
Abstract
Background
Traditional CRISPR/Cas9 systems that rely on U6 or U3 snRNA promoters (RNA polymerase III-dependent promoters) can only achieve constitutive gene editing in plants, hampering the functional analysis of specifically expressed genes. Ribozyme-mediated CRISPR/Cas9 systems increase the types of promoters which can be used to transcribe sgRNA. Therefore, such systems allow specific gene editing; for example, transcription of the artificial gene Ribozyme-sgRNA-Ribozyme (RGR) is initiated by an RNA polymerase II-dependent promoter. Genetic transformation is indispensable for editing plant genes. In certain plant species, including pyrethrum, genetic transformation remains challenging to do, limiting the functional verification of novel CRISPR/Cas9 systems. Thus, this study’s aim was to develop a simple Agrobacterium rhizogenes-mediated hairy root transformation system to analyze the function of a ribozyme-mediated CRISPR/Cas9 system in pyrethrum.
Results
A hairy root transformation system for pyrethrum is described, with a mean transformation frequency of 7%. Transgenic hairy roots transformed with the pBI121 vector exhibited significantly increased beta-glucuronidase staining as a visual marker of transgene expression. Further, a ribozyme-based CRISPR/Cas9 vector was constructed to edit the TcEbFS gene, which catalyzes synthesis of the defense-related compound (E)-β-farnesene in pyrethrum. The vector was transferred into the hairy roots of pyrethrum and two stably transformed hairy root transgenic lines obtained. Editing of the TcEbFS gene in the hairy roots was evaluated by gene sequencing, demonstrating that both hairy root transgenic lines had DNA base loss at the editing target site. Gas chromatography–mass spectrometry showed that the (E)-β-farnesene content was significantly decreased in both hairy root transgenic lines compared with the empty vector control group. Altogether, these results show that RGR can be driven by the CaMV35S promoter to realize TcEbFS gene editing in pyrethrum hairy roots.
Conclusion
An A. rhizogenes-mediated hairy root transformation and ribozyme-mediated CRISPR/Cas9 gene editing system in pyrethrum was established, thereby facilitating gene editing in specific organs or at a particular developmental stage in future pyrethrum research.
Similar content being viewed by others


Background
CRISPR systems can modify target genes in plants by substituting, removing, inserting, or knocking out DNA [1]. In these systems, a single guide RNA (sgRNA) forms a complex with the Cas9 protein and guides the Cas9 protein to bind to a specific target DNA for gene editing [2]. Since the first successful application of a CRISPR system [3], this technology has been continuously refined and optimized to improve editing efficiency and accuracy [4, 5]. In recent years, CRISPR systems have been successfully applied to food crops, such as maize (Zea mays L.) [6]; cash crops, such as cotton (Gossypium hirsutum) [7]; fruits, including strawberry (Fragaria ananassa) [8]; and medicinal crops, including Chinese wolfberry (Lycium ruthenicum) [9], and is considered a new and effective tool for use in plant gene function research [10].
In the CRISPR/Cas9 system, the transcription of sgRNA is key. Transcription in eukaryotes is conducted by three RNA polymerase enzymes [11]: RNA polymerase I transcribes large rRNAs; RNA polymerase II synthesizes mRNAs; and RNA polymerase III transcribes small noncoding RNAs, such as the U6 snRNA [12]. Accordingly, their corresponding promoters were also divided into three types, namely RNA polymerase I-dependent promoter (pol I promoter), RNA polymerase II-dependent promoter (pol II promoter) and RNA polymerase III-dependent promoter (pol III promoter) [13,14,15]. Specific gene silencing requires specific promoters [16]; however, specific promoters are usually driven by RNA polymerase II and cannot be directly used for sgRNA transcription because the RNA undergoes extensive processing and modification at both ends to become a mRNA [17]. Previous researchers have used a root-cap-specific promoter or stomatal lineage-specific promoter (both are pol II promoters) to initiate Cas9 mRNA transcription, with the AtU6-26 promoter (a pol III promoter belonging to non-tissue specific constitutive promoters [17]) used to initiate sgRNA transcription, and thereby achieved tissue-specific gene editing in Arabidopsis [18]. Yet the mRNA can be transported long distances to other tissues [19], and constitutive expression of sgRNA, driven by the U6 or U3 snRNA promoters, may bind to the Cas9 protein outside of the specific tissue of interest, resulting in gene editing occurring in unintended locations.
The development of the ribozyme-mediated CRISPR/Cas9 system represents a remarkable improvement, because it can theoretically enhance gene editing’s specificity by using the pol II promoter. The common feature of this system is that ribozyme sequences, such as the hammerhead-type ribozyme (HH) [20] or hepatitis delta virus ribozyme (HDV) [21], are added at the ends of an sgRNA sequence, and when transcribed into RNA, the covalent bond of RNA is broken via the self-cleavage property of such ribozymes, with additional structures at both ends of RNA separated with ribozyme. In the first report [17], an ADH1 promoter (a pol II promoter of yeast) was used to drive the expression of a ribozyme-sgRNA-ribozyme (RGR) artificial gene, and an sgRNA was generated by ribozyme self-cleavage, facilitating site-specific gene editing.
Pyrethrum (Tanacetum cinerariifolium) is a perennial herbaceous plant belonging to the Asteraceae family that has been cultivated for centuries to extract botanical insecticides from its dried flowers [22]. (E)-β-farnesene is a volatile organic compound expressed in different tissues of pyrethrum that is considered to be related to insect resistance [23,24,25]. Although the (E)-β-farnesene synthase gene (TcEbFS) has been identified, and (E)-β-farnesene synthase expressed in vitro can catalyze (E)-β-farnesene synthesis [24], it has been postulated that TcEbFS has diverse functions in different pyrethrum tissues [23,24,25]. Therefore, further characterization of TcEbFS function and tissue-specific gene editing in pyrethrum is desirable.
To conduct gene editing research, an efficient transformation system is essential; however, no transformation system suitable for pyrethrum has yet been developed. Agrobacterium rhizogenes-mediated hairy root transformation is considered a rapid, simple, and highly efficient method that enables rapid characterization of improved CRISPR/Cas9 systems in hairy roots [26,27,28,29]. This report describes an A. rhizogenes-mediated hairy root transformation system that can quickly verify the effect of gene editing in pyrethrum, and tests the gene editing effect of a ribozyme-mediated CRISPR/Cas9 system using a pol II promoter in this context. Compared with traditional gene editing methods that rely on U6 or U3 snRNA promoters, this versatile method has unique advantages for use in pyrethrum breeding and gene function research that could be applied in future studies.
Results
A strategy for A. rhizogenes-mediated hairy root transformation system in pyrethrum
Leaves from pyrethrum tissue culture plantlets were used as explants, infected with A. rhizogenes, and roots appeared on explants after 20 days (Fig. 1a). Root tips were excised and placed on culture medium with the root tip upward (Fig. 1b). When the root tips were placed upside down for 7 days, two kinds of roots were identified: one type exhibiting geotropism and another without geotropism (Fig. 1c). Specific primers for the rol B gene on the Ri plasmid were used for PCR amplification of DNA extracted from the two types of root as template, and roots without geotropism were identified as containing the rol B gene (Fig. 1). Therefore, in the present study, geotropism was used to select hairy roots carrying the Ri plasmid.

Induction of pyrethrum hairy roots. a Twenty days after transformation, roots emerged from the infected leaf explants. b Root tips were placed on culture medium and culture dishes oriented vertically to ensure that the root tips were upward. c After a week of cultivation, two different root types appeared: one exhibiting geotropism and the other without geotropism. d PCR detection of rol B in the two types of root. M, marker; 1, bacterial plasmid DNA control; 2–4, DNA from roots exhibiting geotropism; 5–7, DNA from roots without geotropism. Scale bars in a–c, 1.0 cm
After screening for roots without geotropism, root tips were cut off and placed in kanamycin screening medium to select hairy roots carrying Ti plasmids (Fig. 2a). The resulting proportion of explants inducing roots was 51% ± 4% (100 explants, four replicate experiments), that of roots without geotropism was approximately 26% (10 roots, five replicate experiments), and the mean proportion of resistant roots was 7% (50 roots, four replicate experiments). Next, resistant roots were cut into 1-cm-long segments and inoculated in the same medium. These resistant roots continued to grow and could grow lateral roots (Fig. 2b). To further determine whether T-DNA was transferred into the hairy roots, resistant roots were stained with X-gluc reagent and Lines 1–5 were dyed blue (Fig. 2c). Next, DNA samples were extracted from these roots for PCR analysis. All roots were found to contain the NPTII gene fragment from the pBI121 vector (Fig. 2d). These results indicated that hairy roots stably transformed with pBI121 could be obtained via geotropism and kanamycin screening, and that the CaMV35S promoter was suitable for high level foreign gene expression in pyrethrum hairy roots.

Transformation of pyrethrum mediated by A. rhizogenes harboring the pBI121 vector. a Resistant roots grown on selective medium containing kanamycin (10 mg/L) for 20 days. b Resistant hairy roots were excised and transferred onto selective medium containing kanamycin (10 mg/L) to propagate for approximately 60 days. c Transgenic roots stained with X-gluc reagent. d PCR detection of NPT II in samples from 60-day-old resistant roots. M, marker; +, bacterial plasmid DNA control; −, wild type plant control. e Magnified image of dyed roots from Line 5. Numbers represent different lines, as follows: 1, Line 1; 2, Line 2; 3, Line 3; 4, Line 4; 5, Line 5. Scale bars in a–c and e, 5.0 mm
Analysis of gene editing in transgenic hairy roots
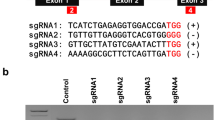
The control group (CK) hairy root line (Fig. 3a), in which the CaMV35S promoter drives GUS; and the resistant hairy root lines, line A (Fig. 3b) and line B (Fig. 3c), in which the CaMV35S promoter drives RGR and Cas9, were obtained. DNA was extracted from resistant roots, and PCR analysis showed that an RGR fragment of approximately 229 bp was amplified from the positive control, as well as lines A and B, whereas the amplicon was not detected in CK hairy root samples. These results confirm that lines A and B were transgenic roots carrying the CRISPR/Cas9 vector (Fig. 3d). Furthermore, PCR amplification followed by restriction enzyme digestion was used to detect editing of target sites in lines A and B. In the control group, two fragments of 162 and 272 bp were produced after the complete digestion by BstbI, while the TcEbFS gene fragments amplified from lines A and B were not completely cleaved by BstbI (Fig. 3e). These findings indicated that no TcEbFS gene fragments in the CK group were edited and contained the BstbI restriction site (TT/CGAA), while the TcEbFS gene fragments from lines A and B contained edited TcEbFS gene fragments, in which the restriction site was destroyed. The TcEbFS gene fragment amplified from the CK group, and the BstbI resistant TcEbFS gene fragments from lines A and B were then sequenced. The results showed that the TcEbFS sequence of the CK group was identical to the published TcEbFS sequence (MF682058), whereas the BstbI-resistant TcEbFS gene fragments from lines A and B carried mutations, including nucleotide insertions and deletions causing amino acid deletions and frame shifts (Fig. 3f).

Analysis of gene editing of transgenic hairy roots 1 month after subculture. a Control group (CK). b Line A. c Line B. d Detection of the RGR sequence by PCR. M, marker; +, ribozyme-based CRISPR/Cas9 vector; −, CK; 1, line A; 2, line B. e PCR amplification followed by restriction enzyme digestion detection of target sites: M, marker; 1−, CK TcEbFS amplicon; 1+, CK TcEbFS fragments following digestion with by BstbI; 2−, Line A TcEbFS amplicon; 2+, Line A TcEbFS fragments following digestion with BstbI; 3−, Line B TcEbFS amplicon; 3+, Line B TcEbFS fragments following digestion with BstbI. f DNA sequencing confirming TcEbFS gene mutations in pyrethrum hairy roots. The net length of insertions and/or deletions are presented in the column to the left. Numbers in parentheses indicate the number of clones. g Diagram showing the TcEbFS genomic locus with the sgRNA targeting site and primer locations (arrows). Scale bars in a–c, 1.0 cm
(E)-β-farnesene content in hairy roots
The (E)-β-farnesene content of CK, line A, and line B was determined by GC–MS. That of the CK group was 87.19 ± 2.89 µg/g (fresh weight (FW)) (Fig. 4b), that of line A was 49.77 ± 1.05 µg/g (FW) (Fig. 4c), and that of the line B was 57.13 ± 0.52 µg/g (FW) (Fig. 4d). These results showed that pyrethrum hairy roots’ (E)-β-farnesene content was very high, being significantly lower in lines A and B s than the CK group (Fig. 4e).

(E)-β-farnesene content in hairy roots. a (E)-β-farnesene standard (1 ng/µl). b Control group (CK). c Line A. d Line B. e (E)-β-farnesene content in CK, line A, and line B. f 7.850 min mass spectra
Discussion
A rapid and highly efficient A. rhizogenes-mediated hairy root transformation system for pyrethrum
To date, there has been only one report of pyrethrum transformation using an A. tumefaciens-mediated system based on leaf explants; however, such methods are unsuitable for functional verification of novel CRISPR/Cas9 systems, because they are time-consuming and extremely inefficient [30]. Therefore, in the present study a new pyrethrum transformation method for the rapid verification of novel CRISPR/Cas9 systems was established. It achieved a high hairy root transformation frequency (7%) and the process took only 49 days (2 days of co-culture, 20 days of root formation, 7 days of screening for roots without geotropism, and 20 days of screening for kanamycin-resistant roots).
Transgenic hairy roots are transformed by pRi and pTi [31]; therefore, both plasmids’ characteristics were used to screen for transgenic hairy roots. In a study of Brassica napus, pRi-transformed roots were less sensitive to gravity than normal roots [32]. This phenomenon of reduced geotropism was also reported in sweet potato hairy roots [33] and is caused by aux genes carried on pRi that interfere with hairy root gravitropism [34]. Therefore, this characteristic loss of gravitropism was employed here to screen for hairy roots transformed by pRi. Kanamycin resistance was used to screen for transgenic hairy roots transformed by pTi. Due to the co-transformation of pRi and pTi, transgenic hairy roots are chimeras (Additional file 1: Fig. S1). This phenomenon has also been reported in Arachis hypogaea, Taraxacum hybernum, and Althaea officinalis [35,36,37]. Nevertheless, this method is suitable only for qualitative, but not for quantitative, research; hence, gene editing efficiency could not be determined in the later experiments.
Advantages of the ribozyme-mediated CRISPR/Cas9 system for pyrethrum research
High expression of sgRNA in cells is critical to the success of gene editing [38]. In general, sgRNA is expressed from the U6 or U3 promoter in the CRISPR/Cas9 system; however, as in most plants, no U6 or U3 promoters have been identified in pyrethrum, making it difficult to select a functional U6 or U3 promoter for use in CRISPR/Cas9 systems [17]. Therefore, gene editing methods using a pol II promoter were sought. Post-transcriptional modification of RNA at both ends influences the ability of an sgRNA to guide Cas9 to cut a target site [39, 40]. Hence, separation of the modified structures at both ends of an sgRNA and conversion of the mRNA into sgRNA is desirable. Using the characteristic of ribozyme self-cleavage, an sgRNA with editing ability can be separated from the cap and tail structures of mRNA. This method is referred to as the RGR strategy and was first successfully applied to yeast [17], and later to zebrafish (Danio Rerio) [41], human cells [42], mice [43], Plasmodium yoelii [44], Arabidopsis thaliana [45], and Oryza sativa [46]. Moreover, some pol II promoters are more efficient than the U6 or U3 promoter [47]. The results reported here are similar to those of other studies, suggesting the RGR strategy is generally applicable to eukaryotes. Applying the RGR strategy reduces the difficulty of promoter selection for use in pyrethrum gene editing.
In addition to the easy availability of the promoter, ribozyme-based CRISPR/Cas9 technology has other advantages. U6 and U3 promoters mediate constitutive expression in plants, and the CRISPR/Cas9 technology that relies on U6 or U3 to initiate sgRNA transcription cannot be used for specific editing [46]. The RGR strategy increases the types of promoters, allowing CRISPR/Cas9 system to use a wide variety of pol II promoters besides U6 and U3 promoters [17]. Therefore, this technology could greatly promote pyrethrum’s gene function research. For example, pyrethrum has three types of (E)-β-farnesene with reported differences in release mode, as follows: large release after leaf damage, large release during flower development, and sustained and stable low-level release [23, 24]. The (E)-β-farnesene released in these three modes can interfere with one another, rendering it challenging to study the biological significance of (E)-β-farnesene in each mode. The best way to investigate the particular biological functions of these types of (E)-β-farnesene is to silence each separately, and applying the RGR strategy could theoretically realize such tissue-specific gene editing [17].
This technology is also promising for breeding of pyrethrum disease resistance. The most damaging foliar disease of pyrethrum is ray blight caused by Stagonosporopsis tanaceti [48]. The emergence of CRISPR/Cas9 technology provides a new approach to address this problem, and this technology has worked to achieve resistance to pathogens in various crops [49,50,51]; however, to reduce the negative effects of a transgene on the whole plant, the most efficient way to confer resistance is to restrict its expression only to infected cells. Pathogen-induced promoters are unique and valuable tools for engineering resistance to plant disease [52, 53]. Therefore, integration of the CRISPR/Cas9 system and pathogen-induced promoters (pol II promoter) to develop a pathogen-inducible gene editing system is of great significance for devising pyrethrum resistance to S. tanaceti, as this method can restrict gene editing only to infected cells and reduce metabolic disruption caused by gene editing, thus minimizing the impact on plant growth and development.
Conclusions
This work established a hairy root transformation system mediated by A. rhizogenes in pyrethrum. Using this system, a self-cleaving ribozyme-mediated genome editing method using the CaMV35S promoter was evaluated. This system increased the types of promoters capable of transcribing sgRNA and can potentially be expanded to other RNA polymerase II-dependent promoters with strong capacity for expression in specific tissues or under certain conditions, to achieve site-specific gene editing in pyrethrum.
Methods
Plant material
Pyrethrum clone W99 was obtained from the Key Laboratory for Biology of Horticultural Plants, Ministry of Education, College of Horticulture & Forestry Sciences, Huazhong Agricultural University. Clones were inoculated on Murashige and Skoog (MS) solid medium for 30 days after subculture, and their leaves used for transformation. Cultures were maintained at 25 °C ± 2 °C, with a photoperiod of 16/8 h at light intensity 40 μmol m−2 s−1.
Construction of ribozyme-based CRISPR/Cas9 vectors
According to Liang et al.’s methods [54], a target site with a PAM sequence in exon 2 of the TcEbFS gene was screened (Fig. 3g). The target site contained a BstBI (TT/CGA) restriction site, which can be used for an editing analysis by the restriction enzyme site-loss method [55]. The plasmids, pRGEB32-GhU6.7-NPT2 and pGTR4, were gifted from the National Key Laboratory of Crop Genetic Improvement, Huazhong Agricultural University [56]. Ribozyme-based CRISPR/Cas9 vectors were constructed using the TcEbFS gene target sequence, based on pRGEB32-GhU6.7-NPT2. The process of vector construction is illustrated in Fig. 5. The primers used in the experiment are listed in Additional file 2: Table S1. A plasmid with the correct sequence was transferred into A. rhizogenes MSU440.

Ribozyme-based CRISPR/Cas9 vector construction process. The hammerhead-type ribozyme sequence is labeled HH, and the hepatitis delta virus ribozyme sequence is labeled HDV. Step 1: The 229-bp fragment encompassing the ‘SacI + HH + sgRNA + HDV + BamHI’ fragment was obtained. Next, this fragment was digested by SacI and BamHI. Step 2: The 288-bp fragment encompassing the ‘BamHI + NOS + Aaul’ fragment was obtained. Next, this fragment was digested by BamHI. Step 3: The 853-bp fragment encompassing the ‘HindIII + 35S + SacI’ fragment was obtained. Next, this fragment was digested by SacI. Step 4: Their digestion products were mixed at the same molar concentration and ligated. Next, using the ligated DNA mixture as a template, a 1346-bp fragment encompassing the ‘HindIII + 35S + HH + sgRNA + HDV + NOS + Aaul’ was obtained. Step 5: The pRGEB32-GhU6.7-NPT2 vector and the ‘HindIII + 35S + HH + sgRNA + HDV + NOS + Aaul’ fragment were ligated. Next, the plasmid was digested by Aaul. Step 6: The 853 bp fragment ‘Aaul + 35S + Aaul’ was obtained. Next, this fragment was digested by Aaul. Step 7: The digested plasmid in step 5 and the digested fragment ‘Aaul + 35S + Aaul’ were connected
Establishment of a stable hairy root transformation system
Leaves were excised from pyrethrum clone W99 and infected with A. rhizogenes MSU440 containing the pBI121 vector. The process of Agrobacterium infection is now well described in the literature [30]. First, hairy roots stably transformed with Ri plasmids were screened. Then, infected leaves were placed on solid 1/2 MS medium containing 400 mg/L cefotaxime. After 20 days of cultivation in the dark, roots appeared in the pyrethrum leaves and root tips were excised, placed horizontally on culture medium, and culture dishes oriented vertically, to ensure that the root tip was pointing upward. After 1 week of cultivation in the dark, two different hairy roots appeared: those exhibiting geotropism and those showing no geotropism. DNA was extracted from these two different types of root, and three agrobacterium-free samples extracted from each type of root. PCR-based amplification of vir genes was used to test for agrobacterium contamination [57, 58]; primers specific for the TetR gene were used, where detection of TetR indicated agrobacterium contamination and the sample was discarded.
Uncontaminated samples were used to analyze the rol B gene. Specific primers, rol B-up and rol B-low, were designed based on the rol B gene sequence of the A. rhizogenes’ Ri plasmid, according to the description by Xiao et al. [59]; theoretically, these primers amplify a 450-bp gene fragment from hairy roots stably transformed with Ri plasmids. Next, hairy roots stably transformed with Ti plasmids were screened. Roots without geotropism were placed horizontally in 1/2 MS medium containing kanamycin (10 mg/L) and cefotaxime (400 mg/L). After 20 days in the dark, numbers of elongated roots were counted. Kanamycin-resistant roots were inoculated in 1/2 MS medium containing kanamycin (10 mg/L) and cefotaxime (400 mg/L), and sub-cultured every 60 days. Kanamycin-resistant roots were histochemically stained using the beta-glucuronidase (GUS) Staining Kit (Coolaber, China) to determine the stability of the transformation and visualize transgene expression.
The specific primers, NPTII-up and NPTII-low, were designed based on the pBI121 plasmid NPTII gene sequence. DNA from kanamycin resistant roots served as a template for the PCR analysis. Theoretically, a 750-bp gene fragment could be amplified from hairy roots stably transformed with the pBI121 vector. Introduction of the TcEbFS ribozyme-based CRISPR/Cas9 vector was conducted under the same conditions (Fig. 6). To confirm the presence of transgenes in putatively transformed hairy roots, HH-sgRNA-HDV sequence specific primers (A-end and A-low) were designed. A PCR product of 229 bp was expected from positive hairy roots, and positive samples according to the PCR analysis were used for subsequent experiments.

Pyrethrum hairy root transformation workflow using leaf explants. At 0 days: Agrobacterium infection. Agrobacterium cell density with an OD600 = 0.5. Co-cultivation began. At 2 days: Co-cultivation was over. Roots’ induction began. At 22 days: Roots’ induction was over. Screening of roots transformed with Ri began. At 29 days: Screening of roots transformed with Ri was over. Screening of roots transformed with Ti began. At 49 days: Screening of roots transformed with Ti was over. Sub-cultivation began
Detection of TcEbFS editing
A TcEbFS gene fragment (approximately 456 bp) was amplified using specific primers (F-up and F-low) across the editing site. As the fragment contained the TT/CGAA restriction site, the TcEbFS gene amplification product from the control group could be recognized and completely digested into 162-bp and 272-bp fragments by BstbI; however, amplified TcEbFS gene products from edited transgenic hairy roots contained fragments could not be recognized and cleaved by BstbI. Undigested bands were ligated into the pCloneEZ vector, and the vector was transferred into Escherichia coli DH5α. Clones were randomly selected and sequenced to detect gene mutations, and eight clones were selected from each line. Hairy roots transformed with the pBI121 vector alone served as the control.
Detection of (E)-β-farnesene in gene edited hairy roots
Gas chromatography-mass spectrometry (GC–MS) was used to analyze secondary metabolites of hairy root lines containing the ribozyme-based CRISPR/Cas9 vector grown on 1/2 MS medium for 60 days. After freezing in liquid nitrogen and grinding using a mortar and pestle, hairy roots (500 mg) were transferred into a 5-mL tube, and 1 mL of methyl-tert-butyl ether (MTBE) containing tetradecane (0.01 ng/mL) added as an internal standard. The tube was vortexed for 3 min at maximum speed, incubated at 24 °C with a rotation speed of 50 rpm, then dried using Na2SO4. For GC–MS analysis, 1-µL sample aliquots were injected into a GC/MS-QP 2010 Ultra instrument (Shimadzu Corporation, Japan) with an HP-5 MS column. The relative concentration of (E)-β-farnesene was calculated by the area ratio of the (E)-β-farnesene peak to that of the internal standard.
Hairy roots transformed with the pBI121 vector were used as a control. Helium (1.4 mL/min) was used as a carrier gas. The injection temperature was set at 240 °C. The oven temperature program went as follows: initial temperature 50 °C, followed by a ramp from 50 to 150 °C at a rate of 20 °C/min, held for 1 min, and then from 150 to 180 °C at 20 °C/min, held for 1 min, and finally increased to 300 °C at 30 °C/min, held for 5 min. Identification of volatiles was conducted by comparing their retention times and mass fragmentation values with those reported in the literature, and the NIST (2017) and PESTEI_3.lib databases. Spectral data were also compared to (E)-β-farnesene standards (Sigma, Germany), these diluted to 1 ng/μL with MTBE.
Statistical analysis
Data obtained from all experiments are presented as means ± standard error, and separations performed using the least significant difference (LSD) test. Percentage data were subjected to angular transformation and evaluated by ANOVA using SPSS software v17.0. Values with different letters show significant differences at P ≤ 0.05 (LSD).
Availability of data and materials
The datasets generated and analyzed in the current study are available from the corresponding author on reasonable request.
References
Ali Z, Abul-faraj A, Piatek M, Mahfouz MM. Activity and specificity of TRV-mediated gene editing in plants. Plant Signal Behav. 2015;10(10):e1044191.
Nishimasu H, Ran FA, Hsu PD, Konermann S, Shehata SI, Dohmae N, et al. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell. 2014;156(5):935–49.
Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao YJ, Pirzada ZA, et al. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 2011;471:602–7.
Chen SM, Yao YF, Zhang YC, Fan GF. Discovery, development and off-target detection. Cell Signal. 2020;70:109577.
Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157:1262–78.
Gentzel IN, Park CH, Bellizzi M, Xiao GQ, Gadhave KR, Murphree C, et al. A CRISPR/dCas9 toolkit for functional analysis of maize genes. Plant Methods. 2020;16:9.
Ramadan M, Alariqi M, Ma Y, Li Y, Liu Z, Zhang R, et al. Efficient CRISPR/Cas9 mediated pooled-sgRNAs assembly accelerates targeting multiple genes related to male sterility in cotton. Plant Methods. 2021;17:16.
Wilson FM, Harrison K, Armitage AD, Simkin AJ, Harrison RJ. CRISPR/Cas9-mediated mutagenesis of phytoene desaturase in diploid and octoploid strawberry. Plant Methods. 2019;15:45.
Wang W, Liu J, Wang H, Li T, Zhao H. A highly efficient regeneration, genetic transformation system and induction of targeted mutations using CRISPR/Cas9 in Lycium ruthenicum. Plant Methods. 2021;17:71.
Lyzenga WJ, Pozniak CJ, Kagale S. Advanced domestication: harnessing the precision of gene editing in crop breeding. Plant Biotechnol J. 2021;19:660–70.
Roeder RG, Rutter W. Multiple forms of DNA-dependent RNA polymerases in eukaryotic organisms. Nature. 1969;224:234.
Cramer P, Armache KJ, Baumli S, Benkert S, Brueckner F, Buchen C, Damsma GE, Dengl S, Geiger SR, Jasiak AJ, et al. Structure of eukaryotic RNA polymerases. Annu Rev Biophys. 2008;37:337–52.
Lewis BA, Burlingame AL, Myers SA. Human rna polymerase II promoter recruitment in vitro is regulated by o-linked n-acetylglucosaminyltransferase (OGT). J Biol Chem. 2016;291:14056–61.
Trivedi A, Vilalta A, Gopalan S, Johnson DL. TATA-binding protein is limiting for both TATA-containing and TATA-lacking RNA polymerase III promoters in drosophila cells. Mol Cell Biol. 1996;16:6909–16.
Brenz Verca MS, Weber P, Mayer C, Graf C, Refojo D, Kuhn R, et al. Development of a species-specific RNA polymerase I-based shRNA expression vector. Nucl Acids Res. 2007;35:e10.
Nakatsuka T, Pitaksutheepong C, Yamamura S, Nishihara M. Induction of differential flower pigmentation patterns by RNAi using promoters with distinct tissue-specific activity. Plant Biotechnol Rep. 2007;1:251–7.
Gao YB, Zhao YD. Self- processing of ribozyme-flanked RNAs into guide RNAs in vitro and in vivo for CRISPR-mediated genome editing. J Integr Plant Biol. 2014;56:343–9.
Decaestecker W, Buono RA, Pfeiffer ML, Vangheluwe N, Jacobs TB. CRISPR-TSKO: a technique for efficient mutagenesis in specific cell types, tissues, or organs in Arabidopsis. Plant Cell. 2019;31:2868–87.
Okita TW, Choi SB. mRNA localization in plants: targeting to the cell’s cortical region and beyond. Curr Opin Plant Biol. 2002;5:553–9.
Pley HW, Flaherty KM, Mckay DB. Three-dimensional structure of a hammerhead ribozyme. Nature. 1994;372(6501):68–74.
Ferré-D’Amaré AR, Zhou K, Doudna J. Crystal structure of a hepatitis delta virus ribozyme. Nature. 1998;395:567.
Pavela R. History, presence and perspective of using plant extracts as commercial botanical insecticides and farm products for protection against insects—a review. Plant Protect Sci. 2016;52:229–41.
Kikuta Y, Ueda H, Nakayama K, Katsuda Y, Ozawa R, Takabayashi J, et al. Specific regulation of pyrethrin biosynthesis in Chrysanthemum cinerariaefolium by a blend of volatiles emitted from artificially damaged conspecific plants. Plant Cell Physiol. 2011;52:588–96.
Li J, Hu H, Mao J, Yu L, Stoopen G, Wang M, et al. Defense of pyrethrum flowers: repelling herbivores and recruiting carnivores by producing aphid alarm pheromone. New Phytol. 2019;223:1607–20.
Liu F, Wang Q, Xu P, Andreazza F, Valbon WR, Bandason E, et al. A dual-target molecular mechanism of pyrethrum repellency against mosquitoes. Nat Commun. 2021;12:2553.
Carrijo J, Illa-Berenguer E, LaFayette P, Torres N, Aragao FJL, Parrott W, et al. Two efficient CRISPR/Cas9 systems for gene editing in soybean. Transgenic Res. 2021;30:239–49.
Di YH, Sun XJ, Hu Z, Jiang QY, Song GH, Zhang B, et al. Enhancing the CRISPR/Cas9 system based on multiple GmU6 promoters in soybean. Biochem Biophys Res Comm. 2019;519:819–23.
Triozzi P, Schmidt HW, Dervinis C, Kirst M, Conde D. Simple, efficient and open-source CRISPR/Cas9 strategy for multi-site genome editing in Populus tremula × alba. Tree Physiol. 2021;41:2216–27.
Zheng N, Li T, Dittman JD, Su JB, Li RQ, Gassmann W, et al. CRISPR/Cas9-based gene editing using egg cell-specific promoters in Arabidopsis and Soybean. Front Plant Sci. 2020;11:800.
Mao J, Cao L-Y, Kong L-F, Jongsma MA, Wang C-Y. An Agrobacterium-mediated transformation system of pyrethrum (Tanacetum cinerariifolium) based on leaf explants. Sci Hortic. 2013;150:130–4.
Bahramnejad B, Naji M, Bose R, Jha S. A critical review on use of Agrobacterium rhizogenes and their associated binary vectors for plant transformation. Biotechnol Adv. 2019;37:14.
Legue V, Vilaine F, Tepfer M, Perbal G. Modification of the gravitropic response of seedling roots of rapeseed (Brassica napus) transformed by Agrobacterium rhizogenes A4. Physiol Plant. 1994;91:559–66.
Otani M, Mii M, Handa T, Kamada H, Shimada T. Transformation of sweet-potato (Ipomoea batatas (L.) Lam.) plants by Agrobacterium rhizogenes. Plant Sci. 1993;94:151–9.
Failla MC, Maimone F, Depaolis A, Costantino P, Cardarelli M. The non-conserved region of cucumopine-type Agrobacterium rhizogenes T-DNA is responsible for hairy root induction. Plant Mol Biol. 1990;15:747–53.
Knyazev A, Kuluev B, Fateryga A, Yasybaeva G, Chemeris A. Aseptic germination and Agrobacterium rhizogenes-mediated transformation of Taraxacum hybernum Steven. Plant Tissue Cult Biotechnol. 2017;27:141–51.
Liu S, Su L, Liu S, Zeng X, Zheng D, Hong L, et al. Agrobacterium rhizogenes-mediated transformation of Arachis hypogaea: an efficient tool for functional study of genes. Biotechnol Biotechnol Equip. 2016;30:869–78.
Parisa T, Akbar SA. Influence of different Agrobacterium rhizogenes strains on hairy root induction and analysis of phenolic and flavonoid compounds in marshmallow (Althaea officinalis L.). Biotech. 2018;8:351.
Xie K, Minkenberg B, Yang Y. Boosting CRISPR/Cas9 multiplex editing capability with the endogenous tRNA-processing system. Proc Natl Acad Sci USA. 2015;112:3570–5.
Haurwitz RE, Jinek M, Wiedenheft B, Zhou KH, Doudna JA. Sequence- and structure-specific RNA processing by a CRISPR endonuclease. Science. 2010;329:1355–8.
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–21.
Ho L, Mei N, Ingham PW. Ribozyme Mediated gRNA Generation for In Vitro and In Vivo CRISPR/Cas9 Mutagenesis. Plos One. 2016;11(11):e0166020.
Gao ZL, Herrera-Carrillo E, Berkhout B. Improvement of the CRISPR-Cpf1 system with ribozyme-processed crRNA. RNA Biol. 2018;15:1458–67.
Xu L, Zhao L, Gao Y, Xu J, Han R. Empower multiplex cell and tissue-specific CRISPR-mediated gene manipulation with self-cleaving ribozymes and tRNA. Nucleic Acids Res. 2017;45:e28.
Walker MP, Lindner SE. Ribozyme-mediated, multiplex CRISPR gene editing and CRISPR interference (CRISPRi) in rodent-infectious Plasmodium yoelii. J Biol Chem. 2019;294:9555–66.
Zhang Y, Zhang Y, Qi Y. Plant gene knockout and knockdown by CRISPR-Cpf1 (Cas12a) systems. In: Qi Y, editor. Methods Mol Biol 2019. p. 245–56.
He Y, Zhang T, Yang N, Xu M, Yan L, Wang L, et al. Self-cleaving ribozymes enable the production of guide RNAs from unlimited choices of promoters for CRISPR/Cas9 mediated genome editing. J Genet Genomics. 2017;44:469–72.
Cermak T, Curtin SJ, Gil-Humanes J, Cegan R, Kono TJY, Konecna E, et al. A multipurpose toolkit to enable advanced genome engineering in plants. Plant Cell. 2017;29:1196–217.
Pethybridge SJ, Gent DH, Groom T, Hay FS. Minimizing crop damage through understanding relationships between pyrethrum phenology and ray blight disease severity. Plant Dis. 2013;97:1431–7.
Prabhukarthikeyan SR, Parameswaran C, Keerthana U, Teli B, Jagannadham PTK, Cayalvizhi B, et al. Understanding the plant-microbe interactions in CRISPR/Cas9 era: indeed a sprinting start in marathon. Curr Genomics. 2020;21:429–43.
Xu ZY, Xu XM, Gong Q, Li ZY, Li Y, Wang S, et al. Engineering broad-spectrum bacterial blight resistance by simultaneously disrupting variable tale-binding elements of multiple susceptibility genes in rice. Mol Plant. 2019;12:1434–46.
Zaynab M, Sharif Y, Fatima M, Afzal MZ, Aslam MM, Raza MF, et al. CRISPR/Cas9 to generate plant immunity against pathogen. Microb Pathog. 2020;141:103996.
Sathoff AE, Dornbusch MR, Miller SS, Samac DK. Functional analysis of medicago-derived pathogen-induced gene promoters for usage in transgenic alfalfa. Mol Breed. 2020;40:66.
Zhen W, Chen X, Liang HB, Hu YL, Gao Y, Lin ZP. Enhanced late blight resistance of transgenic potato expressing glucose oxidase under the control of pathogen-inducible promoter. Chin Sci Bull. 2000;45:1982–6.
Liang G, Zhang HM, Lou DJ, Yu DQ. Selection of highly efficient sgRNAs for CRISPR/Cas9-based plant genome editing. Sci Rep. 2016;6:21451.
Iaffaldano B, Zhang YX, Cornish K. CRISPR/Cas9 genome editing of rubber producing dandelion Taraxacum kok-saghyz using Agrobacterium rhizogenes without selection. Ind Crop Prod. 2016;89:356–62.
Wang P, Zhang J, Sun L, Ma Y, Xu J, Liang S, et al. High efficient multisites genome editing in allotetraploid cotton (Gossypium hirsutum) using CRISPR/Cas9 system. Plant Biotechnol J. 2018;16:137–50.
Siritunga D, Sayre RT. Generation of cyanogen-free transgenic cassava. Planta. 2003;217:367–73.
Trontin JF, Harvengt L, Garin E, Vernaza ML, Arancio L, Hoebeke J, et al. Towards genetic engineering of maritime pine (Pinus pinaster Ait.). Ann Forest Sci. 2002;59:687–97.
Xiao X, Ma F, Chen C-L, Guo W-W. High efficient transformation of auxin reporter gene into trifoliate orange via Agrobacterium rhizogenes-mediated co-transformation. PCTOC. 2014;118:137–46.
Acknowledgements
The authors thank Shuangxia Jin (Huazhong Agricultural University, China) for providing the pRGEB32-GhU6.7-NPT2 and pGTR4 plasmids, and also thank other lab members for their useful discussion and help.
Funding
This work was financially supported by the National Key Research and Development Project (Grant no. 2019YFD1001500), Fundamental Research Funds for the Central Universities (Grant no. 2662019FW016), and National Natural Science Foundation of China (NSFC, Grant no. 32160718).
Author information
Authors and Affiliations
Contributions
JWL, JL, and CYW conceived the research. JWL and TZ wrote the manuscript. QY prepared the materials. JWL, ZZX, and LZ conducted the experiments. TZ analyzed the data. JWL, TZ, ZZX, JJL, HH, RRZ, JL, and CYW revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Fig. S1.
Transgenic roots (pBI121) stained with x-gluc reagent showing transgenic hairy root chimeras.
Additional file 2: Table S1.
Primers and construction process of ribozyme-based CRISPR/Cas9 vectors used in the experiment.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, JW., Zeng, T., Xu, ZZ. et al. Ribozyme-mediated CRISPR/Cas9 gene editing in pyrethrum (Tanacetum cinerariifolium) hairy roots using a RNA polymerase II-dependent promoter. Plant Methods 18, 32 (2022). https://doi.org/10.1186/s13007-022-00863-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13007-022-00863-5