
Abstract
Background
Granular cell or Abrikossoff’s tumors are usually benign however rare malignant forms concern 1 to 3% of cases reported. Pelvic locations are exceptional.
Case presentation
We report a case of a 43-years-old patient who had a benign Abrikossoff’s tumor localized in the right femoral triangle diagnosed at the biopsy. The patient underwent a surgical tumorectomy and inguinal lymph nodes resection. Histologically, the tumor showed enough criteria to give diagnosis of malignancy: nuclear pleomorphism, tumor cell spindling, vesicular nuclei with large nucleoli. Moreover, five lymph nodes were metastatic. Immunohistochemistry findings confirmed the diagnosis of granular cell tumor which is positive for S100 protein and CD68 antibodies. The mitotic index was nevertheless low with a Ki67 labeling index of 1–2%. A large surgical revision with an inguinal curage following radiotherapy were decided on oncology committee. Adjuvant radiotherapy on the tumor bed and right inguinal area of 50 Gy in conventional fractionation was delivered with the aim of reducing local recurrence risk. There was no recurrence on longer follow-up (10 months post radiotherapy).
Conclusions
Adjuvant radiotherapy seems an appropriate therapeutic approach, even if controversial, given that some authors report effectiveness on local disease progression.
Similar content being viewed by others


Background
Granular cell tumors (GCT), a rare entity, were first described in 1926 by Abrikossoff, at tongue location [1,2,3,4,5]. It is now known that their origin is the schwannian cells [6]. Most of them are benign [7]. GCT are most common in the fourth to sixth decade, but can appear at any age, including children and even congenital cases have been reported [8]. GCT are more frequent in women [4]. Since large series, long-term follow-up studies and oncology protocols and clinical trials on treatment of GCT are lacking, it is not possible to draw firm conclusions on optimal treatment and follow-up procedures of GCT. Local surgical excision with clear margins and Mohs micrographic surgery have been utilized for treatment of benign and malignant GCT [9,10,11]. The approach for a malignant lesion may also include regional lymph node dissection. The role of radiotherapy and chemotherapy is still uncertain, most reports describing poor response to these two therapeutic modalities [12].
We present, here an unusual case of a patient who was treated for an atypical GCT localized in the right inguinal region with surgery and radiotherapy due to the malignant lesion and the lymph node metastasis.
Case presentation
The patient is a 43-year-old woman who was admitted for the first time for a progressive non-painful, mobile mass of the right inguinal fold evolving for 7 months. The medical history of the patient included childhood asthma, chronic tonsillitis, seven pregnancies and four children, caesarean section and abortions. Pelvic ultrasound showed a heterogeneous suspicious non-circumscribed mass measuring 5 cm in its longer axis. It was localised in the right inguinal region and showed cutaneous adhesions. CT scan confirmed the presence of this inguinal mass, measuring 5.8 × 4.9 × 3.2 cm and extending within the right femoral triangle in contact with the long adductor muscle, without enhanced contrast, and without locoregional lymph node (Fig. 1).

Initial CT scan. Axial CT scan showing an inguinal non-circumscribed contour 5 cm long axis, mass adhering to skin and in contact with the adductor muscle of the deep lip
The patient underwent a chirurgical biopsy. The pathological analysis diagnosed a granular cell tumor (Abrikossoff’s tumor) without any malignant signs (absence of mitosis, necrosis and cytonuclear atypias). Tumorectomy of this inguinal mass were performed three weeks later. At the gross pathology examination, the tumoral tissue was homogeneous with a greyish stain. Its margin was not well defined and the hypodermic, dermic were involved. One lymph node was discovered and was invaded. The epidermis was not ulcerated (Fig. 2). Histologically, collagen bundles were infiltrated by cords of large, polygonal cells with inconspicuous cell membrane and homogenous finely granular cytoplasm. Nuclei were round or oval and presented large nucleoli, vesicular of dark chromatin and sometime an intranuclear vacuole. Mitosis were rare and the mitotic index was low (1 mitosis/ 10 High Power Field). There was a slight increase of the nucleo-cytoplasmic ratio. We observed no necrosis (Fig. 3). Fanburg-Smith score of malignancy was of 3: nuclear pleomorphism, tumor cell spindling, vesicular nuclei with large nucleoli. Immunohistological finding showed a cell expression of S-100 protein, vimentin, calretinin (slight), α-Inhibin, CD56, CD57, CD68 and neuron specific enolase (NSE). (Fig. 3) Cytokeratin AE1-AE3, EMA, calponin, caldesmon, desmin, smooth muscle actine, myosin, myogenin, chromogranin, synaptphysin, Neurofilament proteins, Glial fibrillary acidic protein, CD1a, renal cell carcinoma antibodies were all negative. Therapeutic marker, estrogen receptors, progesterone receptors, androgen receptor, HER2, CD117 ALK, C-MET, ROS1 and PDL-1 were negative too.

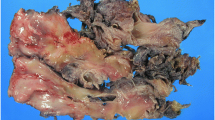
Gross pathology. Poorly defined greyish tumoral mass infiltrating hypodermic and dermic tissues. The epidermis is observed (Arrows)

Microscopy and Immunohistochemistry. Microscopic and immunohistochemical findings: a Hematoxilin and eosing stain. See nuclear pleomorphim and nuclear vacuole (arrow). b Negative expression of cytokeratine AE1-AE3. c–f Positive expression of CD56, CD57, calretinin and α-inhibin antibodies (magnification 60×)
Due to microscopic resection and the nodal status, a large surgical revision with an inguinal curage was then decided at the oncology committee. Pathological evaluation did not reveal tumor tissue in the tumorectomy region but showed metastatic invasion by granular cells in four of twelve lymph nodes, without capsular rupture. Adjuvant chemotherapy was excluded because of very low mitotic activity. To reduce the risk of local recurrence, adjuvant radiotherapy on the tumor bed and right inguinal area of 50 Gy in conventional fractionation was delivered. After 10 months after the end of radiotherapy, the patient is in complete remission. Due to the unpredictable tumor, the follow up strategy is a physical examination with a CT scan every 4 months for the first 2 post-operative years, then every 6 months for up 5 years and yearly thereafter, as a sarcoma.
Discussion
Granular cell tumors are rare (0.019 to 0.03% of all cancers) [5]. GCT are most common in the fourth to sixth decade, but can appear at any age, including in children and even congenital cases have been reported [8]. The male/female proportion varies but GCT are more frequent in women [1, 2, 13]. Granular cell tumors mainly develop at the mucocutaneous region. GCT have been reported in many different locations, most commonly in the head and neck region [14], skin [15], subcutaneous and soft tissue [16, 17] and also in the breast [9], thyroid [18], mediastinum [19], respiratory tract [20], gastrointestinal tract [11, 21], female and male genitalia [22, 23], urinary system [24] and peripheral/central nervous system [25]. Among the few pelvic locations, there are some cases in the vulva [23]. No case of inguinal location or within the femoral triangle has been described in the literature.
Clinically, granular cell tumor is generally a protruding nodule, measuring between 1 and 2 cm, painless and firm on palpation. Overlying skin and mucosa may be normal, greyish, yellowish or ulcerated in case of trauma [26]. Tumors with benign behavior, with or without atypical morphology, represent the majority of cases, while malignant GCT with documented metastatic disease comprise less than 3% of GCT. These tumors grow more rapidly and have the potential to metastasize. The most common sites for metastatic spread are the regional lymph nodes, lung, liver and bones.
Currently, neurogenic origin of granular cell tumors is the most likely hypothesis [27]. Existence of tumor connections with nerve branches, as well as the S100 neuroectodermal protein, NSE and myelin proteins expressions, support this theory [8, 26, 28].
Rare malignant forms represent 1 to 3% of cases, clinically suspected by a size greater than 4 cm, necrotic or hemorrhagic plaques or rapid growth [29]. Histologically, Funburg-Smith et al. have developed a more precise classification with six criteria (tumor necrosis, fusiform cells, vesicular nucleus with large nucleoli, mitotic index greater than 2 for ten fields, high nucleocytoplasmic ratio and pleomorphism) [26, 23]. At least three of these criteria are enough to consider the granular tumor as malignant. In our case, invasion of the locoregional lymph nodes allowed to assert the malignancy.
Other authors suggest that rapid growth, tumor size greater than 4 cm, mitoses and high Ki67 (greater than 20%) are in favor of malignancy [13]. Malignant forms require an extension assessment to search for lymphatic or systemic secondary localizations (lung, liver, bone). Indeed, pulmonary secondary injuries are frequently described, and locoregional lymph nodes involvement goes with most malignant tissue lesions. In our case, extension assessment by chest-abdomen-pelvic computerized tomography showed no secondary injury.
Any suspicious soft tissue lesion should be examined by MRI before surgical resection. Standard treatment for granular cell tumors is surgical, requiring complete resection with healthy margins; incomplete resection requires surgical revision (high risk of tumor recurrence) [30]. In malignant forms, safety margins of 2 to 3 cm are certainly recommended because of the risks of local recurrence (32 to 41% of the cases) and metastatic (50 to 63% of the cases) [23, 31]. Benefit of adjuvant radiotherapy, proposed by some authors, is very controversial [12]. However, some observations report radiotherapy efficiency in case of local recurrence or inoperable metastases [27]. In the previous case reports, radiotherapy was use in adjuvant intention after surgery in 6 cases, [12, 27, 32,33,34,35] and after local recurrence in 3 cases [36,37,38]. After adjuvant radiation 2 patients of 6 had developed a local recurrence in field and 1 of them with distant metastasis.
Due to regional lymph node metastases, our patient received adjuvant radiotherapy with dose of 50 Gy to treat the tumor bed and the lymph node region. Moreover, benefit of chemotherapy is not proven, several cases testify to the low chemosensitivity of granular cell tumor. In this case, radiotherapy combined with surgical excision has produced local control, good cosmetic and preserved function. Adjuvant radiotherapy may be appropriate for patients with GCT thought clinically and pathologically to be at high risk of local recurrence or metastasis.
Conclusion
In conclusion, Abrikossoff’s tumor, although benign in a large majority of cases, must alert as to its potential malignancy. With preferential localizations, this tumor can grow anywhere. Since chemotherapy has not been effective, local adjuvant radiotherapy after a large excision, seem to be an appropriate therapeutic approach in malignant granular cell tumors.
Abbreviations
- Gy:
-
Gray
- NSE:
-
neuron specific enolase
References
Fanburg-Smith JC, Meis-Kindblom JM, Fante R, Kindblom LG. Malignant granular cell tumor of soft tissue: diagnostic criteria and clinicopathologic correlation. Am J Surg Pathol juill. 1998;22(7):779–94.
Tsuchida T, Okada K, Itoi E, Sato T, Sato K. Intramuscular malignant granular cell tumor. Skeletal Radiol févr. 1997;26(2):116–21.
Lack EE, Worsham GF, Callihan MD, Crawford BE, Klappenbach S, Rowden G, et al. Granular cell tumor: a clinicopathologic study of 110 patients. J Surg Oncol. 1980;13(4):301–16.
Billeret Lebranchu V. Granular cell tumor. Epidemiology of 263 cases. Arch Anat Cytol Pathol. 1999;47(1):26–30.
Machado I, Cruz J, Lavernia J, Llombart-Bosch A. Solitary, multiple, benign, atypical, or malignant: the « granular cell tumor » puzzle. Virchows Arch Int J Pathol mai. 2016;468(5):527–38.
Ordóñez NG, Mackay B. Granular cell tumor: a review of the pathology and histogenesis. Ultrastruct Pathol août. 1999;23(4):207–22.
Ordóñez NG. Granular cell tumor: a review and update. Adv Anat Pathol juill. 1999;6(4):186–203.
Yuwanati M, Mhaske S, Mhaske A. Congenital granular cell tumor - a rare entity. J Neonatal Surg juin. 2015;4(2):17.
Papalas JA, Wylie JD, Dash RC. Recurrence risk and margin status in granular cell tumors of the breast: a clinicopathologic study of 13 patients. Arch Pathol Lab Med juill. 2011;135(7):890–5.
Niknam R, Bagheri Lankarani K, Geramizadeh B. Diagnosis and treatment of esophageal granular cell tumor: a case report and review of the literature. Case Rep Gastrointest Med. 2017;2017:1071623.
Chen W, Zheng X, Jin L, Pan X, Ye M. Novel diagnosis and treatment of esophageal granular cell tumor: report of 14 cases and review of the literature. Ann Thorac Surg janv. 2014;97(1):296–302.
Rosenthal SA, Livolsi VA, Turrisi AT. Adjuvant radiotherapy for recurrent granular cell tumor. Cancer 15 févr. 1990;65(4):897–900.
Nasser H, Ahmed Y, Szpunar SM, Kowalski PJ. Malignant granular cell tumor: a look into the diagnostic criteria. Pathol res Pract. 15 mars. 2011;207(3):164–8.
Vered M, Carpenter WM, Buchner A. Granular cell tumor of the oral cavity: updated immunohistochemical profile. J Oral Pathol Med Off Publ Int Assoc Oral Pathol Am Acad Oral Pathol janv. 2009;38(1):150–9.
Lazar AJF, Fletcher CDM. Primitive nonneural granular cell tumors of skin: clinicopathologic analysis of 13 cases. Am J Surg Pathol. juill. 2005;29(7):927–34.
Rekhi B, Jambhekar NA. Morphologic spectrum, immunohistochemical analysis, and clinical features of a series of granular cell tumors of soft tissues: a study from a tertiary referral cancer center. Ann Diagn Pathol juin. 2010;14(3):162–7.
Imanishi J, Yazawa Y, Saito T, Shimizu M, Kawashima H, Ae K, et al. Atypical and malignant granular cell tumors in Japan: a Japanese musculoskeletal oncology group (JMOG) study. Int J Clin Oncol août. 2016;21(4):808–16.
Baloch ZW, Martin S, Livolsi VA. Granular cell tumor of the thyroid: a case report. Int J Surg Pathol juill. 2005;13(3):291–4.
Ponce FA, Killory BD, Wait SD, Theodore N, Dickman CA. Endoscopic resection of intrathoracic tumors: experience with and long-term results for 26 patients. J Neurosurg Spine mars. 2011;14(3):377–81.
Joung M-K, Lee YJ, Chung C-U, Lee JE, Jung SS, Kim SY, et al. A case of granular cell tumor of the trachea. Korean J Intern Med juin. 2007;22(2):101–5.
Dusoleil A, Lahoud S, Condat B, Bellanger J, Fritsch J, Berthelot P, et al. Granular cell tumor of the biliary tract and bile ducts. Gastroenterol Clin Biol. sept 1999;23(8–9):993–4.
Laskin WB, Fetsch JF, Davis CJ, Sesterhenn IA. Granular cell tumor of the penis: clinicopathologic evaluation of 9 cases. Hum Pathol mars. 2005;36(3):291–8.
Cheewakriangkrai C, Sharma S, Deeb G, Lele S. A rare female genital tract tumor: benign granular cell tumor of vulva: case report and review of the literature. Gynecol Oncol mai. 2005;97(2):656–8.
Olaya M, Vicioso L, Hierro I, Quiñonero A, Matilla A, López-Beltrán A. Granular cell tumor of the bladder: a case report. Anal Quant Cytopathol Histopathol oct. 2013;35(5):289–93.
Papachristou DJ, Palekar A, Surti U, Cieply K, McGough RL, Rao UNM. Malignant granular cell tumor of the ulnar nerve with novel cytogenetic and molecular genetic findings. Cancer Genet Cytogenet mai. 2009;191(1):46–50.
Brown AC, Audisio RA, Regitnig P. Granular cell tumour of the breast. Surg Oncol juin. 2011;20(2):97–105.
Tawfiq N, Sabri S, Saiss K, Bouchbika Z, Benchekroun N. Jouhadi H, et al. [granular cell tumor: report of a complicated vulvar localization of pulmonary metastases]. Cancer Radiother J Soc Francaise Radiother Oncol. nov. 2013;17(7):671–4.
Tison C, Doubremelle M, Le Rhun M, Cuillière P. Le Borgne J [Rectal Abrikossof tumor: a rare location] Gastroenterol Clin Biol mars. 2005;29(3):289–90.
Rodríguez MS, Reija MFG, Rodilla IG. Primary clear cell carcinoma of parotid gland: case report and review of literature. J Oral Maxillofac Pathol JOMFP janv. 2013;17(1):101–5.
Tawfiq N, Sabri S, Saiss K, Bouchbika Z, Benchekroun N. Jouhadi H, et al. [granular cell tumor: report of a complicated vulvar localization of pulmonary metastases]. Cancer Radiother J Soc Francaise Radiother Oncol nov. 2013;17(7):671–4.
Aksoy S, Abali H, Kilickap S, Harputluoglu H, Erman M. Metastatic granular cell tumor: a case report and review of the literature. Acta Oncol Stockh Swed. 2006;45(1):91–4.
Simsir A, Osborne BM, Greenebaum E. Malignant granular cell tumor: a case report and review of the recent literature. Hum Pathol août. 1996;27(8):853–8.
Bouraoui S, Letaief H, Mestiri H, Chadly-Debbiche A, Ben Zineb S. Haouet S, et al. [malignant granular cell tumors. Report of a case of anal localization]. Ann Pathol avr. 1999;19(2):151–2.
Ramos PC, Kapp DS, Longacre TA, Teng NNH. Malignant granular cell tumor of the vulva in a 17-year-old: case report and literature review. Int J Gynecol Cancer Off J Int Gynecol Cancer Soc sept. 2000;10(5):429–34.
Berg JC, Tewari KS, Del Rosario R, Berman ML. Unusual presentation of a malignant granular cell tumor of the pelvis: case report and literature review. Gynecol Oncol juill. 2003;90(1):224–30.
Gartmann H. Malignant granular cell tumor. Hautarzt Z Dermatol Venerol Verwandte Geb janv. 1977;28(1):40–4.
Urabe A, Imayama S, Yasumoto S, Nakayama J, Hori Y. Malignant granular cell tumor. J Dermatol mars. 1991;18(3):161–6.
Wang J, Zhu X-Z. Zhang R-Y. [malignant granular cell tumor: a clinicopathologic analysis of 10 cases with review of literature]. Zhonghua Bing Li Xue Za Zhi déc. 2004;33(6):497–502.
Availability of data and materials
The datasets used and analysed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
Authors’ contributions: CM, SSV wrote the manuscript. CG: reviewed the pathology. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Written informed consent was obtained from the patient for publication of this case report and accompanying images. A copy of the written consent is available for review by Editor-in-Chief of this journal.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Marchand Crety, C., Garbar, C., Madelis, G. et al. Adjuvant radiation therapy for malignant Abrikossoff’s tumor: a case report about a femoral triangle localisation. Radiat Oncol 13, 115 (2018). https://doi.org/10.1186/s13014-018-1064-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13014-018-1064-4