
Abstract
Background
Neuropathic pain that caused by lesion or dysfunction of the nervous system is associated with gene expression changes in the sensory pathway. Long noncoding RNAs (lncRNAs) have been reported to be able to regulate gene expression. Identifying lncRNA expression patterns in the spinal cord under normal and neuropathic pain conditions is essential for understanding the genetic mechanisms behind the pathogenesis of neuropathic pain.
Results
Spinal nerve ligation (SNL) induced rapid and persistent pain hypersensitivity, characterized by mechanical allodynia and heat hyperalgesia. Meanwhile, astrocytes and microglia were dramatically activated in the ipsilateral spinal cord dorsal horn at 10 days after SNL. Further lncRNA microarray and mRNA microarray analysis showed that the expression profiles of lncRNA and mRNA between SNL and sham-operated mice were greatly changed at 10 days. The 511 differentially expressed (>2 fold) lncRNAs (366 up-regulated, 145 down-regulated) and 493 mRNAs (363 up-regulated, 122 down-regulated) were finally identified. The expression patterns of several lncRNAs and mRNAs were further confirmed by qPCR. Functional analysis of differentially expressed (DE) mRNAs showed that the most significant enriched biological processes of up-regulated genes in SNL include immune response, defense response, and inflammation response, which are important pathogenic mechanisms underlying neuropathic pain. 35 DE lncRNAs have neighboring or overlapping DE mRNAs in genome, which is related to Toll-like receptor signaling, cytokine–cytokine receptor interaction, and peroxisome proliferator-activated receptor signaling pathway.
Conclusion
Our findings uncovered the expression pattern of lncRNAs and mRNAs in the mice spinal cord under neuropathic pain condition. These lncRNAs and mRNAs may represent new therapeutic targets for the treatment of neuropathic pain.
Similar content being viewed by others

Background
Neuropathic pain is one of the most common chronic pain in humans and characterized by an increase in the responsiveness of nociceptive neurons in the peripheral and central nervous system (CNS) [1]. Peripheral and central sensitization represents the altered functional status of nociceptive neurons and results from changes of a vast amount of functional protein and signaling pathways in the neuron and glial cell [2, 3]. Recent pharmaceutical research and discovery activities focus on well-characterized molecular targets, such as ion channels, G-protein-coupled receptors, and kinases in neurons and glial cells localized along the nociceptive pathways, which are regarded as direct contributors to the sensitization of pain signaling systems [4, 5]. However, the transcriptional or translational regulatory mechanisms underlying the expression and functional changes of these molecules are poorly defined.
RNAs that do not code for a protein (noncoding RNAs, ncRNAs) consist of two major classes: the small ncRNAs, which include microRNAs (miRNAs) and other noncoding transcripts of less than 200 nucleotides, and long noncoding RNAs (lncRNAs), which are a novel class of non-protein coding transcripts longer than 200 nucleotides [6]. LncRNAs were initially considered as transcriptional by-products, but recent data suggest that lncRNAs can regulate gene expression via interfering with transcription, post-transcriptional processing, chromatin remodeling, miRNA sequestration, and generating small ncRNAs [7, 8]. Also, lncRNAs are involved in various aspects of cell biology and disease etiology, such as development [9], immune [10], cardiovascular disease [11], oncogenesis [12], and neurological disease [13]. LncRNAs are highly expressed in the CNS, and their expression profiles are associated with specific neuroanatomical regions, cell types, or subcellular compartments suggesting their potential functional roles in the nervous system [14–16]. It was reported that sciatic nerve resection induced differential expression of lncRNAs in dorsal root ganglia (DRG) [17]. Moreover, Zhao et al. have recently identified a functional lncRNA Kcna2, which contributed to neuropathic pain by silencing Kcna2 in DRG neurons [18]. These findings indicate the involvement of lncRNAs in neuropathic pain.
The spinal cord is responsible for receiving input from nociceptors and projecting to the brain, and plays an important role in the integration and modulation of pain-related signals. To clarify the molecular mechanisms underlying neuropathic pain and explore novel approaches for analgesic strategies, herein, we investigated the genome-wide expression of lncRNAs in the spinal cord following L5 spinal nerve ligation (SNL)-induced neuropathic pain. We found a large number of differentially expressed (DE) lncRNAs and mRNAs in the spinal cord after SNL. Among them, 39 correlated lncRNA-mRNA pairs, consisting of DE lncRNAs and mRNAs with adjacent or overlapping position relationship, were screened out. Our findings will provide new insights into the roles of lncRNAs in the regulation of neuropathic pain-associated genes.
Results
Model identification of neuropathic pain
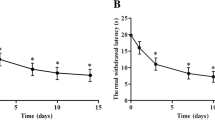
The SNL model has been widely used in the investigation of the mechanisms underlying neuropathic pain [19]. Here we also found that SNL induced rapid (1 d) and persistent (>21 d) mechanical allodynia (Figure 1a) and heat hyperalgesia (Figure 1b) in mice. We then harvested the spinal cord at 10 days (maintenance phase) after SNL and checked the expression of astrocytic marker GFAP and microglial marker IBA-1, which are known to be upregulated in the spinal cord under neuropathic pain condition [20, 21]. As shown in Figure 1c, d, GFAP expression and IBA-1 expression were both increased in the ipsilateral dorsal horn in SNL animals but not in sham-treated animals, indicating that glial activation was induced in the spinal cord by SNL.

SNL induces persistent neuropathic pain and glial activation in the spinal cord. SNL-induced rapid and persistent mechanical allodynia (a) and heat hyperalgesia (b). Data are expressed as mean ± SEM (n = 5 for each group). ***P < 0.001, two-way repeated measures ANOVA. c, d Representative images of GFAP and IBA-1 immunofluorescence in the L5 spinal cord from sham and SNL mice. GFAP and IBA-1 immunoreactive were very low in sham-treated mice, but significantly increased in the ipsilateral superficial dorsal horn at 10 days after SNL.
Overview of lncRNAs and mRNA expression profiles after SNL
We then detected the expression profiles of lncRNAs and mRNAs in the L5 spinal cord at 10 days after SNL by microarray. First, we obtained a graphically overview of the expression signatures of lncRNAs and mRNAs by using scatter plot and hierarchical clustering analyses. The scatter plots showed that a large number of lncRNAs and mRNAs were differentially expressed between SNL and sham-operated mice (Figure 2a, b). Hierarchical cluster analysis of all lncRNAs or mRNA showed that the 3 sham or 3 SNL samples were clustered together respectively, and signal intensity was consistent in sham or SNL group (Figure 2c, d). The heatmap of DE lncRNAs or mRNAs whose expression were up-regulated or down-regulated by twofold were magnified (Figure 2e, f), indicating the high level of concordance in either SNL or sham samples. These data suggest that neuropathic pain is associated with the changes of lncRNAs and mRNAs in the spinal cord.

SNL results in the expression profiling changes of lncRNA and mRNA. Scatter plot comparing global lncRNA (a) or mRNA (b) gene expression profiles in the spinal cord between the SNL and sham mice. Green lines indicate twofold differences in either direction in lncRNA and mRNA expression. Heat map showing hierarchical clustering of overall lncRNAs (c) or mRNA (d) expression pattern of reliably measured probe sets. Heat map showing hierarchical clustering of LncRNAs (e) or mRNA (f), whose expression changes were more than twofold. In clustering analysis, up- and down-regulated genes are colored in red and green, respectively.
Differentially expressed lncRNAs and mRNAs
We further analyzed differentially expressed (DE) lncRNAs using significance analysis of microarrays method, following the criteria q-value <0.05, and fold change >2. The results showed that 511 lncRNAs, containing 366 up-regulated and 145 down-regulated, were significantly changed in SNL group, comparing with the sham group. The most up-regulated lncRNAs were: uc009egw.1, Speer7-ps1, MM9LINCRNAEXON12113+, ENSMUST00000118074, and uc009nzx.1, of which uc009egw.1 showed the largest up-regulation (Log2 fold change = 7,332.4243). The most down-regulated lncRNAs were: AK045739, AK020832, AK047380, ENSMUST00000171761 and uc008dwx.1, of which AK045739 showed the largest down-regulation (Log2 fold change = −45.320816). Detailed information including the top 20 up-regulated and 20 down-regulated lncRNAs was listed in Table 1.
In the DE mRNAs, there are 493 genes whose mRNA change was more than twofold, and the number of up-regulated (363) mRNAs was larger than down-regulated (122) mRNAs in SNL. These DE mRNAs contain many known genes involving in pain processing, including Cacna1g (calcium channel, voltage-dependent, T type, alpha 1G subunit, 16.0978 fold increase) [22], Trpv1 (transient receptor potential cation channel, subfamily V, member 1, 9.31-fold increase) [23], Ccl5 (chemokine (C-C motif) ligand 5, 3.93-fold increase) [24], Cx3cr1 (chemokine (C-X3-C) receptor 1, 2.51-fold increase) [25], and Irf5 (interferon regulatory factor 5) [26]. Besides, a lot of other genes, whose roles in pain have not been identified, were dramatically changed. Further analysis showed that 39 genes whose expression were changed >tenfold, including 38 up-regulated genes and 1 down-regulated gene, such as Sprr1a (small proline-rich protein 1A, 148.7-fold), Anxa10 (annexin A10, 76.3-fold), and Kng1 (kininogen 1, 38.4-fold); 66 genes whose expression was changed between 5- and 10-fold, including 64 up-regulated and 2 down-regulated genes. Detailed information about the top 20 up-regulated and 20 down-regulated mRNAs was listed in Table 2.
Real-time quantitative PCR (qPCR) validation of lncRNA and mRNA expression
To validate the reliability of the microarray results and also analyze the temporal changes of lncRNA and mRNA expression after SNL, the up-regulated lncRNAs including Speer7-ps1 and uc007pbc.1, the down-regulated lncRNAs, including ENSMUST00000171761 and ENSMUST00000097503, the up-regulated mRNA Cyp2d9, and the down-regulated mRNA Mnx1 were randomly selected and analyzed by qPCR. The spinal cord tissues were collected from naïve animals, and SNL animals at 1, 3, 10, and 21 days. Speer7-ps1 and uc007pbc.1, which are intergenic lncRNAs, were both significantly increased at 10 days and peaked at 21 days (Figure 3a, b). ENSMUST00000171761 and ENSMUST00000097503 are antisense overlap and bidirectional lncRNA with matching gene Tagap (T-cell activation Rho GTPase-activating protein) and Zfp236 (zinc finger protein 236). They were significantly decreased at 10 days and persisted to 21 days (Figure 3c, d). Cyp2d9, a member of cytochrome P450, family 2, subfamily d, was increased more than 12-fold at 10 days (Figure 3e). Mnx1 is a sequence-specific DNA binding transcription factor. It decreased from 1 to 21 days (Figure 3f). In addition, the fold changes of these lncRNAs and mRNAs detected by qPCR at SNL 10 days were consistent with the results from microarray (Figure 3g), further supporting the reliability of the array data.

QPCR validations of four deregulated lncRNAs and two deregulated mRNA in the spinal cord from SNL mice. The expressions of lncRNA Speer7-ps1 (a), lncRNA Uc007pbc.1 (b), lncRNA ENSMUST00000171761 (c), and lncRNA ENSMUST00000097503 (d) were significantly deregulated at 10 and 21 days after SNL. e The expression of Cyp2d9 mRNA was markedly up-regulated at 10 days after SNL. f The expression of Mnx1 mRNA was significantly down-regulated at 1, 3, 10 and 21 days after SNL. One-way ANOVA followed by Tukey’s multiple comparison test. *P < 0.01, **P < 0.01, ***P < 0.001. g Log 10 value of signal intensity detected by microarray.
Class distribution of changed LncRNAs
lncRNAs were shown to regulate the expression of adjacent or overlapping mRNAs in genome [18, 27, 28]. Thus, the associations of DE lncRNAs with coding genes were analyzed and classified according to the method described by Li et al. [29]. LncRNAs are classified into four groups: intergenic lncRNAs (lncRNAs are located and transcribed from intergenic regions, and do not overlap with known protein coding genes or other types of genes in genome. It is also called lincRNAs), antisense lncRNAs (LncRNA exon is transcribed from the antisense strand and overlaps with a coding transcript exon), sense lncRNAs (LncRNA exon overlaps with a coding transcript exon on the same genomic strand), and bidirectional lncRNAs (LncRNA is oriented head to head with a coding transcript within 1,000 bp). As shown in Figure 4, among the DE lncRNAs, intergenic lncRNAs were the largest category, with 236 up-regulated and 90 down-regulated lncRNAs. The other DE lncRNAs included 100 antisense lncRNAs (78 up-regulated and 22 down-regulated), 59 sense lncRNAs (37 up-regulated and 22 down-regulated), and 26 bidirectional lncRNAs (15 up-regulated and 11 down-regulated).

Distribution of various types of DE lncRNAs. Four classes (sense overlap lncRNAs, antisense overlap lncRNAs, bidirectional lncRNAs and intergenic lncRNAs) were analyzed.
Functional prediction of DE mRNAs in SNL
To explore the molecular mechanism in neuropathic pain, we further did GO and pathway analysis of deregulated genes in SNL versus sham. The GO results showed that the most significant enriched molecular function of up-regulated genes in SNL was chemokine activity, CCR chemokine receptor binding, chemokine receptor binding, and cysteine-type endopeptidase inhibitor activity (Figure 5a). The most significant enriched biological processes of up-regulated genes in SNL were immune response, immune system process, defense response, and regulation of immune system process (Figure 5b). The most noteworthy enriched cellular components of up-regulated genes in SNL were extracellular region, extracellular space, extracellular region part, and external side of plasma membrane (Figure 5c). The most significant enriched molecular function of down-regulated genes in SNL were binding, receptor binding, calcium ion binding, and tropomyosin binding (Figure 5d). The most significant enriched biological processes of down-regulated genes in SNL were regulation of ATPase activity, monovalent inorganic cation transport, glucosamine-containing compound catabolic process, and amino sugar catabolic process (Figure 5e). The most significant enriched cellular components of down-regulated genes in SNL were extracellular region, striated muscle thin filament, extracellular space, and cell part (Figure 5f).

Biological functions of differentially expressed mRNAs with fold changes >2. a–c The significant molecular function, biological process and cellular component of up-regulated mRNAs. d–f The significant molecular function, biological process and cellular component of down-regulated mRNAs.
Similarly, different genes were analyzed in KEGG. The results showed that the up-regulated genes in SNL are involved in complement and coagulation cascades, Toll-like receptor signaling pathway, chemokine signaling pathway, cytosolic DNA-sensing pathway, and cytokine–cytokine receptor interaction, Changas disease, and NOD-like receptor signaling pathway (Figure 6a). Down-regulated genes in SNL are involved in amyotrophic lateral sclerosis (ALS), prostate cancer, citrate cycle, glutamatergic synapse, osteoclast differentiation and NOD-like receptor signaling pathway (Figure 6b).

Pathway analysis for 366 up-regulated and 127 down-regulated mRNAs with fold changes >2. a The significant pathways for up-regulated genes in SNL group. b The significant pathways for down-regulated genes in SNL group.
Comparison of our DE mRNAs with previously published microarrays
Previous studies have shown differential gene expression profile in the spinal cord in rats with neuropathic pain [30, 31]. In order to compare neuropathic pain-associated gene expression patterns in mice and rats, we did the overlap analysis between other’s microarray data from rat [30] and our current data from mice (Figure 7a). LaCroix-Fralish et al. reported that 88 genes were upregulated and 83 genes were downregulated in the spinal cord 7 days after L5 nerve root ligation in rats [30]. Surprisingly, compared to 361 up-regulated genes and 119 down-regulated genes in mouse, only 1 gene (Cd74) was upregulated and 2 genes (Nefm, Aco2) were downregulated in both rats and mice (Figure 7b). In addition, we compared our array data with 79 significantly regulated genes which were identified by meta-analysis from 20 independent microarray experiments from rats and mice after tissue inflammation or nerve injury [2]. We observed an overlap of 15 genes with the meta-analysis dataset (Figure 7c). These genes included 14 up-regulated genes (Ctss, C1qb, C1qc, Npy, Cd74, Gal, Aif1, Calca, Cxcl10, Atf3, Ccl2, Ctsh, Fcgr2b and Sprr1a) and 1 down-regulated gene (Nefm) (Figure 7d).

Gene overlap analysis between the present data and previously published microarrays in pain model. a Venn diagram showing the number of common up- and down-regulated genes in our present mice model (mice-up, mice-down) and previously published rat model (rat-up, rat-down) after SNL. Only three genes were shared with the same tendency between the two microarray experiments. b The detailed information of the overlap genes that were significantly regulated in both the mice and rat spinal cord. c Venn diagram showing the overlap between gene-sets of our present data and previously published microarrays (Up-P up-regulated genes of the previous studies, Down-P down-regulated genes of the previous studies, Up-A up-regulated genes of the author’s data, Down-A down-regulated genes of the author’s data). d The detailed information of 14 up-regulated and 1 down-regulated overlapped genes between our present data and previously published microarrays.
Relational analysis of lncRNAs and mRNAs
As some lncRNAs have been suggested to play key roles in regulating the expression of their neighboring or overlapping genes in genome wide, we further screened out DE mRNAs related to DE lncRNAs based on their location distributions on mouse chromosomes by UCSC Genome Browser. In the spinal cord, there are 39 DE lncRNA-mRNA pairs for 35 DE lncRNAs and 35 DE mRNAs. Among them, 32 pairs exhibited coordinated expression changes, and 7 pairs were non-coordinated, which may suggest a complex and various regulatory mechanisms across different lncRNAs and their target mRNAs. Intriguingly, all the seven non-coordinated lncRNA-mRNA pairs belong to intergenic lncRNA-mRNA pairs (Table 3). Further GO and pathway analysis showed that the high enriched molecular functions include pheromone binding, chemokine activity, high-density lipoprotein binding, and phosphatidylcholine-sterol O-acyltransferase activator activity (Figure 8a). Based on gene-pathway network graph analysis, we found that the DE mRNAs from lncRNA-mRNA pairs, such as Cxcl9 (chemokine (C-X-C motif) ligand 9), Cxcl10 (chemokine (C-X-C motif) ligand 10), Cxcl11 (chemokine (C-X-C motif) ligand 11), Trhr (thyrotropin releasing hormone receptor), and Apoa2 (apolipoprotein A-II), might involve in toll-like receptor signaling pathway, calcium signaling pathway, and PPAR signaling pathway (Figure 8b; Table 3), which have been proven to be involved in neuropathic pain pathogenesis [32–34].

Function prediction of DE lncRNAs with related mRNAs. a Molecular function enrichment analysis of DE lncRNAs-related mRNAs. The enrichment scores (−log10 (P-value)) of the GO molecular function were shown in the histogram. b Gene-pathway network graph of DE lncRNAs-related mRNAs from Table 3. The DE lncRNAs-related genes and the corresponding pathways were shown in the circles and boxes, respectively. The color of pathway terms is defined by the enrichment P value.
Discussion
Chronic neuropathic pain is a somatosensory disorder caused by nerve injury or disease that affects the nervous system [35]. Evidence suggested that the particular patterns of gene expression at different levels of the nociceptive system play important roles in the development and maintenance of neuropathic pain [2, 36]. Over the past decades, the molecular mechanisms underlying neuropathic pain have been extensively studied; however, the pathophysiological process of pain is still vague. LncRNAs were recently shown to regulate gene expression [37] and traffic cellular protein complexes, genes, and chromosomes to appropriate locations [8]. Their function in regulating gene expression switching in the maintenance phase of neuropathic pain is poorly understood. In this study, we for the first time identified the global expression changes in lncRNAs and analyzed their characteristics and possible relation with coding genes in the spinal cord under neuropathic pain condition. The 24,833 lncRNAs were detected in the spinal cord of mice. Among them, 366 lncRNAs were up-regulated and 145 lncRNAs were down-regulated at 10 days after SNL. These DE lncRNAs are consistently altered in a high percentage of analyzed spinal cords from SNL and sham mice, suggesting that lncRNAs may be involved in neuropathic pain processing. So far, most DE lncRNAs have not been functionally characterized. Although it was still too early to translate this knowledge into the development of novel analgesic agents for better pain relief, these findings may likely provide novel insight into the molecular basis of pain.
In this study, the expression profiles of mouse genome-wide mRNAs were also detected using lncRNA Microarray Chip at the same time. Among DE mRNAs, the up-regulated mRNAs are far more numerous than the down-regulated in SNL samples, which reflects the emergence of new biology processes and pathways in pathological conditions. A number of reported pain-related genes, including Cacna1g, Trpv1, Ccl5, Cx3cr1 and Irf5 were dramatically increased after SNL. Moreover, a lot of other mRNAs, such as Sprr1a, Anxa10, Kng1, and Gpr151 (G-protein-coupled receptor 151), whose functions are unclear in the spinal cord were also screened out. As the expression changes for some genes may be related to nerve damage and homeostatic responses to denervation, further studies are needed to identify whether they are involved in neuropathic pain processing.
Based on the GO term enrichment analyses of DE mRNA, we found that significantly enriched molecular functions and biological processes of up-regulated gene in SNL vs sham were mainly involved in chemokine activity, inflammation, and immunity. These findings are consistent with previous studies showing that neuroinflammation, manifested as infiltration of immune cells [38], activation of glial cells [39] and production of inflammatory mediators [40] in the peripheral and CNS, plays an important role in the induction and maintenance of chronic pain [41]. Additionally, our immunostaining of GFAP and IBA-1 showed dramatic glial activation in the spinal cord at 10 days after SNL. From significant pathway analyses of DE gene, the third most significant enriched pathway of the up-regulated genes in SNL vs sham is the toll-like receptor signaling pathway. Indeed, Tlr2 [42], Tlr4 [43], and Tlr7 [44] have been implicated as potential therapeutic targets in neuropathic and other pain models. The data collectively indicate that anti-neuroinflammation may be an effective strategy for the treatment of neuropathic pain.
Previous studies utilizing cDNA microarrays to analyze gene expression profiles primarily focus on pain models in rats, rarely in mice [2]. The overlap analysis showed little overlap between rat and mice spinal cord gene expression patterns under neuropathic pain states, suggesting the species difference in gene expression. However, we found that there were 15 overlap genes between our current data and meta-analysis results reported by LaCroix-Fralish et al. [2]. These overlap genes including Atf3, Sprr1al and Nefm can be induced by nerve damage, which contribute to chronic pain [45–47]. In addition, gene ontology-based functional annotation clustering analyses of the previous gene chip study revealed strong evidence for regulation of immune-related genes in pain states, which was consistent with our data.
Although lncRNAs play important roles in the regulation of gene expression [48], there is a large gap between the number of existing lncRNAs and their known association with a particular molecular or cellular function [49]. Regulatory mechanisms and major functional principles of lncRNAs are complex and quite obscure. Unlike microRNA, there are no common languages that can be used to predict lncRNAs’ target genes and function by their sequence information or secondary structure. Accumulating evidence suggests that a number of lncRNAs function locally to activate or repress their neighboring or overlapping genes’ expression [18, 27, 50]. In this study, we found that intergenic lncRNAs (lincRNAs) were the largest category in all DE lncRNAs after SNL. In reality, lincRNAs are found to be conserved across multiple vertebrate species [51] and perform important functions in many cellular processes, from cell proliferation to cancer progression [52]. Furthermore, lincRNAs can function through different types of mechanisms, including cis or trans transcriptional regulation, translational control, splicing regulation, and other post-transcriptional regulation [33]. We examined whether their neighboring or overlapping protein-coding genes in the genome are simultaneously DE in the spinal cord after SNL, and found that there are 39 DE lncRNA-mRNA pairs. Our further analysis showed that an up-regulated lincRNA, MM9LINCRNAEXON10576− in the spinal cord after SNL was found to be located near Cxcl10, Cxcl9 and Cxcl11 gene cluster in mice chromosome 5. All the four RNAs have the same expression trends and increased more than twofold after SNL. Recently, studies using animal models have shown that upregulation of chemokines in the spinal cord play a vital role in the development and maintenance of chronic pain [41, 53, 54]. Indeed, recent research found that Cxcl10 and its receptor Cxcr3 were involved in inflammatory pain and cancer pain [55–57]. Therefore, lncRNA MM9LINCRNAEXON10576− may contribute to neuropathic pain through regulation of chemokines Cxcl10, Cxcl9 and Cxcl11.
In our microarray results, 12 DE mRNA have their corresponding DE sense-overlap lncRNAs, and the change patterns of these lncRNA were same as that of their accompanying protein-coding genes. Di et al. found that a sense-overlap lncRNA arising from the CCAAT/enhancer-binding protein alpha (Cebpa) gene locus can bind to DNA methyltransferase 1 (DNMT1) and prevent Cebpa gene locus methylation, then to increase the expression of Cebpa gene. Their deep sequencing of transcripts associated with DNMT1 combined with genome-scale methylation and expression profiling extend the generality of this finding to numerous gene loci. [27]. Given that the 12 DE mRNA and their DE sense-overlap lncRNAs were both increased after SNL, it’s possible that the DE sense-overlap lncRNAs regulate the expression of their sense-overlapping mRNAs via demethylation after SNL.
Conclusion
Our results demonstrated that lncRNA transcripts were highly enriched and hundreds of lncRNAs were differentially expressed in the spinal cord after SNL. Dozens of DE lncRNAs were observed to have neighboring or overlapping DE mRNAs in genome. These lncRNAs may locally regulate their related protein-genes expression and play key roles in the pathogenesis of neuropathic pain. Further studies are required to clarify the molecular and cellular functions of DE lncRNAs and determine whether they can serve as novel analgesic targets in neuropathic pain.
Methods
Animals and surgery
Adult male ICR mice (male, 8 weeks) were maintained on a 12:12 light–dark cycle at a room temperature of 22 ± 1°C with free access to food and water. The experimental procedures were approved by the Animal Care and Use Committee of Nantong University and performed in accordance with the guidelines of the International Association for the Study of Pain. To produce a SNL, animals were anesthetized with isoflurane and the L6 transverse process was removed to expose the L4 and L5 spinal nerves. The L5 spinal nerve was then isolated and tightly ligated with 6-0 silk threads [58]. For sham operations, the L5 spinal nerve was exposed but not ligated.
Behavioral test
Animals were habituated to the testing environment daily for at least 2 days before baseline testing. The room temperature remained stable for all experiments. For testing mechanical sensitivity, animals were put in boxes on an elevated metal mesh floor and allowed 30 min for habituation before examination. The plantar surface of each hindpaw was stimulated with a series of von Frey hairs with logarithmically incrementing stiffness (0.02–2.56 g, Stoelting, Wood Dale, IL, USA), presented perpendicular to the plantar surface (2–3 s for each hair). The 50% paw withdrawal threshold was determined using Dixon’s up-down method [59]. For testing heat sensitivity, animals were put in plastic boxes and allowed 30 min for habituation. Heat sensitivity was tested by radiant heat using Hargreaves apparatus (IITC Life Science Inc., Woodland Hills, CA, USA) and expressed as paw withdrawal latency (PWL). The radiant heat intensity was adjusted so that basal PWL is between 10 and 14 s, with a cutoff of 18 s to prevent tissue damage.
Immunohistochemistry
At 10 days after SNL or sham-operation, animals were deeply anesthetized with isoflurane and perfused through the ascending aorta with PBS followed by 4% paraformaldehyde with 1.5% picric acid in 0.16 M PB. After the perfusion, the L4–L5 spinal cord segments were removed and postfixed in the same fixative overnight. Spinal cord sections (30 μm, free-floating) were cut in a cryostat. The sections were first blocked with 5% goat serum for 2 h at room temperature. The sections were then incubated overnight at 4°C with the following primary antibodies: GFAP antibody (mouse, 1:6,000; Millipore, Billerica, MA, USA), IBA-1 antibody (Mouse, 1:3,000, Serotec, Kidlington, UK). The sections were then incubated for 2 h at room temperature with FITC-conjugated secondary antibodies (1:1,000, Jackson ImmunoResearch). The stained sections were examined with a Leica fluorescence microscope, and images were captured with a CCD Spot camera.
Tissue collection and RNA isolation
We prepared nine mice for SNL and nine mice for sham-operation. At 10 days after operation, the animals were deeply anesthetized with isoflurane and perfused through the ascending aorta with saline. After the perfusion, the L4–L5 spinal cord segments were collected. Total RNA was extracted from the spinal cord dorsal horn tissue using Trizol reagent (Invitrogen, Carlsbad) according to the manufacturer’s protocol. The RNA concentration and purity were assayed by the absorbance values at 260 and 280 nm using the NanoDrop 1000 Spectrophotometer (Thermo). RNA integrity was checked by electrophoresis on 2% (m/v) agarose gels. After these testing, equal mRNA from three mice under the same treatment was mixed as one sample. Therefore, six samples (3 for SNL and 3 for sham) were sent for microarray analysis.
Microarray assay
The gene chip of the mouse lncRNA microarray V2.0 (8 × 60K, Arraystar), which includes 25,376 lncRNA probes and 31,423 coding gene probes, was used in the experiments. The total RNAs of sham and SNL groups were individually hybridized with gene chips. Briefly, RNA was purified from 1 μg total RNA after removing rRNA. The RNA sample was then transcribed into fluorescent cRNA along the entire length of the transcripts without 3′ bias utilizing random primers. The labeled cRNAs were hybridized to mouse lncRNA microarray. Finally, arrays were scanned by Agilent Scanner G2505B. The array images were analyzed by Agilent Feature Extraction software (version 10.7.3.1). The GeneSpring GX v11.5.1 software package (Agilent Technologies) was utilized to analyze quintile normalization and subsequent data processing. The microarray hybridization was carried out by Kangchen Bio-tech, Shanghai, China.
Bioinformatics analysis
Differentially expressed lncRNAs and mRNAs with statistical significance were identified through Volcano Plot filtering. The threshold used to screen up- or down-regulated RNAs was fold-change >2.0 (P < 0.05). Hierarchical clustering was carried out by Cluster 3.0, and the heat maps were generated in Java Treeview. The DE mRNAs which were adjacent to or overlap with the DE lncRNAs were recognized as DE lncRNAs related mRNAs using UCSC Genome Browser. The differentially expressed mRNAs or DE lncRNAs related mRNAs were analyzed by pathway annotation and gene ontology (GO) functional enrichment using CapitalBio® Molecule Annotation System V3.0 (MAS3.0). The −log10 (P-value) of the GO and pathway results were shown in the histogram.
Real-time reverse transcription-polymerase chain reaction (RT-PCR)
The microarray results were confirmed by RT-PCR. Total RNA was extracted from the spinal cord tissue as described above and total RNA was reverse transcribed using random hexamers primer (TaKaRa Bio Inc) according to the manufacturer’s description. The expression level of six genes was checked, including Speer7-ps1, uc007pbc.1, ENSMUST00000171761, ENSMUST00000097503, Cyp2d9, and Mnx1. The Gapdh was used as house-keeping gene. The sequences of all primers were shown in Table 4. RT-PCR was performed using the Fast Start Universal SYBR Green Master (TaKaRa Bio Inc) with 20-μl reaction system, according to the manufacturer’s protocol, in a Rotor-Gene 6000 instrument (Hamburg, Germany). The melting-curve analysis was performed in order to monitor the specificity of production. All experiments were replicated three times. The gene expression levels in the sham and SNL groups were analyzed with the 2−∆∆CT method.
Statistical analysis
The behavioral data were analyzed by two-way analysis of variance. The RT-PCR results were reported as mean ± SEM and analyzed by the one-way analysis of variance followed by Tukey’s multiple comparison test. The criterion for statistical significance was P < 0.05.
Abbreviations
- Anxa10 :
-
annexin A10
- Apoa2 :
-
apolipoprotein A-II
- Atf3 :
-
activating transcription factor 3
- Cacna1g :
-
calcium channel, voltage-dependent, T type, alpha 1G subunit
- Ccl5 :
-
chemokine C-C motif ligand 5
- Cebpa :
-
CCAAT/enhancer-binding protein alpha
- CNS:
-
central nervous system
- Cyp2d9 :
-
cytochrome P450, family 2, subfamily d, polypeptide 9
- Cx3cr1 :
-
chemokine (C-X3-C) receptor 1
- DE:
-
differentially expressed
- Dnmt1 :
-
DNA methyltransferase 1
- GO:
-
gene ontology
- Gpr151 :
-
G-protein-coupled receptor 151
- Irf5 :
-
interferon regulatory factor 5
- Kng1 :
-
kininogen 1
- miRNA:
-
microRNA
- lncRNA:
-
long non-coding RNA
- Mnx1 :
-
motor neuron and pancreas homeobox 1
- Nefm :
-
neurofilament, medium polypeptide
- PPAR:
-
peroxisome proliferator-activated receptor
- SNL:
-
spinal nerve ligation
- Sprr1a :
-
small proline-rich protein 1A
- Tagap :
-
T cell activation Rho GTPase-activating protein
- Trhr :
-
thyrotropin releasing hormone receptor
- Trpv1 :
-
transient receptor potential cation channel, subfamily V, member 1
- Zfp236 :
-
zinc finger protein 236
References
Latremoliere A, Woolf CJ. Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J Pain. 2009;10:895–926.
LaCroix-Fralish ML, Austin JS, Zheng FY, Levitin DJ, Mogil JS. Patterns of pain: meta-analysis of microarray studies of pain. Pain. 2011;152:1888–98.
Schaible HG. Peripheral and central mechanisms of pain generation. In: Stein C, editor. Analgesia. Handbook of experimental pharmacology. Berlin: Springer; 2007. p. 3–28.
Chamorro A, Meisel A, Planas AM, Urra X, van de Beek D, Veltkamp R. The immunology of acute stroke. Nat Rev Neurol. 2012;8:401–10.
Alvarez-Rueda N, Desselle A, Cochonneau D, Chaumette T, Clemenceau B, Leprieur S, et al. A monoclonal antibody to O-acetyl-GD2 ganglioside and not to GD2 shows potent anti-tumor activity without peripheral nervous system cross-reactivity. PLoS One. 2011;6:e25220.
Kapranov P, Cheng J, Dike S, Nix DA, Duttagupta R, Willingham AT, et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science. 2007;316:1484–8.
Bali KK, Kuner R. Noncoding RNAs: key molecules in understanding and treating pain. Trends Mol Med. 2014;20:437–48.
Batista PJ, Chang HY. Long noncoding RNAs: cellular address codes in development and disease. Cell. 2013;152:1298–307.
Zhang B, Arun G, Mao YS, Lazar Z, Hung G, Bhattacharjee G, et al. The lncRNA Malat1 is dispensable for mouse development but its transcription plays a cis-regulatory role in the adult. Cell Rep. 2012;2:111–23.
Wang P, Xue Y, Han Y, Lin L, Wu C, Xu S, et al. The STAT3-binding long noncoding RNA lnc-DC controls human dendritic cell differentiation. Science. 2014;344:310–3.
Vausort M, Wagner DR, Devaux Y. Long noncoding RNAs in patients with acute myocardial infarction. Circ Res. 2014;115:668–77.
Huarte M, Guttman M, Feldser D, Garber M, Koziol MJ, Kenzelmann-Broz D, et al. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell. 2010;142:409–19.
Faghihi MA, Modarresi F, Khalil AM, Wood DE, Sahagan BG, Morgan TE, et al. Expression of a noncoding RNA is elevated in Alzheimer’s disease and drives rapid feed-forward regulation of β-secretase. Nat Med. 2008;14:723–30.
Qureshi IA, Mehler MF. Emerging roles of non-coding RNAs in brain evolution, development, plasticity and disease. Nat Rev Neurosci. 2012;13:528–41.
Ramos AD, Diaz A, Nellore A, Delgado RN, Park K-Y, Gonzales-Roybal G, et al. Integration of genome-wide approaches identifies lncRNAs of adult neural stem cells and their progeny in vivo. Cell Stem Cell. 2013;12:616–28.
Mercer TR, Dinger ME, Sunkin SM, Mehler MF, Mattick JS. Specific expression of long noncoding RNAs in the mouse brain. Proc Natl Acad Sci. 2008;105:716–21.
Yu B, Zhou S, Hu W, Qian T, Gao R, Ding G, et al. Altered long noncoding RNA expressions in dorsal root ganglion after rat sciatic nerve injury. Neurosci Lett. 2013;534:117–22.
Zhao X, Tang Z, Zhang H, Atianjoh FE, Zhao JY, Liang L, et al. A long noncoding RNA contributes to neuropathic pain by silencing Kcna2 in primary afferent neurons. Nat Neurosci. 2013;16:1024–31.
Chung JM, Kim HK, Chung K. Segmental spinal nerve ligation model of neuropathic pain. Methods Mol Med. 2004;99:35–45.
Zhuang ZY, Wen YR, Zhang DR, Borsello T, Bonny C, Strichartz GR, et al. A peptide c-Jun N-terminal kinase (JNK) inhibitor blocks mechanical allodynia after spinal nerve ligation: respective roles of JNK activation in primary sensory neurons and spinal astrocytes for neuropathic pain development and maintenance. J Neurosci. 2006;26:3551–60.
Jin SX, Zhuang ZY, Woolf CJ, Ji RR. P38 mitogen-activated protein kinase is activated after a spinal nerve ligation in spinal cord microglia and dorsal root ganglion neurons and contributes to the generation of neuropathic pain. J Neurosci. 2003;23:4017–22.
Jarvis MF, Scott VE, McGaraughty S, Chu KL, Xu J, Niforatos W, et al. A peripherally acting, selective T-type calcium channel blocker, ABT-639, effectively reduces nociceptive and neuropathic pain in rats. Biochem Pharmacol. 2014;89:536–44.
Park CK, Lu N, Xu ZZ, Liu T, Serhan CN, Ji RR. Resolving TRPV1- and TNF-alpha-mediated spinal cord synaptic plasticity and inflammatory pain with neuroprotectin D1. J Neurosci. 2011;31:15072–85.
Ramaglia V, Daha MR, Baas F. The complement system in the peripheral nerve: friend or foe? Mol Immunol. 2008;45:3865–77.
Morgan BP, Gasque P, Singhrao SK, Piddlesden SJ. Role of complement in inflammation and injury in the nervous system. Exp Clin Immunogenet. 1997;14:19–23.
Ferrari S, Morbin M, Nobile-Orazio E, Musso A, Tomelleri G, Bertolasi L, et al. Antisulfatide polyneuropathy: antibody-mediated complement attack on peripheral myelin. Acta Neuropathol. 1998;96:569–74.
Di Ruscio A, Ebralidze AK, Benoukraf T, Amabile G, Goff LA, Terragni J, et al. DNMT1-interacting RNAs block gene-specific DNA methylation. Nature. 2013;503:371.
Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458:223–7.
Li D, Chen G, Yang J, Fan X, Gong Y, Xu G, et al. Transcriptome analysis reveals distinct patterns of long noncoding RNAs in heart and plasma of mice with heart failure. PLoS One. 2013;8:e77938.
Lacroix-Fralish ML, Tawfik VL, Tanga FY, Spratt KF, DeLeo JA. Differential spinal cord gene expression in rodent models of radicular and neuropathic pain. Anesthesiology. 2006;104:1283–92.
Levin ME, Jin JG, Ji RR, Tong J, Pomonis JD, Lavery DJ, et al. Complement activation in the peripheral nervous system following the spinal nerve ligation model of neuropathic pain. Pain. 2008;137:182–201.
Sauvageau M, Goff LA, Lodato S, Bonev B, Groff AF, Gerhardinger C, et al. Multiple knockout mouse models reveal lincRNAs are required for life and brain development. eLife. 2013;2:e01749.
Ulitsky I, Bartel DP. lincRNAs: genomics, evolution, and mechanisms. Cell. 2013;154:26–46.
Akay C, Cooper M, Odeleye A, Jensen BK, White MG, Vassoler F, et al. Antiretroviral drugs induce oxidative stress and neuronal damage in the central nervous system. J Neurovirol. 2014;20:39–53.
Ochoa JL. Neuropathic pain: redefinition and a grading system for clinical and research purposes. Neurology. 2009;72:1282–3.
Imai S, Ikegami D, Yamashita A, Shimizu T, Narita M, Niikura K, et al. Epigenetic transcriptional activation of monocyte chemotactic protein 3 contributes to long-lasting neuropathic pain. Brain. 2013;136:828–43.
Guttman M, Rinn JL. Modular regulatory principles of large non-coding RNAs. Nature. 2012;482:339–46.
Costigan M, Moss A, Latremoliere A, Johnston C, Verma-Gandhu M, Herbert TA, et al. T-Cell infiltration and signaling in the adult dorsal spinal cord is a major contributor to neuropathic pain-like hypersensitivity. J Neurosci. 2009;29:14415–22.
Zhang J, Shi XQ, Echeverry S, Mogil JS, De Koninck Y, Rivest S. Expression of CCR2 in both resident and bone marrow-derived microglia plays a critical role in neuropathic pain. J Neurosci. 2007;27:12396–406.
Guo W, Wang H, Watanabe M, Shimizu K, Zou SP, LaGraize SC, et al. Glial-cytokine-neuronal interactions underlying the mechanisms of persistent pain. J Neurosci. 2007;27:6006–18.
Ji RR, Xu ZZ, Gao YJ. Emerging targets in neuroinflammation-driven chronic pain. Nat Rev Drug Discov. 2014;13:533–48.
Vacca V, Marinelli S, Luvisetto S, Pavone F. Botulinum toxin A increases analgesic effects of morphine, counters development of morphine tolerance and modulates glia activation and mu opioid receptor expression in neuropathic mice. Brain Behav Immun. 2013;32:40–50.
Christianson CA, Dumlao DS, Stokes JA, Dennis EA, Svensson CI, Corr M, et al. Spinal TLR4 mediates the transition to a persistent mechanical hypersensitivity after the resolution of inflammation in serum-transferred arthritis. Pain. 2011;152:2881–91.
Park CK, Xu ZZ, Berta T, Han Q, Chen G, Liu XJ, et al. Extracellular microRNAs activate nociceptor neurons to elicit pain via TLR7 and TRPA1. Neuron. 2014;82:47–54.
Seijffers R, Mills CD, Woolf CJ. ATF3 increases the intrinsic growth state of DRG neurons to enhance peripheral nerve regeneration. J Neurosci. 2007;27:7911–20.
Bonilla IE, Tanabe K, Strittmatter SM. Small proline-rich repeat protein 1A is expressed by axotomized neurons and promotes axonal outgrowth. J Neurosci. 2002;22:1303–15.
Rodriguez Parkitna J, Korostynski M, Kaminska-Chowaniec D, Obara I, Mika J, Przewlocka B, Przewlocki R. Comparison of gene expression profiles in neuropathic and inflammatory pain. J Physiol Pharmacol. 2006;57:401–14.
Pauli A, Rinn JL, Schier AF. Non-coding RNAs as regulators of embryogenesis. Nat Rev Genet. 2011;12:136–49.
Fritah S, Niclou SP, Azuaje F. Databases for lncRNAs: a comparative evaluation of emerging tools. RNA. 2014;20:1655–65.
Lai F, Orom UA, Cesaroni M, Beringer M, Taatjes DJ, Blobel GA, et al. Activating RNAs associate with mediator to enhance chromatin architecture and transcription. Nature. 2013;494:497–501.
Ulitsky I, Shkumatava A, Jan CH, Sive H, Bartel DP. Conserved function of lincRNAs in vertebrate embryonic development despite rapid sequence evolution. Cell. 2011;147:1537–50.
Beckedorff FC, Ayupe AC, Crocci-Souza R, Amaral MS, Nakaya HI, Soltys DT, et al. The intronic long noncoding RNA ANRASSF1 recruits PRC2 to the RASSF1A promoter, reducing the expression of RASSF1A and increasing cell proliferation. PLoS Genet. 2013;9:e1003705.
Diamond B, Volpe BT. A model for lupus brain disease. Immunol Rev. 2012;248:56–67.
Zhang ZJ, Cao DL, Zhang X, Ji RR, Gao YJ. Chemokine contribution to neuropathic pain: respective induction of CXCL1 and CXCR2 in spinal cord astrocytes and neurons. Pain. 2013;154:2185–97.
Wang Y, Gehringer R, Mousa SA, Hackel D, Brack A, Rittner HL. CXCL10 controls inflammatory pain via opioid peptide-containing macrophages in electroacupuncture. PLoS One. 2014;9:e94696.
Ye D, Bu H, Guo G, Shu B, Wang W, Guan X, et al. Activation of CXCL10/CXCR3 signaling attenuates morphine analgesia: involvement of Gi protein. J Mol Neurosci. 2014;53:571–9.
Bu H, Shu B, Gao F, Liu C, Guan X, Ke C, et al. Spinal IFN-gamma-induced protein-10 (CXCL10) mediates metastatic breast cancer-induced bone pain by activation of microglia in rat models. Breast Cancer Res Treat. 2014;143:255–63.
Kim SH, Chung JM. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain. 1992;50:355–63.
Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63.
Authors’ contributions
BCJ designed the microarray experiment, analyzed the data, and drafted the manuscript. WSX participated in the data analysis and prepared the figures. LNH did the real-time PCR analysis. DLC did the immunostaining. ZJZ prepared SNL model and did the behavioral test. YJG designed and supervised the overall experiment, revised the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We thank Dr. Fu-Lu Dong (Institutes of Biology and Medical Sciences, Soochow University) for aiding the technical assistance. This study was supported by the National Natural Science Foundation of China (NSFC 31371121, 31171062, and 81400915), the National Science Foundation for Young Scientists of Jiangsu Province (BK20140427), the Natural Science Foundation of the Jiangsu Higher Education Institutions (13KJB180016), the Natural Science Foundation of Nantong University (13040443), and the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Compliance with ethical guidelines
Competing interests The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Additional information
Bao-Chun Jiang and Wen-Xing Sun have contributed equally to this work
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Jiang, BC., Sun, WX., He, LN. et al. Identification of lncRNA expression profile in the spinal cord of mice following spinal nerve ligation-induced neuropathic pain. Mol Pain 11, 43 (2015). https://doi.org/10.1186/s12990-015-0047-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12990-015-0047-9