
Abstract
Background
Perivascular epithelioid cell neoplasms (PEComas) are a family of mesenchymal tumors with features of both smooth muscle and melanocytic differentiation. A subset of PEComas demonstrate rearrangements involving the TFE3 (Xp11) locus. Xp11 translocation PEComa is a rare neoplasm with special clinicopathological features and a more aggressive behavior. We recently encountered a case of Xp11 translocation PEComa occurring in the testis, with SFPQ⁃TFE3 rearrangement.
Case presentation
A 57-year-old male touched a mass in his testis incidentally. MRI revealed a 10 mm diameter mass in the right testis. The patient underwent radical orchiectomy. Gross examination revealed a well-demarcated mass from the surrounding testicular tissue. Microscopically, the tumor mainly displayed nested or sheet-like architecture separated by delicate fibrovascular septa. The tumor cells exhibited marked nuclear atypia and pleomorphism. Immunohistochemistry showed that the tumor cells were strongly positive for cathepsin-K, HMB45 and TFE3. Molecular analysis revealed SFPQ⁃TFE3 gene fusion. Thus, it was diagnosed as primary Xp11 translocation PEComa of the testis.
Conclusions
The present case reports primary Xp11 translocation PEComa of the testis for the first time, which to our knowledge has not been described in the literature in this anatomic site, where it could potentially be problematic in diagnosis.
Similar content being viewed by others


Introduction
Perivascular epithelioid cell tumors (PEComas) are a group of rare mesenchymal neoplasms composed of perivascular epithelioid cells exhibiting both melanocytic and muscular differentiation. The majority of PEComas show genetic alterations in TSC2 (the result of a loss of heterozygosity in the TSC2 gene) and, less commonly, TSC1 [1]. A small subset of PEComas harbour TFE3 (Xp11) gene fusions, which show unique characteristics, as initially described by Argani et al. [2] in 2010 and considered to represent an unusual variant of PEComa. PEComas of the urinary system and male genital organs are extremely rare mesenchymal neoplasms that have been described mainly in the kidney but may also occur in other locations. Approximately 40 cases of PEComas of the urinary bladder and 4 cases of PEComas of the prostate have been reported in the literature; only 9 cases of bladder PEComas and 1 case of prostatic PEComas with TFE3 rearrangement have been described in the literature [3,4,5]. We present a case of Xp11 translocation PEComa occurring in the testis harboring SFPQ⁃TFE3 rearrangement in a 57-year-old male, which, to the best of our knowledge, represents the first case of primary Xp11 translocation PEComa of the testis.
Case presentation
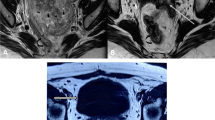
A 57-year-old male touched a mass in his testis incidentally by himself a month ago, without pain, redness and swelling or urinary irritation symptoms. There was no evidence of family history, tuberous sclerosis complex (TSC) or tumor elsewhere on comprehensive and systematic work-up. Magnetic resonance imaging (MRI) of the pelvic cavity revealed a 10 mm diameter mass in the right testis (Fig. 1A). No distant organ or lymph node metastases were detected. Radical orchiectomy was performed. Grossly, the mass was well demarcated from the surrounding testicular tissue and had a solid, yellow-tan and fleshy cut surface. Microscopically, the tumor showed limited infiltration into the surrounding testicular tissue. The tumor mainly displayed nested or sheet-like architecture separated by delicate fibrovascular septa (Fig. 1B). Many of the tumor cells exhibited marked nuclear atypia and pleomorphism (Fig. 1C), significant variation in cell size and multinucleation. Multinucleate giant cells (Fig. 1D) and spider-like cells (Fig. 1E) were readily seen, with abundant granular epsinophilic cytoplasm and prominent nucleoli. A few giant cells had coarse cytoplasmic stippling (basophilic to purple cytoplasmic granules). The tumor cells in the local region of the tumor had clear-to-granular cytoplasm, distinct cell borders and round or oval nuclei with small visible nucleoli (Fig. 1F). Thick stromal collagen bands could be seen between some neoplastic cell nests. Melanin pigment was focally identified. The mitotic count was up to 2 per 10 high-power fields, and necrosis was not found.

A MRI revealed a mass in the right testis (red arrow). B The tumor showed limited infiltration into the surrounding testicular tissue and displayed nested or sheet-like architecture separated by delicate fibrovascular septa. C Epithelioid tumor cells exhibited marked nuclear atypia and pleomorphism, with abundant clear to granular epsinophilic cytoplasm. D Multinucleate giant tumour cell with melanin pigment (red arrow). E Spider-like tumour cell (red arrow). F The tumor cells had clear-to-granular cytoplasm, distinct cell borders and round or oval nuclei with small visible nucleoli
Immunohistochemical staining showed that the tumor cells were strongly positive for cathepsin-K (Fig. 2A), HMB45 (Fig. 2B) and TFE3 (Fig. 2C) and completely negative for PAX8, AE1/AE3, EMA, CD10, CK7, CK20, calretinin, inhibin, Melan A, S100, WT1, SMA, desmin, MyoD1, chromogranin, synaptophysin, CD56, CD68, D2-40, hepatocyte, GS and SALL4. The Ki-67 index was approximately 5%. The histomorphological findings and immunohistochemical profile were found to be consistent with Xp11 translocation PEComa.

The tumor cells were diffusely strongly positive for cathepsin K (A) and HMB45 (B). (C) The tumor nuclei were diffusely strongly positive for TFE3. (D) The fusion probe assay using probes centromeric to PSF and telomeric to TFE3 indicated an abnormal signal pattern consistent with the SFPQ-TFE3 fusion
To confirm the presence of a TFE3 gene rearrangement, the break-apart fluorescence in situ hybridization (FISH) assay was performed on formalin-fixed, paraffin-embedded tissue sections. The case exhibited TFE3 rearrangement with Xp11.2 translocation. The fusion probe assay using probes centromeric to SFPQ and telomeric to TFE3 revealed fusion signals (Fig. 2D), thus confirming the diagnosis of Xp11 translocation PEComa.
Since the tumor was so small and the cut margin of the resected sample was tumor-free, adjuvant chemotherapy was not planned. Although the follow-up time was relatively limited, the patient was alive and well without local recurrence and distant metastasis 18 months after the surgery.
Discussion and conclusions
The TFE3 gene, which is located on the short arm of chromosome Xp11.2, is a member of the micropathalima-associated transcription factor family along with TFEB, TFEC, and MiTF. The TFE3-rearranged tumors mainly include Xp11 translocation renal cell carcinoma (RCC), alveolar soft part sarcoma (ASPS), PEComas/melanotic Xp11 neoplasm or Xp11 neoplasm with melanocytic differentiation, a subset of epithelioid hemangioendotheliomas and some ossifying fibromyxoid tumors [6]. Xp11 translocation PEComa reported in the literature occurred in younger patients, with a female predominance. The tumor arose in a wide range of anatomic sites, such as the kidney, ovary, pelvis, nasal cavity, adrenal gland, stomach, appendix, cervix, prostate, soft tissue, small intestine, uterus, eye and mesentery [3,4,5, 7,8,9,10,11]. The tumor sizes ranged from 1 to 17 cm (mean: 5.5 cm). Our case occurred in a relatively older male, which was the first case of a primary Xp11 translocation PEComa of the testis. The tumor size of the present case was small, with the largest dimension measuring 1.0 cm.
Because of its rarity along with nonspecific clinical presentations and imaging features, Xp11 translocation PEComa is very difficult to accurately diagnose preoperatively. It also causes diagnostic challenges to practical pathologists. Attention to its characteristic morphology, including epithelioid cells with clear or granular eosinophilic cytoplasm arranged in nested or sheet-like architectures and separated by delicate vascular networks, with the judicious use of proper immunohistochemical markers, including melanocytic markers (HMB45/MelanA), PAX8, SMA, TFE3 and cathepsin K, can help to arrive at the correct diagnosis. However, an accurate diagnosis requires molecular genetic analyses to confirm the presence of TFE3 rearrangement. The SFPQ-TFE3 gene fusion has been found to be the most common gene fusion in this rare entity. A much smaller proportion of cases have been reported with NONO-TFE3 fusion [5, 12]. Rare examples demonstrated a DVL2, MED15 and RBMX fusion partner [8,9,10, 13].
The main differential diagnosis of our case included Leydig cell tumor (LCT), metastatic Xp11 translocation RCC and ASPS. LCT is a neoplasm composed of cells with abundant eosinophilic cytoplasm, uniform and round tumor cells, round nuclei with prominent central nucleoli and minimal cytological atypia. Focal areas of the present tumor morphologically resembled LCT. LCT is typically positive for inhibin, calretinin, CD99 and melan A. However, nuclear TFE3 is absent. Another important differential diagnosis is with metastatic Xp11 translocation RCC, which has similar morphological features and identical gene fusion. However, Xp11 translocation PEComa is a mesenchymal tumor different from Xp11 translocation RCC. Immunophenotypic differences (absence of renal tubular markers) can distinguish the neoplasm from Xp11 translocation RCC. However, not all Xp11 translocation RCC labels renal tubular markers (including PAX8), and the clinical history is important (evidence of tumors involving kidney). ASPS shares similar clinical characteristics and morphological features with Xp11 translocation PEComa. ASPS also occurs mainly in adolescents and young adults, with a female predominance. ASPS commonly involves deep soft tissues of the extremities. ASPS is composed of epithelioid cells with eosinophilic to clear cytoplasm arranged in a uniform organoid or nest-like pattern delineated by delicate fibrovascular septa. The tumor cells show central discohesion resulting in the characteristic alveolar pattern. ASPS also shows immunopositivity for cathepsin K and nuclear immunoreactivity for TFE3. However, ASPS is negative for melanocytic markers. Cytogenetically, ASPS is defined by a specific alteration, der(17)t(X;17)(p11;q25), which results in ASPSCR1-TFE3 gene fusion. ASPSCR1–TFE3 translocation is known to be the characteristic diagnostic gene fusion for ASPS among sarcomas.
Xp11 translocation PEComa has special clinicopathological features compared to conventional PEComas, such as alveolar/nested growth pattern of large epithelioid cells with clear to eosinophilic cytoplasm, lack of spindle cell or fat components, negative for SMA, TFE3 gene rearrangement and absence of TSC1/2 genetic alterations. Xp11 translocation PEComa had a more aggressive behavior. In the study by Rao et al., they summarized 24 cases of this unique entity in the literature: 15 of 24 (62.5%) cases experienced recurrence and/or metastases or died of disease [9]. A recent study on the largest series of melanotic Xp11 neoplasms showed local recurrence and distant metastatic rates of 31.8% and 31.8%, respectively [10]. Xp11 translocation PEComa with a novel RBMX-TFE3 gene fusion in a pediatric kidney presented with a fulminant clinical course (multiple pulmonary metastases at diagnosis and died of disease 3 months later) [13]. While optimal treatment for Xp11 translocation PEComa has not been established, surgical resection appears to be the mainstay of the therapy. PEComa patients with TSC mutations with locally aggressive or metastatic disease can use palliative therapy with mTOR inhibitors [14]. Recently, Schmiester et al. [15] reported a clinically aggressive TSC1-mutated PEComa with concomitant TFE3 activation. It is unclear whether TFE3-associated PEComa patients can respond to mTOR inhibitors. TFE3 transcription triggers Met receptor tyrosine kinase by direct transcriptional upregulation, leading to the activation of downstream pathways, such as the PI3K/AKT/mTOR pathway. It is possible that patients with Xp11 translocation PEComa might benefit from tyrosine kinase inhibitors.
In conclusion, we described the first case of a primary Xp11 translocation PEComa arising within the testis in a relatively older man harboring the SFPQ-TFE3 gene fusion by FISH. It is difficult to predict the prognosis, and a treatment strategy has not been established. The accumulation of further cases and long-term follow-up data are desirable.
Availability of data and materials
Not applicable.
Abbreviations
- PEComa :
-
Perivascular epithelioid cell tumor
- RCC :
-
Renal cell carcinoma
- ASPS :
-
Alveolar soft part sarcoma
- LCT :
-
Leydig cell tumor
References
WHO Classification of Tumours Editorial Board. WHO classification of Tumours·5th Edition: soft tissue and bone tumours [M]. Lyon: IARC Press; 2020. pp. 312–4.
Argani P, Aulmann S, Illei PB, et al. A distinctive subset of PEComas harbors TFE3 gene fusions. Am J Surg Pathol. 2010;34:1395–406.
Neumann NM, Haffner MC, Argani P, et al. Histopathologic characterization of bladder perivascular epithelioid cell neoplasms (PEComa): a series of 11 cases with a subset having TFE3 rearrangements. Am J Surg Pathol. 2021;45:169–77.
Vannucchi M, Minervini A, Salvi M, et al. TFE3 gene rearrangement in perivascular epithelioid cell neoplasm (PEComa) of the genitourinary tract. Clin Genitourin Cancer. 2020;18:e692–7.
Wang XT, Xia QY, Ni H, et al. Xp11 neoplasm with melanocytic differentiation of the prostate harbouring the novel NONO–TFE3 gene fusion: report of a unique case expanding the gene fusion spectrum. Histopathology. 2016;69:450–8.
Pinto K, Chetty R. Gene of the month: TFE3. J Clin Pathol. 2020;73:691–4.
Argani P, Aulmann S, Karanjawala Z, et al. Melanotic Xp11 translocation renal cancers: a distinctive neoplasm with overlapping features of PEComa, carcinoma, and melanoma. Am J Surg Pathol. 2009;33:609–19.
Argani P, Zhong M, Reuter VE, et al. TFE3-fusion variant analysis defines specific clinicopathologic associations among Xp11 translocation cancers. Am J Surg Pathol. 2016;40:723–37.
Rao Q, Shen Q, Xia QY, et al. PSF/SFPQ is a very common gene fusion partner in TFE3 rearrangement-associated perivascular epithelioid cell tumors (PEComas) and melanotic Xp11 translocation renal cancers: clinicopathologic, immunohistochemical, and molecular characteristics suggesting classification as a distinct entity. Am J Surg Pathol. 2015;39:1181–96.
Wang XT, Fang R, Zhang RS, et al. Malignant melanotic Xp11 neoplasms exhibit a clinicopathological spectrum and gene expression profiling akin to alveolar soft part sarcoma: a proposal for reclassification. J Pathol. 2020;251:365–77.
Utpatel K, Calvisi DF, Köhler G, et al. Complexity of PEComas: a diagnostic approach, molecular background, clinical management. Pathologe. 2020;41(suppl 1):9–19.
McGregor SM, Alikhan MB, John RA, et al. Melanotic PEComa of the Sinonasal Mucosa with NONO-TFE3 Fusion: an elusive Mimic of Sinonasal Melanoma. Am J Surg Pathol. 2017;41(5):717–22.
Argani P, Zhang L, Sung YS, et al. A novel RBMX-TFE3 gene fusion in a highly aggressive pediatric renal perivascular epithelioid cell tumor. Genes Chromosomes Cancer. 2019;13:10.1002/gcc.22801. https://doi.org/10.1002/gcc.22801.
Fabbroni C, Sbaraglia M, Sanfilippo R. Medical treatment of advanced malignant perivascular epithelioid cell tumors. Curr Opin Oncol. 2020;32(4):301–6.
Schmiester M, Dolnik A, Kornak U, et al. TFE3 activation in a TSC1-altered malignant PEComa: challenging the dichotomy of the underlying pathogenic mechanisms. J Pathol Clin Res. 2021;7:3–9.
Acknowledgements
The authors thank Xiaotong Wang for gene rearrangement assistance.
Funding
The authors did not receive support from any organization for the submitted work.
Huizhi Zhang, Suying Wang, Lingli Meng.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. HZ Z designed the research study and wrote the paper. SY W collected the clinicopathological data and performed some research studies. LL M performed the research and revised the paper. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The data reported here were obtained in agreement with the Ethics Committee of the Ningbo Clinical Pathology Diagnosis Center.
Consent for publication
Consent for publication was waived because the submission did not include images that might identify the recipients.
Competing interests
The authors state that there are no conflicts of interest with respect to the research, authorship, and/or publication of this article.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, H., Wang, S. & Meng, L. Primary Xp11 translocation PEComa of the testis with SFPQ⁃TFE3 rearrangement: a case report and review of the literature. Diagn Pathol 18, 6 (2023). https://doi.org/10.1186/s13000-023-01288-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13000-023-01288-x