+ T cell infiltration after RT is pivotal for tumour control. However, in a radiation-resistant mouse model, RT did not induce CD8+ T cell infiltration. CCL5 is involved in the recruitment of CD8+ T cells, which has been confirmed in solid tumours [75]. Studies have shown that CCL5 expression is much higher in parental tumour tissue than in radioresistant tumour tissue [76]. This finding highlights the role of CCL5 in CD8+ T cell infiltration.
Low CD8+ T cell infiltration and high programmed death ligand 1 (PD-L1) expression in pancreatic cancer predicts poor outcomes. Radiation combined with vaccination upregulates the expression of the chemokines CXCL10 and CCL5 in the tumour, along with increased CD8+ T cell infiltration. The use of anti-PD-L1 antibodies further enhanced the efficacy of the combination [77]. AZD6738 is an ATR inhibitor that has been investigated in early phase clinical trials. AZD6738 combined with RT significantly improved the efficacy of radiation. Increased infiltration of CD3+ and NK cells was observed in the tumour. In vitro and in vivo data demonstrated that ATRi plus RT promoted the production of CCL3, CCL5, and CXCL10 in tumour cells [78]. The released chemokines may contribute to the infiltration of antitumour immune cells. These results indicated that the presence of CCL5 promotes antitumour immunity and predicts a better response to treatment.
CCL5 and radiation-induced injury
As mentioned previously, CCL2 contributes to radiation-induced EMT in HPAEpiC cells. CCL5 mediates this process, indicating that CCL5 may contribute to radiation-induced injury [42]. In human irradiated arteries, the inflammasome-associated chemokines CCL2 and CCL5 were elevated. In a mouse model of radiation-induced injury, the same effect was observed. Treatment with the IL-1 receptor antagonist anakinra significantly reduced CCL2 and CCL5 levels in mice. This finding indicated that IL-1 blockade could be a treatment for RT-induced vascular disease [79]. However, CCL5 not only contributes to radiation-induced injury but also protects normal tissue. Haematopoietic regeneration deficiency is a common side effect of RT. In mice with CCR5 deficiency in haematopoietic cells, haematopoietic regeneration was delayed. Further investigation showed that CCL5 was an endothelial cell-secreted haematopoietic growth factor that could bind to CCR5 on haematopoietic cells. CCL5 treatment accelerated haematopoietic regeneration. Mechanistically, CCL5 could promote haematopoietic cell cycling and survival [80]. These two experiments exhibited seemingly contrary results: CCL5 exerted a protective effect on haematopoietic regeneration, but in RT-induced cardiovascular disease, CCL5 promoted injury. This difference may be due to the different effects of CCL5 in different contexts. CCL5 binds to CCR5 on haematopoietic cells but not immune cells, thus promoting haematopoietic regeneration. In irradiated arteries, CCL5 may bind specific receptors on immune cells, thus promoting the development of inflammation.
CCL8
CCL8 is also known as MCP-2. CCL8 can bind to CCR1, CCR2, CCR3, and CCR5 [81]. The main function of CCL8 is to recruit monocytes to inflammatory sites. In addition, CCL8 can attract eosinophils and Tregs [82]. In tumours, CCL8 can promote cancer cell proliferation [83] and migration [84, 85]. The role of CCL8 in radiation-related events is poorly understood. However, CCL8 may participate in lung metastasis. In a Lewis lung cancer model, thorax irradiation with 5 Gy X-ray dramatically increased the number of tumours in the lungs. The administration of nicaraven, a radioprotective agent, significantly reduced the number of tumours in the lungs. Moreover, radiosensitivity was not affected by nicaraven. Further investigation of the underlying mechanism revealed that nicaraven effectively inhibited CCL8 expression and macrophage recruitment [86]. Cranial irradiation can lead to cognitive impairments in patients with brain tumours. Radiation-induced inflammation may be the primary reason for these deficits. It has been reported that CCR2 is critical for macrophage trafficking in the brain. Gene expression analysis revealed radiation-induced gene expression of CCR2 ligands, including CCL8, in the hippocampus. CCR2 deficiency could reduce this induction and prevent hippocampus-dependent spatial learning and memory impairments [47]. This protective effect may be attributed to decreased infiltration of macrophages attracted by CCL8. In human irradiated arteries, CCL8 is dysregulated. This effect may be attributed to NF-κB activation [61]. However, the direct role of CCL8 in radiation-induced cardiovascular disease still needs further investigation.
CCL11
CCL11 is a member of the eotaxin family and is also called eotaxin-1. CCL11 can recruit eosinophils by binding to the receptor CCR3 [7]. CCL11 is a ligand for CCR5 [87]. CCR3 was shown to be expressed in tumour cells, and activation of this receptor on cancer cells could increase proliferation and migration [88, 89]. Moreover, CCL11 could promote cancer cell apoptosis resistance [90] and angiogenesis [91]. A study showed that severe lung toxicity was associated with lower levels of eotaxin at 24 h after irradiation compared with that in patients who did not develop severe lung toxicity [92]. CCL11 could be a predictive marker of liver toxicity [93]. High-dose irradiation led to skin injury, and this effect was eosinophil-associated. Eosinophil-related Th2 cytokines, including CCL11, were upregulated in irradiated skin and HUVECs. Blocking IL-33 suppressed cytokine secretion in a coculture system of THP-1 cells and irradiated HUVECs, indicating that IL-33 may be a key molecule that regulates this process [94]. Similarly, in the context of radiation-induced brain tissue damage, inflammation-related genes, including CCL11, were strongly induced in endothelial cells [95]. In human dermal fibroblasts, the expression of eotaxin was upregulated by radiation, which may be an important step leading to eosinophilia in patients with radiation exposure [96]. Radiation-induced intestinal fibrosis (RIF) is a serious complication after abdominal RT. Excessive eosinophil accumulation is associated with RIF in both humans and mice. Irradiation increases extracellular adenosine triphosphate, which induces the expression of CCL11 by pericryptal α-SMA+ cells. Eosinophil-deficient mice showed marked amelioration of RIF. Blocking CCR3 by genetic deficiency or neutralizing antibodies suppressed eosinophil accumulation after irradiation in mice, suggesting a role for eosinophils in mice [97].
Other chemokines
CCL7 is involved in the anti-inflammatory response and antitumour immunity [98, 99]. In fibrosis-sensitive C57BL/6 mouse model, total lung RNA was extracted at 26 weeks. CCL7 expression was upregulated, indicating a role in radiation-induced lung fibrosis [100]. CCL22 and CCL17 play pivotal roles in various type-2 T helper cell-dominant diseases. In a rat model of radiation pneumonitis/pulmonary fibrosis, CCL22 and CCL17 were upregulated in irradiated lung tissues. CCL22 and CCL17 were localized primarily to alveolar macrophages, as shown by immunohistochemistry. This finding indicated that macrophages are the primary producers of CCL22 and CCL17. Similarly, bronchoalveolar lavage fluid from patients with idiopathic pulmonary fibrosis exhibited elevated levels of CCL22. The corresponding receptor CCR4 was detected on alveolar lymphocytes and macrophages [101]. Thus, CCL22 and CCL17 are released by alveolar macrophages and may act on CCR4+ type-2 T helper cells and alveolar macrophages to promote pulmonary fibrosis. One of the biological functions of CCL20 is to attract immature DCs and lymphocytes to tissues. Injecting pcDNA3.1/MIP-3α into LLC subcutaneous tumours after radiation significantly delayed tumour growth. The accumulation of DCs was observed in tumour tissues. The local infiltrating lymphocytes after the treatment were predominantly CD8+ T cells [102]. Whole-body irradiation (WBI) depleted DCs in the epidermis and dermis. However, this effect was not due to DC apoptosis. Increased mRNA levels of CCL19/CCL21 and CCR7 in lymph nodes and skin were observed after radiation. CCR7 and its ligands CCL19/CCL21 mediate cDC migration to the draining lymph node. Thus, the radiation-induced depletion of DCs was due to DC migration to the lymph node. In vitro experiments showed that the number of DCs that migrated to CCL19-containing medium increased, indicating that chemokines regulated the migration of DCs [103]. However, a study showed that γ-ray irradiation could significantly inhibit the production of PEG2 and the expression of CCR7 induced by LPS. Eventually, the migration of DCs towards CCL19 is impaired in vitro and in vivo [104]. Low-dose RT (LD-RT) is known to exert an anti-inflammatory effect. In a coculture system of the EC line EA.hy.926 and polymorphonuclear neutrophils (PMNs), irradiation with a low dose between 0.5 and 1 Gy resulted in a significant reduction in CCL20 expression. Moreover, PMN/EA.hy.926 EC adhesion was significantly decreased [105]. In nasopharyngeal carcinoma, CCL22 was significantly increased in patient-derived xenograft tumours or cell lines upon radiation. In serum collected from patients who received RT, this effect was confirmed. Further experiments revealed that CCL22 could recruit CCR4+ CD8 T cells, which could enhance antitumour immunity [106]. Radiation could induce CCL27 production in the human keratinocyte cell line HaCaT. This effect depended on crosstalk between TNF-α and ROS. CCL27 production promotes skin immune and inflammatory reactions. However, the mechanism of CCL27 still needs investigation [107].
Conclusion
CC chemokines mainly exert chemotactic effects, attracting monocytes, DCs, and T lymphocytes, which are involved in inflammatory reactions and antiviral effects. Radiation stimulates the production of these chemokines (Table 1). In this review, we mainly focused on the chemokines CCL2, CCL3, CCL5, CCL8, CCL11, CCL20, and CCL22. Multiple types of cells can produce CC chemokines, including tumour cells, endothelial cells, fibroblasts, and MSCs. The released chemokines bind to the corresponding receptors in an autocrine or paracrine manner. The most common effects of released chemokines were the recruitment of immune cells, including lymphocytes, macrophages, neutrophils and eosinophils, to irradiated sites, which could form an inflammatory environment. In normal tissues, this recruitment usually leads to acute and late radiation side effects. Radiation-induced chemokines can be detected in serum and thus could be used as predictive biomarkers. However, the time at which to measure serum levels of specific chemokines may differ in different cancer types. The optimal cut-off values of the predictive chemokines need to be critically evaluated. Several studies have identified relevant factors that inhibit specific chemokines to alleviate radiation-induced injury. This finding indicated that CC chemokines could be promising targets to inhibit the side effects of radiation. However, it should be noted that these strategies may influence the systematic inflammatory response, leading to serious side effects.
In tumour tissues, the infiltration of these immune cells, especially macrophages, can promote metastasis or radioresistance. However, in some cases, anticancer effector cells are recruited to facilitate tumour control. Some chemokines, such as CCL2 and CCL5, exert both protumour and antitumour effects. Thus, it is difficult to target these chemokines as a treatment strategy. The protective effects of specific chemokines have been reported, which could exert induce antitumour immunity. To maximize the antitumour effects of chemokines, radiation doses, fractions or techniques should be taken into consideration. The patterns of chemokine release may differ in response to different doses or fractions. Both preclinical experiments and clinical trials are needed to confirm the optimal doses to boost antitumoural chemokines.
Although the role of chemokines in radiation is complex, investigation of the responses of the chemokines to RT may lead to the development of novel treatments.
Availability of data and materials
Not applicable.
Abbreviations
- cGAS-STING:
-
Cyclic GMP-AMP synthase stimulator of interferon genes
- CAF:
-
Cancer-associated fibroblasts
- CRT:
-
Conventional radiotherapy
- DC:
-
Dendritic cells
- DNA:
-
Deoxyribonucleic acid
- DSB:
-
Double-strand breaks
- EMT:
-
Epithelial-mesenchymal transition
- FGF-P:
-
Fibroblast growth factor 2
- HUVECs:
-
Human umbilical vein ndothelial cells
- IFN:
-
Interferon
- iNOS:
-
Nitric oxide synthase
- MCP-1:
-
Monocyte chemoattractant protein 1
- MDSCs:
-
Myeloid-derived suppressor cells
- MIP-1:
-
Macrophage inflammatory protein
- MIP-1α:
-
Macrophage inflammatory protein-1α
- MSC:
-
Mesenchymal stem cells
- MRT:
-
Synchrotron microbeam radiation therapy
- NF-B:
-
Nuclear factor kappa-light-chain-enhancer of activated B cells
- NPC:
-
Nasopharyngeal carcinoma
- PDAC:
-
Pancreatic ductal adenocarcinoma
- PD-L1:
-
Programmed death ligand 1
- TAMs:
-
Tumor-associated macrophages
- TAN:
-
Tumor-associated neutrophils
- TILs:
-
Tumor-infiltrating lymphocytes
- Treg:
-
Regulatory T lymphocytes
- RT:
-
Radiotherapy
- TNF:
-
Tumor necrosis factor-alpha
- RANTES:
-
Regulated on activation, normally T cell expressed and secreted
- RIF:
-
Radiation-induced intestinal fibrosis
- ROS:
-
Reactive oxygen species
References
Rodriguez-Ruiz ME, Vitale I, Harrington KJ, Melero I, Galluzzi L. Immunological impact of cell death signaling driven by radiation on the tumor microenvironment. Nat Immunol. 2020;21(2):120–34.
Alizadeh E, Orlando TM, Sanche L. Biomolecular damage induced by ionizing radiation: the direct and indirect effects of low-energy electrons on DNA. Annu Rev Phys Chem. 2015;66:379–98.
Eriksson D, Stigbrand T. Radiation-induced cell death mechanisms. Tumour Biol. 2010;31(4):363–72.
Barker HE, Paget JT, Khan AA, Harrington KJ. The tumour microenvironment after radiotherapy: mechanisms of resistance and recurrence. Nat Rev Cancer. 2015;15(7):409–25.
Portella L, Scala S. Ionizing radiation effects on the tumor microenvironment. Semin Oncol. 2019;46(3):254–60.
Jagodinsky JC, Harari PM, Morris ZS. The Promise of Combining Radiation Therapy With Immunotherapy. Int J Radiat Oncol Biol Phys. 2020;108(1):6–16.
Hughes CE, Nibbs RJB. A guide to chemokines and their receptors. FEBS J. 2018;285(16):2944–71.
Zlotnik A, Yoshie O. Chemokines: a new classification system and their role in immunity. Immunity. 2000;12(2):121–7.
Viola A, Sarukhan A, Bronte V, Molon B. The pros and cons of chemokines in tumor immunology. Trends Immunol. 2012;33(10):496–504.
Dagouassat M, Suffee N, Hlawaty H, Haddad O, Charni F, Laguillier C, et al. Monocyte chemoattractant protein-1 (MCP-1)/CCL2 secreted by hepatic myofibroblasts promotes migration and invasion of human hepatoma cells. Int J Cancer. 2010;126(5):1095–108.
Monteclaro F, Charo I. The amino-terminal domain of CCR2 is both necessary and sufficient for high affinity binding of monocyte chemoattractant protein 1. Receptor activation by a pseudo-tethered ligand. J Biol Chem. 1997;272(37):23186–90.
Sun W, Li WJ, Wei FQ, Wong TS, Lei WB, Zhu XL, et al. Blockade of MCP-1/CCR4 signaling-induced recruitment of activated regulatory cells evokes an antitumor immune response in head and neck squamous cell carcinoma. Oncotarget. 2016;7(25):37714–27.
Blanpain C, Migeotte I, Lee B, Vakili J, Doranz B, Govaerts C, et al. CCR5 binds multiple CC-chemokines: MCP-3 acts as a natural antagonist. Blood. 1999;94(6):1899–905.
Parody TR, Stone MJ. High level expression, activation, and antagonism of CC chemokine receptors CCR2 and CCR3 in Chinese hamster ovary cells. Cytokine. 2004;27(1):38–46.
Lu B, Rutledge BJ, Gu L, Fiorillo J, Lukacs NW, Kunkel SL, et al. Abnormalities in monocyte recruitment and cytokine expression in monocyte chemoattractant protein 1-deficient mice. J Exp Med. 1998;187(4):601–8.
Deshmane SL, Kremlev S, Amini S, Sawaya BE. Monocyte chemoattractant protein-1 (MCP-1): an overview. J Interferon Cytokine Res. 2009;29(6):313–26.
Proost P, Wuyts A, Van Damme J. Human monocyte chemotactic proteins-2 and -3: structural and functional comparison with MCP-1. J Leukoc Biol. 1996;59(1):67–74.
Ohta M, Kitadai Y, Tanaka S, Yoshihara M, Yasui W, Mukaida N, et al. Monocyte chemoattractant protein-1 expression correlates with macrophage infiltration and tumor vascularity in human esophageal squamous cell carcinomas. Int J Cancer. 2002;102(3):220–4.
Koide N, Nishio A, Sato T, Sugiyama A, Miyagawa S. Significance of macrophage chemoattractant protein-1 expression and macrophage infiltration in squamous cell carcinoma of the esophagus. Am J Gastroenterol. 2004;99(9):1667–74.
Desai S, Kumar A, Laskar S, Pandey BN. Cytokine profile of conditioned medium from human tumor cell lines after acute and fractionated doses of gamma radiation and its effect on survival of bystander tumor cells. Cytokine. 2013;61(1):54–62.
Dutta P, Sarkissyan M, Paico K, Wu Y, Vadgama J. MCP-1 is overexpressed in triple-negative breast cancers and drives cancer invasiveness and metastasis. Breast Cancer Res Treat. 2018;170(3):477–86.
Han E, Lee J, Ryu S, Choi C. Tumor-conditioned Gr-1(+)CD11b(+) myeloid cells induce angiogenesis through the synergistic action of CCL2 and CXCL16 in vitro. Biochem Biophys Res Commun. 2014;443(4):1218–25.
Jia XH, Du Y, Mao D, Wang ZL, He ZQ, Qiu JD, et al. Zoledronic acid prevents the tumor-promoting effects of mesenchymal stem cells via MCP-1 dependent recruitment of macrophages. Oncotarget. 2015;6(28):26018–28.
Mohamed M, El-Ghonaimy E, Nouh M, Schneider R, Sloane B, El-Shinawi M. Cytokines secreted by macrophages isolated from tumor microenvironment of inflammatory breast cancer patients possess chemotactic properties. Int J Biochem Cell Biol. 2014;46:138–47.
Chang A, Miska J, Wainwright D, Dey M, Rivetta C, Yu D, et al. CCL2 produced by the glioma microenvironment is essential for the recruitment of regulatory T cells and myeloid-derived suppressor cells. Can Res. 2016;76(19):5671–82.
Gabrusiewicz K, Rodriguez B, Wei J, Hashimoto Y, Healy LM, Maiti SN, et al. Glioblastoma-infiltrated innate immune cells resemble M0 macrophage phenotype. JCI Insight. 2016;1(2): e85841.
Zhou SL, Zhou ZJ, Hu ZQ, Huang XW, Wang Z, Chen EB, et al. Tumor-associated neutrophils recruit macrophages and T-regulatory cells to promote progression of hepatocellular carcinoma and resistance to sorafenib. Gastroenterology. 2016;150(7):1646-1658 e17.
Silzle T, Kreutz M, Dobler M, Brockhoff G, Knuechel R, Kunz-Schughart L. Tumor-associated fibroblasts recruit blood monocytes into tumor tissue. Eur J Immunol. 2003;33(5):1311–20.
Subramaniam KS, Tham ST, Mohamed Z, Woo YL, Mat Adenan NA, Chung I. Cancer-associated fibroblasts promote proliferation of endometrial cancer cells. PLoS ONE. 2013;8(7): e68923.
Yang X, Lin Y, Shi Y, Li B, Liu W, Yin W, et al. FAP promotes immunosuppression by cancer-associated fibroblasts in the tumor microenvironment via STAT3-CCL2 signaling. Cancer Res. 2016;76(14):4124–35.
Bravata V, Minafra L, Forte GI, Cammarata FP, Russo G, Di Maggio FM, et al. Cytokine profile of breast cell lines after different radiation doses. Int J Radiat Biol. 2017;93(11):1217–26.
Wang P, Guo F, Han L, Wang X, Li J, Guo Y, et al. X-ray-induced changes in the expression of inflammation-related genes in human peripheral blood. Int J Mol Sci. 2014;15(11):19516–34.
Dehai C, Bo P, Qiang T, Lihua S, Fang L, Shi J, et al. Enhanced invasion of lung adenocarcinoma cells after co-culture with THP-1-derived macrophages via the induction of EMT by IL-6. Immunol Lett. 2014;160(1):1–10.
Krneta T, Gillgrass A, Ashkar AA. The influence of macrophages and the tumor microenvironment on natural killer cells. Curr Mol Med. 2013;13(1):68–79.
Wang X, Yang X, Tsai Y, Yang L, Chuang KH, Keng PC, et al. IL-6 mediates macrophage infiltration after irradiation via up-regulation of CCL2/CCL5 in non-small cell lung cancer. Radiat Res. 2017;187(1):50–9.
Kalbasi A, Komar C, Tooker GM, Liu M, Lee JW, Gladney WL, et al. Tumor-derived CCL2 mediates resistance to radiotherapy in pancreatic ductal adenocarcinoma. Clin Cancer Res. 2017;23(1):137–48.
Liang H, Deng L, Hou Y, Meng X, Huang X, Rao E, et al. Host STING-dependent MDSC mobilization drives extrinsic radiation resistance. Nat Commun. 2017;8(1):1736.
Simon PS, Bardhan K, Chen MR, Paschall AV, Lu C, Bollag RJ, et al. NF-kappaB functions as a molecular link between tumor cells and Th1/Tc1 T cells in the tumor microenvironment to exert radiation-mediated tumor suppression. Oncotarget. 2016;7(17):23395–415.
Mondini M, Loyher PL, Hamon P, GerbedeThore M, Laviron M, Berthelot K, et al. CCR2-dependent recruitment of tregs and monocytes following radiotherapy is associated with TNFalpha-mediated resistance. Cancer Immunol Res. 2019;7(3):376–87.
Guo SS, Liu R, Wen YF, Liu LT, Yuan L, Li YX, et al. Endogenous production of C-C motif chemokine ligand 2 by nasopharyngeal carcinoma cells drives radioresistance-associated metastasis. Cancer Lett. 2020;468:27–40.
Wiesemann A, Ketteler J, Slama A, Wirsdorfer F, Hager T, Rock K, et al. Inhibition of radiation-induced Ccl2 signaling protects lungs from vascular dysfunction and endothelial cell loss. Antioxid Redox Signal. 2019;30(2):213–31.
Zhong Y, Lin Z, Lu J, Lin X, Xu W, Wang N, et al. CCL2-CCL5/CCR4 contributed to radiation-induced epithelial-mesenchymal transition of HPAEpiC cells via the ERK signaling pathways. Am J Transl Res. 2019;11(2):733–43.
Ebrahimian T, Le Gallic C, Stefani J, Dublineau I, Yentrapalli R, Harms-Ringdahl M, et al. Chronic gamma-irradiation induces a dose-rate-dependent pro-inflammatory response and associated loss of function in human umbilical vein endothelial cells. Radiat Res. 2015;183(4):447–54.
Groves AM, Johnston CJ, Williams JP, Finkelstein JN. Role of infiltrating monocytes in the development of radiation-induced pulmonary fibrosis. Radiat Res. 2018;189(3):300–11.
Giaid A, Lehnert SM, Chehayeb B, Chehayeb D, Kaplan I, Shenouda G. Inducible nitric oxide synthase and nitrotyrosine in mice with radiation-induced lung damage. Am J Clin Oncol. 2003;26(4):e67-72.
Malaviya R, Gow AJ, Francis M, Abramova EV, Laskin JD, Laskin DL. Radiation-induced lung injury and inflammation in mice: role of inducible nitric oxide synthase and surfactant protein D. Toxicol Sci. 2015;144(1):27–38.
Belarbi K, Jopson T, Arellano C, Fike JR, Rosi S. CCR2 deficiency prevents neuronal dysfunction and cognitive impairments induced by cranial irradiation. Cancer Res. 2013;73(3):1201–10.
Lee SW, Haditsch U, Cord BJ, Guzman R, Kim SJ, Boettcher C, et al. Absence of CCL2 is sufficient to restore hippocampal neurogenesis following cranial irradiation. Brain Behav Immun. 2013;30:33–44.
Malik IA, Moriconi F, Sheikh N, Naz N, Khan S, Dudas J, et al. Single-dose gamma-irradiation induces up-regulation of chemokine gene expression and recruitment of granulocytes into the portal area but not into other regions of rat hepatic tissue. Am J Pathol. 2010;176(4):1801–15.
Yang Y, Swierczak A, Ibahim M, Paiva P, Cann L, Stevenson AW, et al. Synchrotron microbeam radiotherapy evokes a different early tumor immunomodulatory response to conventional radiotherapy in EMT6.5 mammary tumors. Radiother Oncol. 2019;133:93–9.
Holler V, Buard V, Gaugler MH, Guipaud O, Baudelin C, Sache A, et al. Pravastatin limits radiation-induced vascular dysfunction in the skin. J Invest Dermatol. 2009;129(5):1280–91.
Zhang L, Sun W, Wang J, Zhang M, Yang S, Tian Y, et al. Mitigation effect of an FGF-2 peptide on acute gastrointestinal syndrome after high-dose ionizing radiation. Int J Radiat Oncol Biol Phys. 2010;77(1):261–8.
Nalla AK, Gogineni VR, Gupta R, Dinh DH, Rao JS. Suppression of uPA and uPAR blocks radiation-induced MCP-1 mediated recruitment of endothelial cells in meningioma. Cell Signal. 2011;23(8):1299–310.
Bedini N, Cicchetti A, Palorini F, Magnani T, Zuco V, Pennati M, et al. Evaluation of mediators associated with the inflammatory response in prostate cancer patients undergoing radiotherapy. Dis Markers. 2018;2018:9128128.
Wolpe SD, Davatelis G, Sherry B, Beutler B, Hesse DG, Nguyen HT, et al. Macrophages secrete a novel heparin-binding protein with inflammatory and neutrophil chemokinetic properties. J Exp Med. 1988;167(2):570–81.
Schaller TH, Batich KA, Suryadevara CM, Desai R, Sampson JH. Chemokines as adjuvants for immunotherapy: implications for immune activation with CCL3. Expert Rev Clin Immunol. 2017;13(11):1049–60.
Ntanasis-Stathopoulos I, Fotiou D, Terpos E. CCL3 Signaling in the tumor microenvironment. Adv Exp Med Biol. 2020;1231:13–21.
Jeong KY, Lee EJ, Yang SH, Seong J. Combination of macrophage inflammatory protein 1 alpha with existing therapies to enhance the antitumor effects on murine hepatoma. J Radiat Res. 2015;56(1):37–45.
Nimalasena S, Gothard L, Anbalagan S, Allen S, Sinnett V, Mohammed K, et al. Intratumoral hydrogen peroxide with radiation therapy in locally advanced breast cancer: results from a phase 1 clinical trial. Int J Radiat Oncol Biol Phys. 2020;108(4):1019–29.
Yang X, Walton W, Cook DN, Hua X, Tilley S, Haskell CA, et al. The chemokine, CCL3, and its receptor, CCR1, mediate thoracic radiation-induced pulmonary fibrosis. Am J Respir Cell Mol Biol. 2011;45(1):127–35.
Halle M, Gabrielsen A, Paulsson-Berne G, Gahm C, Agardh HE, Farnebo F, et al. Sustained inflammation due to nuclear factor-kappa B activation in irradiated human arteries. J Am Coll Cardiol. 2010;55(12):1227–36.
Lopes CO, Callera F. Three-dimensional conformal radiotherapy in prostate cancer patients: rise in interleukin 6 (IL-6) but not IL-2, IL-4, IL-5, tumor necrosis factor-alpha, MIP-1-alpha, and LIF levels. Int J Radiat Oncol Biol Phys. 2012;82(4):1385–8.
Jiang Y, You F, Zhu J, Zheng C, Yan R, Zeng J. Cryptotanshinone ameliorates radiation-induced lung injury in rats. Evid Based Complement Alternat Med. 2019;2019:1908416.
Arango D, Ettarh RR, Brennan PC. BB-10010, an analogue of macrophage inflammatory protein-1 alpha, reduces proliferation in murine small-intestinal crypts. Scand J Gastroenterol. 1999;34(1):68–72.
Arango D, Ettarh RR, Holden G, Moriarty M, Brennan PC. BB-10010, an analog of macrophage inflammatory protein-1alpha, protects murine small intestine against radiation. Dig Dis Sci. 2001;46(12):2608–14.
Shiraishi K, Ishiwata Y, Nakagawa K, Yokochi S, Taruki C, Akuta T, et al. Enhancement of antitumor radiation efficacy and consistent induction of the abscopal effect in mice by ECI301, an active variant of macrophage inflammatory protein-1alpha. Clin Cancer Res. 2008;14(4):1159–66.
Aldinucci D, Borghese C, Casagrande N. The CCL5/CCR5 Axis in Cancer Progression. Cancers (Basel). 2020;12(7):1765.
Korbecki J, Grochans S, Gutowska I, Barczak K, Baranowska-Bosiacka I. CC chemokines in a tumor: a review of pro-cancer and anti-cancer properties of receptors CCR5, CCR6, CCR7, CCR8, CCR9, and CCR10 Ligands. Int J Mol Sci. 2020;21(20):7619.
Jiao X, Nawab O, Patel T, Kossenkov AV, Halama N, Jaeger D, et al. Recent advances targeting CCR5 for cancer and its role in immuno-oncology. Cancer Res. 2019;79(19):4801–7.
Timaner M, Bril R, Kaidar-Person O, Rachman-Tzemah C, Alishekevitz D, Kotsofruk R, et al. Dequalinium blocks macrophage-induced metastasis following local radiation. Oncotarget. 2015;6(29):27537–54.
Okubo M, Kioi M, Nakashima H, Sugiura K, Mitsudo K, Aoki I, et al. M2-polarized macrophages contribute to neovasculogenesis, leading to relapse of oral cancer following radiation. Sci Rep. 2016;6:27548.
Mi S, Qu Y, Chen X, Wen Z, Chen P, Cheng Y. Radiotherapy increases 12-LOX and CCL5 levels in esophageal cancer cells and promotes cancer metastasis via THP-1-derived macrophages. Onco Targets Ther. 2020;13:7719–33.
Zheng Z, Jia S, Shao C, Shi Y. Irradiation induces cancer lung metastasis through activation of the cGAS-STING-CCL5 pathway in mesenchymal stromal cells. Cell Death Dis. 2020;11(5):326.
Willenbrock F, Cox CM, Parkes EE, Wilhelm-Benartzi CS, Abraham AG, Owens R, et al. Circulating biomarkers and outcomes from a randomised phase 2 trial of gemcitabine versus capecitabine-based chemoradiotherapy for pancreatic cancer. Br J Cancer. 2021;124(3):581–6.
Dangaj D, Bruand M, Grimm AJ, Ronet C, Barras D, Duttagupta PA, et al. Cooperation between constitutive and inducible chemokines enables t cell engraftment and immune attack in solid tumors. Cancer Cell. 2019;35(6):885-900 e10.
Chen HY, Xu L, Li LF, Liu XX, Gao JX, Bai YR. Inhibiting the CD8(+) T cell infiltration in the tumor microenvironment after radiotherapy is an important mechanism of radioresistance. Sci Rep. 2018;8(1):11934.
Zheng W, Skowron KB, Namm JP, Burnette B, Fernandez C, Arina A, et al. Combination of radiotherapy and vaccination overcomes checkpoint blockade resistance. Oncotarget. 2016;7(28):43039–51.
Dillon MT, Bergerhoff KF, Pedersen M, Whittock H, Crespo-Rodriguez E, Patin EC, et al. ATR inhibition potentiates the radiation-induced inflammatory tumor microenvironment. Clin Cancer Res. 2019;25(11):3392–403.
Christersdottir T, Pirault J, Gistera A, Bergman O, Gallina AL, Baumgartner R, et al. Prevention of radiotherapy-induced arterial inflammation by interleukin-1 blockade. Eur Heart J. 2019;40(30):2495–503.
Piryani SO, Kam AYF, Vu UT, Chao NJ, Doan PL. CCR5 Signaling promotes murine and human hematopoietic regeneration following ionizing radiation. Stem Cell Reports. 2019;13(1):76–90.
Korbecki J, Kojder K, Siminska D, Bohatyrewicz R, Gutowska I, Chlubek D, et al. CC chemokines in a tumor: a review of pro-cancer and anti-cancer properties of the ligands of receptors CCR1, CCR2, CCR3, and CCR4. Int J Mol Sci. 2020;21(21):8142.
Halvorsen EC, Hamilton MJ, Young A, Wadsworth BJ, LePard NE, Lee HN, et al. Maraviroc decreases CCL8-mediated migration of CCR5(+) regulatory T cells and reduces metastatic tumor growth in the lungs. Oncoimmunology. 2016;5(6): e1150398.
Mou T, Xie F, Zhong P, Hua H, Lai L, Yang Q, et al. MiR-345-5p functions as a tumor suppressor in pancreatic cancer by directly targeting CCL8. Biomed Pharmacother. 2019;111:891–900.
Barbai T, Fejos Z, Puskas LG, Timar J, Raso E. The importance of microenvironment: the role of CCL8 in metastasis formation of melanoma. Oncotarget. 2015;6(30):29111–28.
Farmaki E, Chatzistamou I, Kaza V, Kiaris H. A CCL8 gradient drives breast cancer cell dissemination. Oncogene. 2016;35(49):6309–18.
Yan C, Luo L, Urata Y, Goto S, Li TS. Nicaraven reduces cancer metastasis to irradiated lungs by decreasing CCL8 and macrophage recruitment. Cancer Lett. 2018;418:204–10.
Ogilvie P, Bardi G, Clark-Lewis I, Baggiolini M, Uguccioni M. Eotaxin is a natural antagonist for CCR2 and an agonist for CCR5. Blood. 2001;97(7):1920–4.
Johrer K, Zelle-Rieser C, Perathoner A, Moser P, Hager M, Ramoner R, et al. Up-regulation of functional chemokine receptor CCR3 in human renal cell carcinoma. Clin Cancer Res. 2005;11(7):2459–65.
Zhu F, Liu P, Li J, Zhang Y. Eotaxin-1 promotes prostate cancer cell invasion via activation of the CCR3-ERK pathway and upregulation of MMP-3 expression. Oncol Rep. 2014;31(5):2049–54.
Miyagaki T, Sugaya M, Murakami T, Asano Y, Tada Y, Kadono T, et al. CCL11-CCR3 interactions promote survival of anaplastic large cell lymphoma cells via ERK1/2 activation. Cancer Res. 2011;71(6):2056–65.
Park JY, Kang YW, Choi BY, Yang YC, Cho BP, Cho WG. CCL11 promotes angiogenic activity by activating the PI3K/Akt pathway in HUVECs. J Recept Signal Transduct Res. 2017;37(4):416–21.
Siva S, MacManus M, Kron T, Best N, Smith J, Lobachevsky P, et al. A pattern of early radiation-induced inflammatory cytokine expression is associated with lung toxicity in patients with non-small cell lung cancer. PLoS ONE. 2014;9(10): e109560.
Cuneo KC, Devasia T, Sun Y, Schipper MJ, Karnak D, Davis MA, et al. Serum levels of hepatocyte growth factor and CD40 ligand predict radiation-induced liver injury. Transl Oncol. 2019;12(7):889–94.
Lee EJ, Kim JW, Yoo H, Kwak W, Choi WH, Cho S, et al. Single high-dose irradiation aggravates eosinophil-mediated fibrosis through IL-33 secreted from impaired vessels in the skin compared to fractionated irradiation. Biochem Biophys Res Commun. 2015;464(1):20–6.
Bostrom M, Kalm M, Eriksson Y, Bull C, Stahlberg A, Bjork-Eriksson T, et al. A role for endothelial cells in radiation-induced inflammation. Int J Radiat Biol. 2018;94(3):259–71.
Huber MA, Kraut N, Addicks T, Peter RU. Cell-type-dependent induction of eotaxin and CCR3 by ionizing radiation. Biochem Biophys Res Commun. 2000;269(2):546–52.
Takemura N, Kurashima Y, Mori Y, Okada K, Ogino T, Osawa H, et al. Eosinophil depletion suppresses radiation-induced small intestinal fibrosis. Sci Transl Med. 2018;10(429):eaan0333.
Liu Y, Cai Y, Liu L, Wu Y, Xiong X. Crucial biological functions of CCL7 in cancer. PeerJ. 2018;6: e4928.
Zhang M, Yang W, Wang P, Deng Y, Dong YT, Liu FF, et al. CCL7 recruits cDC1 to promote antitumor immunity and facilitate checkpoint immunotherapy to non-small cell lung cancer. Nat Commun. 2020;11(1):6119.
Johnston CJ, Williams JP, Okunieff P, Finkelstein JN. Radiation-induced pulmonary fibrosis: examination of chemokine and chemokine receptor families. Radiat Res. 2002;157(3):256–65.
Inoue T, Fujishima S, Ikeda E, Yoshie O, Tsukamoto N, Aiso S, et al. CCL22 and CCL17 in rat radiation pneumonitis and in human idiopathic pulmonary fibrosis. Eur Respir J. 2004;24(1):49–56.
Zhu B, Zou L, Cheng X, Lin Z, Duan Y, Wu Y, et al. Administration of MIP-3alpha gene to the tumor following radiation therapy boosts anti-tumor immunity in a murine model of lung carcinoma. Immunol Lett. 2006;103(2):101–7.
Yang Y, Cui J, Gao F, Li B, Liu C, Zhang P, et al. Whole body irradiation induces cutaneous dendritic cells depletion via NF-kappaB activation. Cell Physiol Biochem. 2013;32(1):200–9.
Liu C, Lin J, Zhao L, Yang Y, Gao F, Li B, et al. Gamma-ray irradiation impairs dendritic cell migration to CCL19 by down-regulation of CCR7 and induction of cell apoptosis. Int J Biol Sci. 2011;7(2):168–79.
Rodel F, Hofmann D, Auer J, Keilholz L, Rollinghoff M, Sauer R, et al. The anti-inflammatory effect of low-dose radiation therapy involves a diminished CCL20 chemokine expression and granulocyte/endothelial cell adhesion. Strahlenther Onkol. 2008;184(1):41–7.
Li H, Chen X, Zeng W, Zhou W, Zhou Q, Wang Z, et al. Radiation-enhanced expression of CCL22 in nasopharyngeal carcinoma is associated with CCR4(+) CD8 T cell recruitment. Int J Radiat Oncol Biol Phys. 2020;108(1):126–39.
Zhang Q, Zhu L, Wang G, Zhao Y, Xiong N, Bao H, et al. Ionizing radiation promotes CCL27 secretion from keratinocytes through the cross talk between TNF-alpha and ROS. J Biochem Mol Toxicol. 2017;31(3).
Acknowledgements
Not applicable.
Funding
This work was supported by the following grants: National Natural Science Foundation of China (Grant No. 82072597), Beijing Xisike Clinical Oncology Research Foundation (No. Y-XD2019-214, No. Y-2019Roche-149).
Author information
Authors and Affiliations
Contributions
LW collected published articles and wrote the manuscript. QC put forward the idea of the review. JJ revised the content of the article. YC and QJ edited the manuscript. All author read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, L., Jiang, J., Chen, Y. et al. The roles of CC chemokines in response to radiation. Radiat Oncol 17, 63 (2022). https://doi.org/10.1186/s13014-022-02038-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13014-022-02038-x