
Abstract
Background
Association of particulate matter with adverse health effects has been established in epidemiological studies and animal experiments. Epidemiological studies are difficult to undertake while animal studies are impractical for high-throughput toxicity testing. The ease and rapidity of in vitro tests emphasizes their potential for use in risk assessment of chemicals and particles. We examined the association between in vitro and in vivo responses to ambient particles, to determine the potential of cell-based assays as standalone toxicity screening tools.
Methods
Assays of cytotoxicity and key inflammatory mediators were applied to determine the in vitro biological potency of a panel of urban and mineral particles in J774A.1 macrophages and A549 lung epithelial cells. The particles were also screened for the presence of AhR agonists using the Ah receptor-dependent gene induction assay and for endotoxin using the Limulus amebocyte lysate assay. A subset of the particles with a contrasting in vitro toxicity profile was delivered intratracheally in BALB/c mice to assess their in vivo biological potency. Results from various bioassays were combined within the in vitro and in vivo models. The combined potency measures were examined for associations.
Results
Overall, J774A.1 cells were more sensitive to particle effects than A549 cells. Whereas the combined cytotoxicity estimates were highly correlated between the two cell lines, the combined in vitro inflammatory potency estimates were not, emphasizing functional differences of the two cell types. Secretion of inflammatory markers by J774A.1 cells was correlated with AhR ligand binding profile and endotoxin levels of particles. Particle instillation led to an acute toxicity response in BALB/c mice, with neutrophilia and release of inflammatory mediators. While the combined toxicity estimates were not correlated between in vitro and in vivo models, the combined inflammatory and integrated potency estimates (toxicity and inflammation) approached the threshold for significance (p = 0.052) in a correlation within in vitro and in vivo models, with a ranking of fine particle (DWR1), minerals (TiO2, CRI) and coarse particles (SRM-, EHC-type) from low to high potency.
Conclusion
Integration of in vitro endpoints shows promise in determining adverse outcomes of particle exposures in vivo. The devised data reduction and computational approach will prove useful in the development of models for assessment of hazard potential of particles; however, distinct models may be needed for particles of different type, such as urban particles vs. mineral particles, nanomaterials.
Similar content being viewed by others

Background
Air pollution continues to exert a societal burden by impacting multiple aspects of human health, especially through its association with cardiopulmonary morbidity and mortality [1]. Exposure to airborne particulate matter is epidemiologically associated with increased incidence of cardiopulmonary disease and lung cancer in humans [2, 3]. Moreover, recent studies provide indications that the health impacts of exposure to air pollutants may be more widespread, including epidemiological associations with inflammatory bowel disease and non-specific abdominal pain [4, 5], neuropathologies and cognitive impairment [6, 7], alterations of markers of placental growth and function, and adverse birth outcomes [8, 9], as well as diabetes mellitus [10, 11].
Urban air particles are complex mixtures of chemical and biogenic constituents associated with natural and anthropogenic particle sources and their modification in the atmosphere. Increasing evidence shows that in addition to size, chemical composition of the particles plays an important role in determining their hazard potential. Mitigation of specific constituents of air pollution mixtures and particles with highest hazard potential may present a reasonable regulatory strategy for reduction of adverse population health effects.
Both, in vitro and in vivo approaches are widely utilized for the study of the adverse effects of chemicals and inhaled environmental pollutants by employing individual endpoint assays to determine the adverse effects of particulate materials or to examine specific biological mechanisms of toxicity. Animal studies have been generally accepted as a major approach in the regulatory risk assessment regimes [12–14]. Despite the definitive applicability of in vivo methods to hazard evaluation and risk assessment, issues such as low-throughput, high cost and ethical aspects of animal use hinder their practicality as a toxicity screening tool for large panels of airborne particulates and chemicals. In vitro assays are better suited as screening tools for hazard identification and determination of toxicity, as they are amenable to automation and high-throughput, are cost effective and greatly reduce the dependence on animal use [13, 15, 16]. However, systematic evaluations of the predictive potential of in vitro assays for adverse in vivo impacts of particulate matter are required.
Few attempts have been made to conduct more comprehensive in vitro/in vivo evaluations of panels of particles and particle fractions including gasoline and diesel engine emission organic fractions [17, 18], polyurethane filter-extracted particle suspensions [19], stone particles [20] and nanoparticles [21]. These studies were met with mixed success, whereby the correlations between in vitro and in vivo measurements were poor, with a general lack of rank order of particle hazard potency for organic fractions of gasoline and diesel emissions and for nanoscale particles [17, 18, 21]. In contrast, in vitro inflammatory markers were generally well correlated with in vivo inflammation models of exposure to ambient particles from different emission sources, but not with in vivo respiratory allergy models [19]. Correlations were also observed between in vitro cytokines and in vivo neutrophil counts in lung lavage from rats instilled with mineral stone particles [20]. The lack of consistency between the studies emphasizes the need for further standardization of in vitro approaches, including variables such as culture conditions, time course, choice of a wide range of bioassays, particle types, and the validation against relevant in vivo effects.
The usefulness of in vitro studies is highlighted by demonstrations of coherence of the particle-induced biological effects in cultured cells with in vivo responses from a controlled human exposure study [22] and with concomitant epidemiological observations of associations of Utah Valley hospital respiratory admissions and PM10 emissions from a local steel mill source [23]. Particles extracted from filters collected from the Utah Valley before, during, and after a temporary closure of a steel mill differentially induced neutrophilia and IL-8 production in human subjects instilled with the particles via bronchoscopy. Interleukin-8 production was also stimulated in human airway epithelial cells exposed to the same particles in a corresponding manner, with a comparable potency profile [22].
In the present study, we assessed the toxicity and inflammatory potential of a panel of airborne particulates and reference particles in cell lines in vitro and in BALB/c mice exposed in vivo to particles at 24 h post-intratracheal instillation. An approach was developed to summarize large datasets to facilitate a comparison between the in vitro and in vivo responses to particles and to evaluate the level of concordance between the two models for toxicity testing.
Methods
Particulate materials
The particulate preparations SRM-1648 (St. Louis total suspended particles, TSP; urban particulate matter reference material), SRM-1649a (Washington TSP; organics), SRM-1879 (cristobalite, SiO2 reference material) and SRM-154b (fine titanium dioxide; TiO2 reference material) were obtained from the National Institute of Standards and Technology (Gaithersberg, MD, USA). Cristobalite particles will be referred to as CRI throughout the manuscript. The TiO2 particles were washed in methanol and phosphate buffered saline (PBS) to remove potential naphthalene contaminant [24]. The PM2.5 DWR1 particles were collected in Toronto as described before [24]. The urban TSP preparations EHC-93, EHC-98 and EHC-2000 refer to materials collected in Ottawa as described before [25]. Particulate materials were weighed and resuspended in sterile particle preparation solution (Tween-80, 25 μg/mL; NaCl, 0.19 % w/v) to a concentration of 10 mg/mL using a Dounce glass homogenizer. After vortexing, the particle suspensions were sonicated in ice-cold water for 20 min and homogenized with 25 full strokes of the homogenizer piston. The particle stocks were aliquoted into sterile centrifuge tubes with O-ring seals and sterilized in a water bath at 56 °C for 30 min. These were kept frozen at −80 °C, until use [26]. All materials were analyzed for endotoxin using the chromogenic Limulus amebocyte lysate assay (Lonza, Walkersville, MD, USA).
The materials EHC-93, EHC-98, EHC-2000 (Ottawa), SRM-1648 (St. Louis) and SRM-1649 (Washington) are TSP matter from urban environments, characterized by enrichment in elements including aluminium (20–30 mg/g), calcium (26–128 mg/g), iron (16–30 mg/g), magnesium (9–16 mg/g), lead (5–14 mg/g), titanium (2–3 mg/g) and zinc (2–11 mg/g), in the milligram per gram range. DWR1 is a PM2.5, with 10–100-fold less components from crustal origin, e.g. aluminium (0.8 mg/g), iron (1 mg/g), magnesium (1.4 mg/g), lead (0.05 mg/g), titanium (0.06 mg/g). SiO2 and TiO2 are mineral particles containing a variety of trace elements. Available physicochemical information from in-house analysis and NIST Certificates of Analysis are presented in supplemental information (Additional file 1: Table S1).
Cell culture
A549 human alveolar type II epithelial (Caucasian male, lung carcinoma) and J774A.1 murine monocyte/macrophages (BALB/c female, cell sarcoma) cell lines were from ATCC (Manassas, VA, USA). H1L1.1c2 murine hepatoma cells containing a stably transfected Ah receptor-dependent luciferase reporter gene were described elsewhere [27]. A549 were cultured in M199 with 2 mM L-glutamine and 10 % FBS (Invitrogen-Gibco, Burlington, ON, Canada). J774A.1 were cultured in DMEM medium (Thermo-Fisher, Nepean, ON, Canada) with 2 mM L-glutamine, 4.5 g/L glucose and 10 % FBS. H1L1.1c2 cells were cultured in MEM Alpha medium (Thermo-Fisher) supplemented with 10 % FBS and 2 mM L-glutamine. For all cytotoxicity assays except BrdU incorporation, cells were seeded in 96-well (0.3 cm2 well) plates at cell densities of 4 × 104 (A549) and 8 × 104 cells/cm2 (J774A.1). For BrdU incorporation, cells were seeded at 1 × 104 (A549) and 2 × 104 cells/cm2 (J774A.1). For the LDH assay, A549 and J774A.1 cells were cultured in serum-free media supplemented with 2 mM L-glutamine, 4.5 g/L glucose (J774A.1 cells only) and 6 mg/mL BSA. H1L1.1c2 cells were seeded in 24-well (0.5 cm2 well) plates at a density of 1 × 104 cells per well, in 1 mL of complete medium. The cells were then incubated for 24 h at 37 °C in 5 % CO2 and 100 % relative humidity for attachment.
In vitro exposure to particles
Particle suspensions were thawed to room temperature and sonicated for 20 min in ice-cold, ultrasonic water bath, and diluted (1–2 mg/mL) in complete media devoid of serum for dosing cells. The diluted particle suspensions were sonicated for 5 min in an ice-cold, ultrasonic water bath prior to dosing the cells. A549, J774A.1 and H1L1.1c2 cell monolayers were dosed with a 100 μL suspension of particles resulting in 0, 10, 20, 40, 80 and 100 μg/well (0, 30, 60, 120, 240 and 480 μg/cm2) in a final volume of 200 μL/well, 5 % FBS. The A549 and J774A.1 cell culture plates were incubated at 37 °C for 24 h before the cytotoxicity measurements were carried out. Culture plates containing H1L1.1c2 cells were incubated at 37 °C for 4 h before they were assayed for induction of aryl hydrocarbon response.
Cytotoxicity assessment
Following 24 h incubation with particles, A549 and J774A.1 cells were assayed for cytotoxicity using resazurin reduction assay (Alamar Blue), ATP assay, BrdU incorporation assay and lactate dehydrogenase (LDH) assay, and in H1L1.1c2 cells for Ah receptor-dependent gene induction, as briefly outlined below. Assays were performed using aliquots from cell culture supernatants and lysates that were clarified by centrifugation to remove the remnants of particles that could potentially interfere with the assay reagent chemistry and detection methods. The approach utilized has been previously described [28]. ATP assay was performed by top reading.
Resazurin reduction assay
The spent medium was collected and replaced with 100 μL of fresh complete medium containing a 1:10 dilution of Resazurin (Alamar Blue) reagent (Biosource, Camarillo, CA, USA) and fluorescence was measured at λEx 530/25 nm and λEm 590/35 nm using Cytofluor 2350 fluorometer [28], 5 min and 2 h after incubation at 37 °C, 5 % CO2). Rate of reduction is the delta fluorescence at 2 h – 5 min. Five independent experiments were conducted (n = 5), with triplicate wells within each experiment.
ATP assay
Cellular ATP content was assessed using the ViaLight Plus assay (Lonza, Walkersville, MD, USA). Cells were lysed with 10 μL of the assay cell lysis reagent for 10 min, followed by 25 μL of ATP-monitoring reagent (AMR) containing firefly luciferase, luciferin, pyrophosphate, BSA and magnesium ions. After 2 min incubation in the dark, the cellular ATP content was determined by top reading luminescence measurements using LMax luminometer. Luminescence (rate of photon emission) is directly proportional to concentration of ATP substrates). The ATP assay was performed in three independent experiments (n = 3), with triplicate wells within each experiment.
BrdU incorporation
Following exposure to particles, 5-bromo-2-deoxyuridine (BrdU) was added to each well at 100 μM and the cells were returned to the incubator for 2 h. The culture medium was removed by inversion of the 96-well plates, and the cells were fixed for 30 min at room temperature using the FixDenat solution (Roche Diagnostics, Indianapolis, IN, USA). The cells were washed with blocking buffer (1 % BSA in PBS), followed by an incubation with 100 μL of anti-BrdU antibody conjugated with peroxidase (anti-BrdU-POD, Fab fragments) for 90 min at room temperature. After extensive washing, cells were incubated with 100 μL of the 3,3’,5,5’-tetramethylbenzidine (TMB) chromogenic substrate for 10 min at room temperature in the dark, and the reaction product was quantified at 370 nm using SpectraMax Plus spectrophotometer. Three independent assays (n = 3), with triplicate wells within each assay were conducted.
LDH assay
Culture supernatants were recovered and the cells were lysed by a rapid freeze-thaw cycle. LDH activity was measured in the clarified supernatants and the cell lysates by spectrophotometry at 450 nm (SpectraMax Plus) from production of NADH from lactate coupled with the instantaneous conversion of tetrazolium to a red formazan by NADH diaphorase (CytoTox 96 assay, Promega, Madison, WI, USA). Release of cellular LDH was determined from activity in supernatant over total LDH (supernatant plus lysate). Three (A549, n = 3) or four (J774A.1, n = 4) independent assays, with triplicate wells within each experiment were conducted.
Assay of Ah receptor-dependent gene induction
After dosing with particles, H1L1.1c2 cell plates were incubated at 37 °C for 4 h, followed by two washes in ice-cold PBS, and 15 min lysis on ice using 50 μL/well of luciferase assay system (Promega Corporation) lysis buffer, on a plate shaker (250 rpm). The lysates were transferred to v-bottom plates and centrifuged at 3000 × g (2 min, 4 °C) using a Sorvall Legend RT centrifuge. Clarified cell lysates (25 μL) were transferred into a white, clear-bottomed 96-well plate, 50 μL of luciferase substrate was added by injection using an L-Max luminometer, followed by 15 s integrated luminescence reading per well. Three independent experiments were conducted (n = 3), with triplicates within each experiment.
Cytokine secretion in vitro
Cell culture supernatants of A549 cells were assessed for Interleukin (IL)-1β (0.13 ± 0.01 pg/mL basal level in control cells ± standard error mean), IL-1ra (n.d.; not detected), IL-2 (n.d.), IL-4 (n.d.), IL-5 (n.d.), IL-6 (430 ± 111), IL-7 (n.d.), IL-8 (200 ± 4), IL-9 (n.d.), IL-10 (12.5 ± 1.9), IL-12 (p70) (n.d.), IL-13 (n.d.), IL-15 (n.d.), IL-17 (n.d.), basic Fibroblast Growth Factor (FGF) (n.d.), Eotaxin (n.d.), Granulocyte Colony-Stimulating Factor (G-CSF) (n.d.), Granulocyte Macrophage Colony-Stimulating Factor (GM-CSF) (2.8 ± 0.4), Interferon (IFN)-γ (n.d.), IFN-γ Inducible Protein (IP)-10 (n.d.), Monocyte/Macrophage Chemoattractant Protein (MCP)-1 (701.5 ± 44.8), Macrophage Inflammatory Protein (MIP)-1α (n.d.), MIP-1β (0.9 ± 0.5), Platelet-Derived Growth Factor (PDGF)-BB (n.d.), Regulated on Activation, Normal T cell Expressed and Secreted (RANTES) (n.d.), Tumor Necrosis Factor (TNF)-α (0.1 ± 0.1), and Vascular Endothelial Growth Factor (VEGF) (n.d.), while cell culture supernatants of J774A.1 were assessed for IL-1α (1.2 ± 0.1), IL-1β (5.9 ± 1.8), IL-2 (n.d.), IL-3 (n.d.), IL-4 (n.d.), IL-5 (n.d.), IL-6 (9.2 ± 1.0), IL-9 (n.d.), IL-10 (12.5 ± 1.9), IL-12 (p40) (n.d.), IL-12 (p70) (n.d.), IL-13 (n.d.), IL-17A (n.d.), Eotaxin (n.d.), G-CSF (n.d.), GM-CSF (9.4 ± 1.3), IFN-γ (n.d.), Keratinocyte Chemoattractant (KC) (n.d.), MCP-1 (n.d.), MIP-1α (above assay limit), MIP-1β (n.d.) and RANTES (239 ± 28) using Bio-Plex cytokine panels (Bio-Rad Laboratories, Mississauga, ON, Canada), with a Bio-Plex 200 array system.
In vivo exposures to particles
Seven weeks-old, pathogen-free BALB/c male mice (~25 g) were from Charles-River (Saint-Constant, QC, Canada). Mice were housed in shoe-box plexiglass cages, on woodchip bedding, provided with mouse chow and water ad libitum, maintained under HEPA air filtration at 22 +/− 2 °C, ~50 % humidity and were held to a 12-h light/dark cycle. The animals were acclimatized for at least 1 week prior to experimentation. The animal treatment protocol was reviewed and approved by the Animal Care Committee of Health Canada. The particle preparations were thawed at room temperature, sonicated for 10 min, diluted in saline and instilled intratracheally (50 μL volume) using the MicroSprayer aerosolizer (Penn-Century, Philadelphia, PA, USA) at doses of 0, 50, 100, 250 μg into anaesthetized mice in supine position on an angled stand (n = 5 animals per dose). Mice were euthanized after 24 h, bronchioalveolar lavage (BAL) fluid and blood plasma were obtained as described before [25]. BAL cells were counted and differential cytology (Wright-Giems stain) was determined on CytoSpin preparations (Thermo Shandon, Pittsburgh PA, USA) prepared on glass slides using the Cytospin 3 centrifuge. Cell differential counts of alveolar macrophages, neutrophils, lymphocytes, monocytes, eosinophils, basophils and band cells (immature neutrophils) were determined from a pair of slides for each animal. Fold change (i.e. treatment/control) of the cell counts are presented. The values are based on the percentage of cell type in a differential profile multiplied by the total cell number (calculated as cell # per ml BAL × volume of instilled lavage fluid in the animal) to obtain an estimate of the absolute number of the cell types recovered from lavage.
Biochemical assays in vivo
8-Isoprostaglandin-2a (Isoprostane) was quantitated from blood plasma samples by competitive Enzyme Immuno-Assay (EIA) following manufacturer-recommended procedures (Cayman Chemical, Ann Arbor, MI, USA). LDH in BAL was analyzed essentially as described for cell cultures. Total protein was measured by the Coomassie Plus (Bradford) assay as per manufacturer’s protocol (Pierce, Rockford, IL, USA).
Cytokine secretion in vivo
The profile of IL-1α (0.1 ± 0.1 pg/mL basal level in control cells ± standard error mean), IL-1β (1.3 ± 0.2), IL-4 (n.d.; not detected), IL-5 (0.5 ± 0.5), IL-6 (0.1 ± 0), IL-10 (0.3 ± 0.13), GM-CSF (1.3 ± 0), KC ((3.8 ± 0.9), MIP-1α (25.4 ± 8.8), RANTES (0.3 ± 0.2) and TNF-α (3.7 ± 1.0) was determined by Bio-Plex analyses of BAL samples using Bio-Plex cytokine panels, with a Bio-Plex 200 array system. Basal levels in control cells are in parentheses above and represent pg/mL ± standard error mean measured in cell culture supernatants and n.d. denotes cytokine/chemokine not detected.
Particle potency estimates
The dose–response data for cell viability, cytokine secretion and Ah receptor-dependent gene induction (AhR response was scaled by dividing 20-fold to relate its magnitude to the other endpoints), were normalized within an experiment for all doses (including 0 μg dose control), to the grand mean value of all controls (0 μg dose of particles), to obtain fold-effect (FE) for each particle dose. Potency (β) was derived from
where β is the rate of change of dose with respect to the logarithm of fold-effect for a given endpoint [24]. The dose–response data were fitted using CurveExpert v1.3 (D. Hyams, Hixson, TN, USA).
The following potencies were calculated: potency for viability (βV) in a given assay, potency for Ah receptor-dependent gene induction (βAhR), and potencies for secretion of a cytokine (βI). βI was adjusted for cell viability according to
where βI-V represents an unbiased potency estimate that allows distinction of apparent down-regulation of a cytokine from the absence of cytokine secretion by dead cells, or an apparent up-regulation of cytokine expression in cells simply due to an increase of cell mass in the wells.
The potencies were combined as follows;
-
1)
An average cytotoxic potency in each cell line was calculated using assay specific potencies according to:
-
2)
Biological reactivity (βR) of the particles was defined as any deviation from baseline in cytotoxicity assays. βR CELLS was calculated by averaging the absolute values (to capture any deviation from baseline control levels) of the βV according to:
-
3)
An average potency for inflammation was calculated within each cell line using +1*βI-Vfor chemokines and pro-inflammatory cytokines and −1*βI-V for anti-inflammatory cytokines. Two different estimates of inflammatory potency were calculated.
-
a)
βI-V HI, an average considering the anti-inflammatory effect of IL-10 (−1*βI-V for IL10).
-
b)
βI-V LO, an average considering anti-inflammatory effects of IL-6 and IL-10.
-
a)
-
4)
βR CELLS and one of the two estimates of βI-V were further integrated to calculate cell-specific Integrated Beta potencies (Iβ). For example,
-
5)
βR and βI-V were integrated across the cell lines as follows.
-
6)
βR and one of the two estimates of βI-V across cell lines were further integrated to a grand Integrated potency (Iβ). For example,
In a similar fashion, potency estimates were determined for each in vivo endpoint. A potency estimate for acute lung toxicity, βT BALB/c was determined from the average of potencies for BAL endpoints (protein, LDH, 8-isoprostane, neutrophils, macrophages, band cells, lymphocytes), while a potency estimate for inflammation, βI BALB/c was determined from the potencies of particles for inducing the release of cytokines and chemokines. As described for the in vitro inflammatory potency estimates, βI HI, an average considering the anti-inflammatory effect of IL-10 (−1*βI for IL10) and βI LO, an average considering anti-inflammatory effects of IL-6 and IL-10 were determined. An integrated potency estimate that incorporates all in vivo data, Iβ BALB/c was obtained from the average of βT BALB/c and βI BALB/c.
Statistical analyses
To ensure that assays were sensitive to the dose of particles, fold effect (FE) data were first analyzed by two-way ANOVA with DOSE (0, 10, 20, 40, 80, 160 μg/well) for in vitro data, or (0, 50, 100, 250 μg) for in vivo data, and Particulate Matter (PM) as factors. Datasets not meeting normality and equal variance were subjected to log10, inverse or square root transformations (in the order given) until the assumptions were met, or else rank transformed prior to analyses. Pairwise multiple comparisons were carried out using Tukey’s procedure to elucidate the pattern of significant effects (α = 0.05). The analyses were conducted using SigmaPlot, version 12.5 (Systat Software, Inc., San Jose, CA, USA).
Hierarchical clustering of the pattern of cytokine secretion by A549 and J774 in response to particle exposure were conducted using the GenePattern webtool (http://www.broadinstitute.org/cancer/software/genepattern) [29], and visualized as heatmaps using Java TreeView plugin version 1.16.r2 (http://jtreeview.sourceforge.net) [30].
Linear regression between corresponding individual or combined potency estimates in vitro and in vivo was conducted using Sigmaplot v12.5 and depicted using Microsoft Excel 2010 (Microsoft Corp., Redmond, WA, USA). The strength of the relationship between every two variables was described by a correlation coefficient R and the significance of the hypothesis test by the p-value of 0.05 (two-tailed test), or one-tailed test, where applicable (i.e. consistent directionality of the variables).
The correlations presented are performed between in vitro and in vivo matched endpoints across all particles, based on individual particle potencies (Table 1) or combined potency estimates (average of endpoints) for toxicity, inflammation, or integrated inflammation plus toxicity (Tables 2, 3, and 5) all eight particles tested in vitro (EHC-93, EHC-98, EHC-2000, SRM-1648, SRM-1649, DWR1, TiO2, SiO2) or for five particles tested in vivo (EHC-2000, SRM-1649, DWR1, TiO2, SiO2) for Table 5.
In addition, a selection of key potency estimates were ranked and the strength of their relationship was reassessed using Spearman’s rank-order correlation test (one-tailed test), as the selected variables were observed to have a consistent positive relationship, with a significance level of 0.05, using Spearman’s Rho calculator (http://www.socscistatistics.com/tests/spearman/default2.aspx; Social Science Statistics).
Results
Cytotoxicity and Ah receptor-dependent gene induction
Exposures of A549 and J774A.1 cells to particles revealed cytotoxicity manifested by increased release of LDH into cell culture supernatants (Fig. 1a and b) which was particle type- and dose-dependent (two way ANOVA, PM × Dose, p < 0.001), most prominent at the highest doses of the particles. The LDH assay also revealed a significant decrease in LDH release in J774A.1 cells exposed to lower and intermediate doses of urban particles in comparison to baseline levels, but not in response to mineral particles (Fig. 1a). This response was not observed in A549 cells exposed to particles (Fig. 1b). Furthermore, we observed reduced metabolic activity manifested by decreased resazurin reduction (Additional file 2: Figure S1A and S1B), cellular ATP levels (Additional file 2: Figure S1C and S1D) as well as attenuated cell proliferation measured by BrdU incorporation assay (Additional file 2: Figure S1E and S1F). The presence of Ah receptor agonists was detected in urban particles, but not mineral particles (two way ANOVA, PM × Dose, p < 0.001) measured by AhR-mediated luciferase reporter gene activity in H1L1.1c2 cells. A small but significant decrease in AhR-mediated gene activity below baseline levels was detected in cells exposed to the mineral particles (Fig. 1c).

Lactate Dehydrogenase (LDH) release into cell culture supernatants of J774A.1 (a) and A549 (b) cells exposed for 24 h to particles, and Aryl hydrocarbon receptor (AhR) induction by particles in the H1L1.1c2 cells after a 4 h exposure (c). For the LDH assay, data represent LDH release, adjusted for total cellular LDH content. For the AhR assay, B[a]P was used as a positive control (0, 1, 10, 100 nM). All values are presented as average fold-effect (FE) over control ± standard error (J774A.1, n = 4; A549, n = 3; H1L1.1c2, n = 3). Two way ANOVA; J774A.1, PM × Dose interaction, p < 0.001, asterisks (*) represent effects significantly different from Dose 0 control, Tukey test, p < 0.05; A549, PM × Dose interaction, p < 0.001, asterisks (*) represent effects significantly different from Dose 0 control, Tukey test, p < 0.05. Two way ANOVA; AhR, PM × Dose interaction, p < 0.001, asterisks (*) represent effects significantly different from Dose 0 control, Tukey test, p < 0.05; one way ANOVA; B(a)P, Concentration, p = 0.016, Conc. 100 vs. 1 (†), Tukey test, p < 0.05
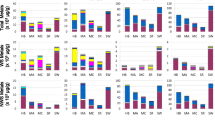
Cytotoxic potency of each particle was represented by βV, the rate of change of dose with respect to the logarithm of fold-effect in a given assay. The potency estimates were summarized in a heatmap (Fig. 2a). In both cell lines, SRM-1648 and SRM-1649 urban particles were more cytotoxic than the other particles, including the EHC-type particles. TiO2 particles were also notably cytotoxic. The PM2.5 DWR1 and the CRI particles showed a cluster of the lowest cytotoxic potency (Fig. 2a). This behavior of CRI in the present series of experiments was atypical, as we expected CRI, based on our experimental data, to be more cytotoxic in vitro than TiO2. In general, there was no correlation of particle effects between the two cell lines based on the corresponding individual assays, with the exception of J774A.1 cell proliferation response which was significantly correlated with A549 cell proliferation estimate (R = 0.792; p = 0.019; Table 1).

Heatmap with hierarchical cluster analysis of cytotoxic potency of particles in J774A.1 and A549 cells based on cytotoxicity assays (LDH release, AB reduction, ATP content, BrdU incorporation) and AhR induction in H1L1.1c2 cells. Within the heatmap, green color represents increased cytotoxic potency of particles (negative β) for majority of endpoints, except the AhR assay, where red color depicts increased potency (positive β) (a). Consensus cytotoxic potency βV CELLS was calculated for each particle as average of all assay potencies, separately, for J774A.1 and A549 cells. AhR induction potencies of the particles in H1L1.1c2 cells are also presented (b)
Next, we combined cytotoxic potencies of the particles from all in vitro endpoints in a given cell line into a single βV value (Fig. 2b). The cell-type specific, combined potency data outlined the cytotoxicity-based ranking of the particles and indicated that overall, J774A.1 cells were more susceptible to particle effects than A549 cells. SRM- and EHC-type urban particles had comparable potency for Ah receptor-dependent gene induction in H1L1.1c2 cells, which was markedly lower for the PM2.5 DWR1 particles, while a weak inhibitory potency was observed in the case of the TiO2 and CRI particles (Fig. 2b). Correlation analysis of the cell type-specific consensus potency βV and biological reactivity of the particles βR (any deviation from baseline control values) indicated that the aggregated cytotoxic responses of A549 and J774A.1 cells were significantly correlated (p ≤ 0.05; Table 2).
Cytokine secretion in vitro
Release of cytokines into the culture medium upon cell exposures was taken as a measure of inflammatory potential of the particles. Significant release of cytokines was induced in response to particle exposures in both cell lines. The observed responses were particle type- and/or dose-specific as exemplified by IL-6 release (Fig. 3a and b; two way ANOVA, PM × Dose, p < 0.001 for J774A.1; Dose main effect, p < 0.001 for A549) and the other cytokines (Additional file 3: Figure S2A to S2F; Additional file 4: Figure S3A to S3G, two way ANOVA, p < 0.05). Cell viability-adjusted inflammatory potency (βI-V; cytokine release) of the particles in the cells was visualized by a heatmap. In both cell lines, the majority of the cytokines were increased in response to particle exposures, except IL-10, which generally remained unchanged or decreased in J774A.1 cells (Fig. 4a and Additional file 3: Figure S2D; two way ANOVA, PM × Dose, p = 0.026) and remained unchanged or showed an increasing trend in A549 cells (Fig. 4b and Additional file 4: Figure S3D; not significant). Interleukin-6 and TNF-α showed the highest fold-change from baseline values in J774A.1 cells (Fig. 3a and Additional file 3: Figure S2F two way ANOVA, PM × Dose, p < 0.001) and clustered separately from the rest of the cytokines (Fig. 4a). In A549 cells, IL-6, IL-8 and TNF-α showed the highest fold-change from controls (Fig. 3b, Additional file 4: Figure S3C and S3G; two way ANOVA, PM × Dose, p < 0.05 for IL8; PM, Dose main effects, p < 0.001 for TNF-α; Dose main effect, p < 0.001 for IL-6) and clustered separately from the remaining cytokines (Fig. 4b).

Interleukin (IL)-6 levels in cell culture supernatants from J774A.1 (a) and A549 (b) cells exposed to particles for 24 h. Values represent mean fold effect (FE) over control ± standard error of the mean (n = 3). Two way ANOVA; J774A.1, PM × Dose interaction, p < 0.001, asterisks (*) represent effects significantly different from Dose 0 control, Tukey test, p < 0.05; A549, Dose main effect, p < 0.001, Dose 160 vs. 0 or 10 (†), Tukey test, p < 0.05

Heatmap with hierarchical cluster analysis of cytotoxic potency of particles in J774A.1 (a) and A549 (b) cells based on cytokine release after 24 h exposure to particles. Inflammatory potency estimate, βI-V for J774A.1 (c) and A549 (d) cells representing the average cytokine potency adjusted for cell viability. The βI-V LO represents the lower estimate (IL-10 and IL-6 potency subtracted from average potency), while βI-V HI is the higher estimate (IL-10 potency subtracted, IL-6 added to average potency). Within the heatmaps, red color represents increased inflammatory potency of particles (increased cytokine/chemokine levels; positive β), whereas green color represents decreased inflammatory potency (negative β)
As was observed for most of the cytotoxicity endpoints, particle effects on corresponding individual cytokine release in A549 and J774A.1 cells were not correlated (Table 1).
Notably, we observed a significant correlation between AhR activity in H1L1.1c2 cells with IL-1β, IL-6 and TNF-α release in J774A.1 cells (Table 3; R > 0.7, p ≤ 0.05), but not with A549-produced cytokines.
Next, inflammatory potency βI-V values for all cytokines were averaged within a cell line to obtain a consensus inflammatory index presented in three scenarios that assumed the general role of specific cytokines as anti- or pro-inflammatory mediators. The lower inflammatory estimate βI-V LO was computed by subtracting potency values for IL-10 and IL-6 from the average estimate, assuming the dual role of IL-6 as a pleiotropic inflammatory mediator [31, 32], while the higher inflammatory estimate, βI-V HI was obtained by subtracting IL-10 potency value only. The inflammatory index revealed a distinct profile of particle effects on cytokine release in A549 and J774A.1 cells, highlighting a difference in particle ranking.
For example, J774A.1 cells were more sensitive to the effects of urban particles, especially EHC-98, EHC-2000 (Fig. 4c) whereas A549 cells were impacted most by CRI and TiO2 mineral particles (Fig. 4d). Based on the inflammatory scenarios, IL-6 release was the dominant response in J774A.1 cells, with highest magnitude of effect under the βI-V LO and βI-V HI inflammatory scenarios, while its impact in A549 cells was minor. Correlation analysis of the consensus βI-V inflammatory potency indices of the particles indicated that the aggregated inflammatory responses of the two cell lines were distinct (Table 2).
The particles were screened for the presence of endotoxin, biogenic material capable of inducing cytokines in vitro and in vivo. Majority of the urban particles, including DWR1 had similar endotoxin content (~70 – 130 EU/μg particles). SRM-1649 particles had noticeably less endotoxin (3.0 EU/μg particles) while mineral particles did not contain endotoxin (Table 4). A correlation of endotoxin with the inflammatory potency estimates in J774A.1 cells (βI-V J774A.1) and A549 cells (βI-V A549) revealed a significant correlation with the inflammatory potency estimate in J774A.1 cells (R = 0.729; p = 0.040), but not A549 cells (Table 5).
In vitro integrated particle potency
To summarize all cytotoxic particle effects in vitro across cell types, biological reactivity βR CELLS was derived by averaging the absolute values of the cytotoxic potencies, βV of the particles in A549 and J774A.1 cells and the AhR activity in H1L1.1c2 cells, assuming biological reactivity of the particles as any deviation from baseline. From the βR CELLS estimate, SRM-1648 and SRM-1649 particles had the highest overall cell potency in contrast to DWR1 and CRI particles which showed the lowest values (Fig. 5a).

Biological reactivity estimate, βR CELLS was derived by averaging the cytotoxic potencies of the particles in A549 and J774A.1 cells, as well as the AhR activity in H1L1.1c2 cells (a). A combined inflammatory estimate, βI-V CELLS, was obtained for each particle by averaging particle inflammatory potency estimates adjusted for cell viability of J774A.1 and A549 cells. The βI-V LO represents the lower estimate (IL-10 and IL-6 potency subtracted from average potency), while βI-V HI is the higher estimate (IL-10 potency subtracted, IL-6 added to average potency) (b). An integrated potency estimate of the particles, Iβ CELLS was determined by averaging the biological reactivity (cytotoxic potency) and inflammatory estimates for each particle. Iβ LO represents the lower estimate (IL-10 and IL-6 potency subtracted from average potency). Iβ HI represents the higher estimate (IL-10 potency subtracted, IL-6 added to average potency) (c)
Similarly, an inflammatory potency estimate βI-V CELLS, was obtained for each particle by averaging the cell viability-adjusted inflammatory potency values of the particles across both cell lines for all cytokines detected, for each inflammatory scenario (Fig. 5b). The estimates impacted the magnitude of particle potency ranking, but the ranking was comparable between the different scenarios, where the urban particles, except EHC-93 were more potent than mineral particles and DWR1.
Lastly, an overall integrated potency estimate, Iβ CELLS of the particles was calculated by averaging the biological reactivity, βR CELLS with inflammatory indices, βI-V CELLSfor each particle (Fig. 5c). The Iβ CELLS profile was similar to the profile of the inflammatory indices βI-V CELLS.
Toxicity of particles in vivo
BALB/c mice were exposed for 24 h to a subset of particles selected based on contrasting in vitro potency (EHC-2000, SRM-1649, CRI, TiO2 and DWR1) by intratracheal instillation (IT). Acute pulmonary toxicity was observed in mice exposed to the high dose (250 μg) of the particles, as shown by a general increase in BAL total protein (Fig. 6a; two way ANOVA, Dose main effect, p < 0.001) and BAL LDH (Additional file 5: Figure S4A; two way ANOVA, PM × Dose, p = 0.035). A particle dose-dependent increase in 8-isoprostane levels in blood plasma (Additional file 5: Figure S4B; two way ANOVA, Dose main effect, p = 0.006) indicated a systemic oxidative stress after exposure to particles. Neutrophil influx (Fig. 6b; two way ANOVA, PM × Dose, p = 0.004) and a drop in lung-resident alveolar macrophages (Additional file 5: Figure S4C; two way ANOVA, Dose main effect, p < 0.001, PM main effect, p = 0.018) were consistent with a particle challenge and clearance after 24 h. Based on the in vivo toxic potency estimates (β from cell differential counts and biochemical endpoints), particles elicited an increase in acute toxicity and cell injury marked by an increase in neutrophils and a decrease in macrophages. SRM-1649, EHC-2000 and CRI clustered separately from less toxic particles DWR1 and TiO2 (Fig. 6c).

Total protein levels (a) in bronchioalveolar lavage (BAL) fluid of BALB/c mice exposed to particles for 24 h by intratracheal (IT) instillation. Neutrophil cell counts (b) were determined from cytospin preparations of BAL. All values are presented as mean fold effect (FE) over control ± standard error (n = 5). Two way ANOVA; Total protein, Dose main effect, p < 0.001, Dose 250 vs. 0, 50 or 100 (†), Tukey test, p < 0.001; Neutrophils, PM × Dose interaction, p = 0.004, asterisks (*) represent effects significantly different from Dose 0 control, Tukey test, p < 0.05. Heatmap with hierarchical cluster analysis of toxicity endpoints (cell differential counts and biochemical endpoints) (c) assessed in BALB/c mice 24 h after exposure to particles by intratracheal instillation, summarized as potency estimates (slopes of the dose response). Within the heatmaps, red color represents increased toxic potency of particles (increased biochemical measures or cell counts; positive β), whereas green color represents decreased toxic potency, with the exception of macrophages, whereby green color representing a loss of cells is a measure of increased particle potency (negative β)
Cytokine secretion in vivo
Significant particle type- and/or dose-dependent increase in inflammatory mediators IL-6, IL-1α, IL-1β, IL-5, KC, MIP-1α, RANTES and TNF-α (Fig. 7a and Additional file 6: Figure S5A to S5I); two way ANOVA, p < 0.05) were measured in BAL fluid collected from mice exposed to particles for 24 h, except IL-10 which was not altered (Additional file 6: Figure S5E, not significant). Individual in vivo inflammatory potency estimates (β from the cytokine dose response) confirmed a particle-induced increase for most of cytokines assessed. IL-6 and TNF-α were highlighted as the most responsive cytokines to particle exposures in vivo (Fig. 7b). SRM-1649 was the most potent particle also based on the inflammatory potency estimates, as it clustered distinctly from the remaining particles (Fig. 7b).

Interleukin (IL)-6 (a) levels in BAL. All values are presented as mean fold effect (FE) over control ± standard error (n = 5). Two way ANOVA; IL-6, PM × Dose interaction, p = 0.036, asterisks (*) represent effects significantly different from Dose 0 control, Tukey test, p < 0.05. Heatmap with hierarchical cluster analysis of the individual inflammatory mediators (cytokines/chemokines) (b) assessed in BALB/c mice 24 h after exposure to particles by intratracheal instillation, summarized as potency estimates (β from the cytokine dose response). Within the heatmaps, red color represents increased inflammatory potency of particles (increased cytokine/chemokine levels; positive β), whereas green color represents decreased inflammatory potency (negative β)
In vivo integrated particle potency
All of the acute toxicity endpoints assessed in vivo were averaged and depicted as βT BALB/c toxicity estimate for each particle. The combined βT BALB/c estimate revealed SRM-1649, EHC-2000 and CRI as highly potent particles, compared to TiO2 and DWR1 (Fig. 8a). An inflammatory potency estimate βI BALB/c was obtained by averaging the dose response of all cytokines measured in BAL fluid. As was done for in vitro cytokines, three scenarios were described, βI LO BALB/c, βI HI BALB/c and their average, based on subtraction of both, IL-10 and IL-6 (βI LO BALB/c), or IL-6 only (βI HI BALB/c). The inflammatory potency estimates βI BALB/c ranked identically to toxicity estimates βT BALB/c, with no impact of the inflammatory potency scenarios on the ranking of particles (Fig. 8b). A final, in vivo integrated potency estimate Iβ BALB/c was calculated by averaging βT BALB/c and βI BALB/c for each particle (Fig. 8c), as a consensus of all endpoints measured in vivo. Iβ BALB/c estimate revealed SRM-1649, EHC-2000 and CRI as more potent than TiO2 and DWR1 respectively, regardless of the integrated potency estimate scenarios (Iβ HI, Iβ LO). Endotoxin content in particle preparations administered to mice was not correlated with the combined in vivo inflammatory potency estimates (βI BALB/c) of the particles (Table 5).

Toxic potency estimate in BALB/c, βT BALB/c was derived by averaging the potencies of the particles across the cell-specific and biochemical measures from BALB/c mice exposed to the particles by intratracheal instillation (a). The combined inflammatory potency estimate, βI BALB/c, was obtained for each particle by averaging the particle potencies for altered cytokine/chemokine levels in bronchioalveolar lavage. The βI LO represents the lower estimate (IL-10 and IL-6 potency subtracted from average potency). The βI HI represents the higher estimate (IL-10 potency subtracted, IL-6 added to average potency) (b). The integrated potency estimate, Iβ BALB/c of the particles was determined by averaging the toxic and inflammatory potency estimates for each individual particle. The Iβ LO represents the lower estimate (IL-10 and IL-6 potency subtracted from average potency), while Iβ HI is the higher estimate (IL-10 potency subtracted, IL-6 added to average potency) (c)
In vitro and in vivo correlation
The relationship between particle effects in cells in vitro and in BALB/c mice exposed to particles by intratracheal instillation was explored by correlating the individual in vitro and in vivo particle potency estimates, as well as the in vitro and in vivo combined particle potency estimates for the five particles tested by both approaches.
Majority of the individual particle potency estimates for the corresponding endpoints were not correlated, with the exception of a single association; the cellular LDH content in J774A.1 cells, significantly correlated with LDH content measured in BALB/c BAL fluid, (Additional file 1: Table S2; R = 0.928, p = 0.023).
When the combined particle potency estimates in vitro and in vivo were assessed by Pearson product–moment correlation analysis for all particles tested, no statistically significant correlations were observed between the corresponding pairs of the key estimates, including the key estimates; βR CELLS versus βT BALB/c (Fig. 9a and Additional file 1: Table S3; R = 0.541, p = 0.173); βI-V CELLS versus βI BALB/c (Fig. 9b and Additional file 1: Table S3; R = 0.639, p = 0.123) and Iβ CELLS versus Iβ BALB/c (Fig. 9c, Additional file 1: Table S3; R = 0.668, p = 0.109). However, a consistent increasing trend between these in vitro and in vivo estimates was observed across the particles, where DWR1 was ranked low, CRI and TiO2 were ranked intermediate and SRM-1649 and EHC-2000 were ranked high in potency (Fig. 9). Based on the observed trend in the ranking of the particles, a directional (one-tailed) Spearman’s rank-order correlation analysis was conducted. The analysis revealed that while the combined potency estimates βR CELLS versus βT BALB/c remained non-correlated (Table 6; R = 0.667, p = 0.109), correlations of βI-V CELLS versus βI BALB/c and the integrated potency estimates Iβ CELLS versus Iβ BALB/c approached the threshold for significance α = 0.05 (Table 6; R = 0.8, p = 0.052). Finally, improved correlation coefficients for the associations of the combined particle potency estimates were observed when mineral particle responses were excluded from the estimates, albeit they were not statistically significant due to low power (Additional file 1: Table S3); βR CELLS versus βT BALB/c (R = 0.862, p = 0.169); βI-V CELLS versus βI BALB/c (R = 0.888, p = 0.153) and Iβ CELLS versus Iβ BALB/c (R = 0.955, p = 0.096). Remarkably, CRI contributed most to the decreased association of in vitro and in vivo potency estimates, as exclusion of CRI only revealed improved correlation comparable to the associations excluding both mineral particles (Fig. 9, Additional file 1: Table S3); βR CELLSversus βT BALB/c (R = 0.865, p = 0.068); βI-V CELLS versus βI BALB/c (R = 0.800, p = 0.100) and Iβ CELLS versus Iβ BALB/c (R = 0.895, p = 0.053). When the Spearman’s rank-order correlation analysis was conducted on the particle ranks while excluding CRI, a statistically significant rank-order was observed for βR CELLS versus βT BALB/c(Table 6; R = 1.0, p < 0.001), while βI-V CELLS versus βI BALB/cand the integrated potency estimates Iβ CELLS versus Iβ BALB/c did not show a significant trend in a correlation due to low power (Table 6; R = 0.8, p = 0.100).

Scatter plots of key corresponding in vitro and in vivo combined particle potency estimates βR CELLS (in vitro assays) with βT BALB/c (in vivo cell counts and biochemical endpoints) (a); βI-V CELLS (in vitro cytokines) with βI BALB/c (in vivo cytokines) (b) and integrated particle potency estimates, Iβ CELLS (averaged in vitro assays and cytokines) with Iβ BALB/c(average of in vivo cell counts, biochemical endpoints and cytokines) (c). Pearson correlation coefficients and p-values (1-tailed) are presented for correlations of all particles (dashed grey line) and particles excluding CRI (solid black line)
Discussion
In this work we examined the level of concordance between in vitro and in vivo responses to particulate matter, to determine the potential of cell-based cytotoxicity assays and cytokine/chemokine mediators in screening the toxicity of ambient particles. The dose response results from the bioassays conducted in cell lines and from intratracheal exposures of mice to particles were summarized as slope-based measures of particle potency for each endpoint. The potency values were combined within each exposure model to obtain model-specific consensus potency estimates of toxicity and inflammatory potential of the particles, which were further combined into an overall integrated potency estimate. The key derived particle potency estimates were then assessed for correlation across the in vitro and in vivo models.
We note that in the present series of experiments we observed an atypically low in vitro cytotoxicity of the CRI sample by comparison to TiO2. In vivo toxicity of the same sample however was high, in accordance with our historical observations and literature data on toxicity of silica. Identity of the CRI samples was reconfirmed by ICP-MS/AES analysis of the archived, diluted working particle stocks that were used in the experiments. We have also carefully validated the datasets, but could not explain this atypical behavior of CRI in the series of in vitro assays presented here, e.g. from computational errors or material switching in assays, and therefore we report the data as observed.
Urban and mineral particles induced toxicity in vitro and in vivo (Figs. 2a and 6c). Cytotoxic responses in vitro based on the individual endpoints were in general different between epithelial cells and macrophages, with the exception of the cell proliferation response (Table 1). The pulmonary alveolar epithelium (represented by the A549 cell line) serves as a barrier/interface with the outside environment, facilitates gas transfer and fluid clearance, reduces surface tension and prevents exposure to particles, toxicants and microbes [33]. It is a resilient interface adapted to the mechanical forces of ventilation and it is involved in pulmonary innate host defense and tissue repair [34]. Alveolar epithelial cells also produce a multitude of factors such as cytokines and chemokines (e.g. MCP-1, GM-CSF, IL-8), which impact the activity of pulmonary macrophages [35]. As well, the cells express a variety of pattern recognition receptors (e.g. NLRs, TLRs) for microbes and particulates [34]. Pulmonary macrophages are phagocytic cells involved in direct response to pathogens, antigen sensing and activation of acquired and innate immunity, and they serve as modulators of inflammation through removal of pathogens, foreign materials and apoptotic cells, and through production of a specific suite of pro- and anti-inflammatory cytokines and chemokines [34]. We used the J774A.1 murine macrophage cell line as a model macrophage. Although the cells originate from peripheral blood, they display the hallmarks common to both blood-borne and lung-resident macrophages, including adherent, cytologic and phagocytic properties [36].
The greater susceptibility of the macrophages to particle exposures compared to the epithelial cells observed in this work (Fig. 3a) highlights the importance of using functionally distinct cell types for hazard screening. The innate phagocytic activity of macrophages results in increased intracellular particle loads and cytotoxic effects that are more pronounced, than in epithelial cells which do not undergo internalization of particles to the same extent. Specifically, J774A.1 cell proliferation was more impacted by particle exposures than A549 cells (Additional file 2: Figure S1E and S1F). A greater decrease in proliferation and increase in apoptosis in J774A.1 cells compared to A549 cells has also been demonstrated after exposures to Mexico City PM10particles [37]. Similarly, we have observed greater sensitivity of J774A.1 cells than A549 cells to carbon nanotube exposures [38].
The impact of increased sensitivity of J774A.1 cells to particle exposures was also revealed by a small decrease of LDH release below control levels in cells exposed to the intermediate doses of the urban particles (Fig. 1a). A combination of the decreased cell proliferation from the effects of the intermediate particle doses and a lack of space for cell division in the nearly confluent wells throughout the exposures likely resulted in an increased cell death. The phenomenon was not observed in the unexposed controls, or in cells exposed to the high doses of the particles where the overt cytotoxicity led to high LDH release, or in A549 cells that were less sensitive to particle exposures.
Nevertheless, when the panel of in vitro toxicity endpoints (viability assays) was combined, the overall particle potency was correlated across the cell lines (Table 2) regardless of their functional difference, suggesting that the cytotoxic profile of the particles reflects a more general outcome (e.g. apoptosis), over different but converging mechanisms of cytotoxicity. Although the particles affect the cells through different pathways, with a sufficiently broad array of assays it appears that the inherent cytotoxic potential of the particles can be captured. This is consistent with our previous observations that cytotoxicity in vitro appears to correlate with time-weighted average exposure limits for particles of occupational health relevance [39].
In the present study, however, inflammatory potential determined from the release of cytokines did not correlate between the two cell lines, or with the overall cytotoxic potency of the particles (Table 2). This reveals that functional impacts of particles on the cells (e.g. inflammatory response) are distinct from overt cytotoxicity and will be sensitive to the cellular model used. Similarly, we have shown previously that the impacts of urban particles and minerals on the respiratory burst function of stimulant-induced primary rat macrophages were not correlated with their cytotoxic effect determined by the water-soluble tetrazolium salt XTT reduction assay [39]. In the present study, the cytokine profiles were more reflective of the functional specificity of the cell types. In this context, A549 epithelial cells were similarly responsive to mineral and urban air particles, in contrast to J774A.1 macrophages which were more impacted by the compositionally-complex urban air particles (Fig. 4). The higher cytotoxicity of minerals in A549 cells compared to J774A.1 cells highlights a potential importance of particle surface interactions with cells that provide a barrier-type function, such as lung epithelial cells. Irregularity of insoluble particle surface at the molecular level can modulate particle cytotoxicity via surface-associated free-radical formation [40]. Assessment of inflammatory mediators was more reflective of organic content of particles. Of note is that the AhR activity in H1L1.1c2 cells was correlated with IL-1β, IL-6 and TNF-α release in J774A.1 cells (Table 3). As shown here, and previously [24], AhR agonists, such as polycyclic aromatic hydrocarbons (PAHs) are a significant component of particulate emissions. Besides its role in xenobiotic metabolism, the Ah receptor is involved in immunity and lung inflammation [41].
It has been shown that 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-activated AhR signaling participates in the up-regulation of IL-1β, IL-6 and IL-8 expression through activation of the nuclear factor (NF-kB) and extra-cellular stimulus-activated kinase (ERK) signaling cascades. Dioxins and PAHs are major components of cigarette smoke. The AhR protein expression was up-regulated in the presence of TNF-α and smoking and exposure to TCDD enhanced rheumatoid arthritis (RA) inflammatory processes in synovial tissue from RA patients [42]. Interestingly, SiO2-induced acute lung inflammation is more severe, while fibrotic response is attenuated in AhR−/− mice compared with C57Bl/6 mice, suggesting a complex role for AhR in regulation of inflammation [43]. The correlation with key pro-inflammatory cytokines produced by J774A.1 cells upon particle exposures in the present study (Table 3) suggests that AhR signaling in lungs may represent an important pathway in regulating inflammation triggered by emission particles. This notion requires further mechanistic testing. Local production of inflammatory mediators is a hallmark of pulmonary response to foreign materials. These observations emphasize the need to capture a diverse set of endpoints that are broadly reflective of the functional specificity of cells and are tied to the particular hazard or route of exposure.
We have observed a small decrease in AhR-mediated gene activity below control levels in cells exposed to CRI and TiO2 particles (Fig. 1c), suggesting an inhibitory effect of the particles in the Ah-dependent gene induction assay. Clarification of supernatants or cell lysates is a recommended approach to facilitate fluorimetric-, colorimetric-, or luminescence-based in vitro toxicological assessments of optically/chemically active materials such as nanoparticles, or their micron-sized counterparts [28]. This assay protocol was adapted by introducing a step to clarify cell lysates from particle residues by centrifugation to prevent potential interference with the luciferase-mediated reaction. Nonetheless, trace amounts of particles may have remained in the cell lysates allowing for the possibility of a small underestimation of AhR-mediated gene activity. Furthermore, a recent publication based on an in silico model for Ah receptor docking by ligands provides some evidence that TiO2-based nanoparticles could directly bind/adsorb to the Ah receptor with very high affinity, although this notion has yet to be experimentally validated in cellular models [44].
Endotoxin levels in urban particles were correlated with the inflammatory index βI-V J774A.1 which is based on the cytokine release by J774A.1 cells, but not with A549-cell based inflammatory index βI-V A549 or the in vivo inflammatory index βI BALB/c (Table 5). It is conceivable that the endotoxin component in urban particles may play a role in cytokine induction by macrophages. Macrophages express lipopolysaccharide-responsive Toll-like receptor 4 on their cell surface [45]. Endotoxin-associated induction of pro-inflammatory cytokines TNF-α, IL-1 and IL-6 can be inhibited by the antimicrobial agent polymyxin B in alveolar macrophages exposed to urban particles [46]. However, only a small amount of endotoxin (~0.1 EU/mg particle) was detected in the urban particles tested (Table 4). The trace levels in EHC-93 particles (a particle used in the present study) do not have appreciable effects on mRNA expression of pro-inflammatory genes in human bronchial epithelial cells [47]. Also, endotoxin at comparable levels did not activate the bone marrow (measure of systemic inflammatory response) of rabbits upon its instillation in lungs of the animals [48]. In addition, in the present study, mineral particles were highly potent in triggering cytokine release both in vitro and in vivo despite the absence of endotoxin (Figs. 4 and 7). Conversely, SRM-1649 particles had ~35-fold lower endotoxin content than the other urban particles, but SRM-1649 induced cytokines at the same magnitude as in cells exposed to the other urban particles. Thus other particle components likely contributed to the release of inflammatory mediators by cells observed in our study. Up-regulation of genes for cytokines and chemokines in cells in vitro (e.g. bronchial epithelial cells) has been observed in response to various subcomponents of particulate matter including carbon-core, transition metals or polyaromatic hydrocarbons [49].
With the exception of the mineral particles, the coarse and fine urban particles tested here represent complex mixtures containing both soluble and insoluble fractions, which will contribute to the potency in vitro to varied extents due to differences in uptake mechanisms, molecular targets of the various particle components, corona formation or differences in the settling rate of the denser particle components. Therefore, homogeneity of particle suspension delivered to the cells was achieved by sonication of the particles prior to delivery. However, it has to be noted that, even with the in vivo delivery of environmental particles, similar differences of soluble and insoluble fractions of the particulate mixture will be encountered. Specifically, the issue of dilution of soluble components of ambient particles in the culture medium has been examined in previous work [24]. By ensuring internal consistency in assay conditions, the biases, if present, will be systematic, affecting all particles studied.
Instillation of particles in lungs of BALB/c mice led to elevated neutrophil counts and cytokine levels in BAL fluid, and generally unremarkable changes in biochemical markers, a profile consistent with mild, acute pulmonary inflammation at 24 h post-exposure. This was accompanied by a decrease in alveolar macrophages in BAL fluid, consistent with macrophage-associated pulmonary clearance of foreign particles (Fig. 6). Determination of neutrophil counts in lung lavage fluid represents a key, sensitive response to pulmonary instillation of urban and mineral particles [50, 51]. Recruitment of neutrophils in response to acute particle exposure is a consequence of a local lung inflammatory cascade aimed at the clearance of the foreign materials and macrophage apoptotic bodies. In our study, production of the inflammatory mediators accompanied the observed cellular differential profile measured in BAL fluid upon instillation with particles (Fig. 7). The in vivo response to particles was dominated by IL-6 and TNF-α release, along with other cytokines, including KC (murine IL-8 homolog), indicators of the presence of acute inflammation. Increases in BAL TNF-α, IL-6 and KC have been also observed by others in C57Bl/6 J mice instilled with fine and coarse urban particles collected from European cities [52].
A European study conducted using particles collected in different seasons from urban centers along with EHC-93 Ottawa urban particles examined the role of physicochemical particle properties in modulating the production of cytokines in vitro and on in vivo inflammation in rat lungs as well as adjuvant potency in allergic mouse models. The in vitro assays of cytokine production (IL-6, IL-8, TNF-α, MIP-2) in macrophage and lung epithelial cells exposed to coarse and fine particles for 20 h correlated with in vivo inflammation endpoints (TNF-α, MIP-2, albumin, Clara cell protein 16) measured in BAL of rats at 24 h after intratracheal exposure to the same particles. In contrast, poor correlation was observed between different endpoints from in vivo allergy models and inflammatory parameters, suggesting that a variety of endpoints are needed to assess the potential hazard of particles [19].
In the present study, we have observed a significant correlation of LDH content in J774A.1 macrophages with LDH in BALB/c lung lavage (Additional file 1: Table S2). Such relationship can be conceptually simpler to understand, i.e. increased potential of particles to perturb macrophage cell membranes in vitro correlates with increased potential to damage cell membranes of particle-laden lung macrophages and neutrophils, however further study is needed to ascertain the mechanisms. Therefore, we have combined the in vitro endpoints in a manner to capture the general cytotoxicity of the particles and the functionally-specific aspects (inflammatory potential) in a meaningful manner that can be subsequently deconstructed to explore the mechanisms of action of the variety of particles, and to facilitate the comparison to in vivo responses.
Our combined estimates of particle potency in vitro and in vivo for toxicity; (βR, βT) were not significantly correlated. However, the combined potency estimates for inflammatory potential (βI-V, β I) and the integrated particle potency estimates (Iβ) that further combined the toxicity and inflammatory bioassay response potency estimates approached the threshold for significance α = 0.05 in a correlation (Table 6; R = 0.8, p = 0.052). The lack of strong associations may be due to insufficient power to obtain statistical significance. Although eight particles (with six urban particles) were screened in vitro, for workload reasons a subset of five particles only (including three urban particles) was tested in vivo, an issue that highlights the impracticality of in vivo assays for high-throughput testing. A better correlation may also have been achieved with additional in vivo time points, capturing different stages of the transient particle-induced in vivo toxicity and inflammation profile. The use of a single species could also have potentially led to higher in vitro/in vivo correlations. However, employing different, routinely used species (mouse and human) enabled the assessment of whether integrating toxicity endpoints originating from widely varied cell types (functionality and species differences) would constitute a robust in vitro surrogate that can be useful in predicting in vivo responses of biological and health relevance. As previously seen, the ranking of potencies of a series of particles was comparable between the mouse macrophage cell line J774A.1 and the human cell line THP-1 (unpublished observations). There are other variables beyond species that affect potency estimates, such as cell seeding or monolayer density at time of exposure, etc.
As illustrated by our results in vitro, toxicological responses of cells to particles of complex composition, such as urban particles may be governed by mechanisms distinct from responses to particles that are more homogeneous, such as minerals. The array of potential mechanisms that may reflect the responses of vastly different particle types may not have been adequately captured in our panel of assays. Nevertheless, the more potent materials that ranked highly in vitro were also highly ranked in vivo, while the less potent particles were ranked low, except TiO2 and CRI particles that were less consistent in vitro and in vivo (Fig. 9). Retesting the model with more particles of comparable physicochemical complexity (i.e. urban particles) and additional relevant assays may provide an improved model performance.
Conclusion
The challenge with investigating the relative risk to health of different particulate source emissions is the reliability of the cytotoxicity models applied, with respect to sensitivity of the cells and potential bias for specific potency determinants. A test system should be sensitive to most if not all potency determinants in particles of different origins and should be relevant to the pathways of toxicity in humans leading to adverse health outcomes. In order to achieve this, computational approaches are needed, to reduce the complex datasets, dose responses, multiple assays and cell lines into meaningful descriptors of potency. Therefore, an approach which combines tissue-specific, functionally distinct cell types with a diverse set of assays that capture convergent toxicity pathways and risk-specific mechanisms forms an ideal basis in a tiered strategy for hazard identification and toxicity testing. It remains difficult to broadly capture most of the key events that underlie adverse pulmonary reactions. This may require the inclusion of a greater variety of assays, such as air-liquid interface models, more cell types and particles, or more relevant particles in the model.
More physiologically relevant cell-cell interaction-based models (e.g. co-cultures, or 3D spheroids grown on reconstituted basement membranes) show promise in capturing additional factors that may narrow the gap between in vitro and in vivo testing [53, 54], the latter representing the currently accepted basis of risk assessment regimes. By design, in vitro-based systems are highly relevant for high-throughput applications (e.g. EPA ToxCast, Tox21 initiatives) and high-content data (genomic, proteomic profiling) which are required to address the current challenge posed by the large number of data-poor chemicals, that must undergo toxicological risk-based prioritization and assessment. More comprehensive evaluations of the predictive potential of in vitro assays for disease-relevant adverse outcomes will be needed for in vitro assays to become a trusted and practical tool in regulatory risk assessment. Although in vitro screening approaches may not yet fully replace the need for in vivo regulatory toxicology testing, they could provide an effective component of a multi-tiered testing approach.
Abbreviations
AhR, aryl hydrocarbon receptor; AMR, ATP-monitoring reagent; ANOVA, analysis of variance; ATCC, American type culture collection; ATP, adenosine triphosphate; BRDU, 5-bromo-2'-deoxyuridine; BSA, bovine serum albumin; CRI, cristobalite; DMEM, Dulbecco’s modified Eagle’s medium; DWR1, Downsview reference 1; EHC, Environmental Health Centre; EIA, enzyme immunoassay; EPA, Environmental Protection Agency; ERK, extra-cellular stimulus-activated kinase; EU, endotoxin units; FBS, fetal bovine serum; FE, fold effect; FGF, fibroblast growth factor; G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte macrophage colony-stimulating factor; HEPA, high-efficiency particulate arrestance; ICP-MS/AES, inductively coupled plasma-mass spectrometry/ atomic emission spectroscopy; IFN, interferon; IP-10, IFN-γ Inducible Protein 10; KC, keratinocyte chemoattractant; LDH, lactate dehydrogenase; M199, medium 199; MCP-1, monocyte/macrophage chemoattractant protein; MEM, minimum essential medium Eagle; MIP-1, macrophage inflammatory protein; NF-kB, nuclear factor kB; NIST, National institute of standards and technology; NLR, nucleotide oligomerization domain-like receptor; PBS, phosphate-buffered saline; PDGF, platelet-derived growth factor; PM, particulate matter; RA, rheumatoid arthritis; RANTES, regulated on activation, normal T cell expressed and secreted; SiNPs, silicon dioxide nanoparticles; SRMs, standard reference materials; TCDD, 2,3,7,8-Tetrachlorodibenzodioxin; TLR, Toll-like receptor; TMB, 3,3',5,5'-tetramethylbenzidine; TNF-α, tumor necrosis factor alpha; TSP, total suspended particulates; VEGF, vascular endothelial growth factor; XTT, 2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide
References
World Health Organization. Burden of disease from Household Air Pollution for 2012. Geneva: World Health Organization; 2014. http://www.who.int/phe/health_topics/outdoorair/databases/FINAL_HAP_AAP_BoD_24March2014.pdf?ua=1. Accessed 5 May 2015.
Cohen AJ, Anderson HR, Ostro B, Pandey KD, Krzyzanowski M, Kuenzli N, Gutschmidt K, Pope CA, Romieu I, Samet JM, et al. Mortality impacts of urban air pollution. In: Ezzati M, Lopez AD, Rodgers A, Murray CUJL, editors. Comparative quantification of health risks: Global and regional burden of disease due to selected major risk factors, vol. 2. Geneva: World Health Organization; 2004. p. 1353–433.
Anderson JO, Thundiyil JG, Stolbach A. Clearing the air: a review of the effects of particulate matter air pollution on human health. J Med Toxicol. 2012;8(2):166–75.
Kaplan GG, Hubbard J, Korzenik J, Sands BE, Panaccione R, Ghosh S, Wheeler AJ, Villeneuve PJ. The inflammatory bowel diseases and ambient air pollution: a novel association. Am J Gastroenterol. 2010;105(11):2412–9.
Kaplan GG, Szyszkowicz M, Fichna J, Rowe BH, Porada E, Vincent R, Madsen K, Ghosh S, Storr M. Non-specific abdominal pain and air pollution: a novel association. PLoS One. 2012;7(10), e47669.
Chen JC, Wang X, Wellenius GA, Serre ML, Driscoll I, Casanova R, McArdle JJ, Manson JE, Chui HC, Espeland MA. Ambient air pollution and neurotoxicity on brain structure: evidence from women's health initiative memory study. Ann Neurol. 2015;78(3):466–76.
Schikowski T, Vossoughi M, Vierkötter A, Schulte T, Teichert T, Sugiri D, Fehsel K, Tzivian L, Bae IS, Ranft U, et al. Association of air pollution with cognitive functions and its modification by APOE gene variants in elderly women. Environ Res. 2015;142:10–6.
Shah PS, Balkhair T. Knowledge synthesis group on determinants of preterm/LBW births. Air pollution and birth outcomes: a systematic review. Environ Int. 2011;37(2):498–516.
van den Hooven EH, Pierik FH, de Kluizenaar Y, Hofman A, van Ratingen SW, Zandveld PY, Russcher H, Lindemans J, Miedema HM, Steegers EA, et al. Air pollution exposure and markers of placental growth and function: the generation R study. Environ Health Perspect. 2012;120(12):1753–9.
Pope 3rd CA, Turner MC, Burnett RT, Jerrett M, Gapstur SM, Diver WR, Krewski D, Brook RD. Relationships between fine particulate air pollution, cardiometabolic disorders, and cardiovascular mortality. Circ Res. 2015;116(1):108–15.
Rao X, Montresor-Lopez J, Puett R, Rajagopalan S, Brook RD. Ambient air pollution: an emerging risk factor for diabetes mellitus. Curr Diab Rep. 2015;15(6):603.
Bottini AA, Amcoff P, Hartung T. Food for thought … on globalisation of alternative methods. ALTEX. 2007;24(4):255–69.
Hartung T, Daston G. Are in vitro tests suitable for regulatory use? Toxicol Sci. 2009;111(2):233–7.
Höfer T, Gerner I, Gundert-Remy U, Liebsch M, Schulte A, Spielmann H, Vogel R, Wettig K. Animal testing and alternative approaches for the human health risk assessment under the proposed new European chemicals regulation. Arch Toxicol. 2004;78(10):549–64.
Riss TL, Moravec RA. Use of multiple assay endpoints to investigate the effects of incubation time, dose of toxin, and plating density in cell-based cytotoxicity assays. Assay Drug Dev Technol. 2004;2(1):51–62.
Zucco F, De Angelis I, Testai E, Stammati A. Toxicology investigations with cell culture systems: 20 years after. Toxicol in Vitro. 2004;18(2):153–63.
Seagrave J, McDonald JD, Gigliotti AP, Nikula KJ, Seilkop SK, Gurevich M, Mauderly JL. Mutagenicity and in vivo toxicity of combined particulate and semivolatile organic fractions of gasoline and diesel engine emissions. Toxicol Sci. 2002;70(2):212–26.
Seagrave J, McDonald JD, Mauderly JL. In vitro versus in vivo exposure to combustion emissions. Exp Toxicol Pathol. 2005;57 Suppl 1:233–8.
Steerenberg PA, van Amelsvoort L, Lovik M, Hetland RB, Alberg T, Halatek T, Bloemen HJ, Rydzynski K, Swaen G, Schwarze P, et al. Relation between sources of particulate air pollution and biological effect parameters in samples from four European cities: an exploratory study. Inhal Toxicol. 2006;18:333–46.
Becher R, Hetland RB, Refsnes M, Dahl JE, Dahlman HJ, Schwarze PE. Rat lung inflammatory responses after in vivo and in vitro exposure to various stone particles. Inhal Toxicol. 2001;13(9):789–805.
Sayes CM, Reed KL, Warheit DB. Assessing toxicity of fine and nanoparticles: comparing in vitro measurements to in vivo pulmonary toxicity profiles. Toxicol Sci. 2007;97(1):163–80.
Devlin RB, Frampton ML, Ghio AJ. In vitro studies: what is their role in toxicology? Exp Toxicol Pathol. 2005;57 Suppl 1:183–8.
Pope 3rd CA. Respiratory disease associated with community air pollution and a steel mill, Utah Valley. Am J Public Health. 1989;79(5):623–8.
Vincent R, Goegan P, Johnson G, Brook JR, Kumarathasan P, Bouthillier L, Burnett RT. Regulation of promoter-CAT stress genes in HepG2 cells by suspensions of particles from ambient air. Fundam Appl Toxicol. 1997;39:18–32.
Vincent R, Bjarnason SG, Adamson IY, Hedgecock C, Kumarathasan P, Guenette J, Potvin M, Goegan P, Bouthillier L. Acute pulmonary toxicity of urban particulate matter and ozone. Am J Pathol. 1997;151:1563–70.
Nadeau D, Fouquette-Couture L, Paradis D, Khorami J, Lane D, Dunnigan J. Cytotoxicity of respirable dusts from industrial minerals: comparison of two naturally occurring and two man-made silicates. Drug Chem Toxicol. 1987;10:49–86.
Garrison PM, Tullis K, Aarts JM, Brouwer A, Giesy JP, Denison MS. Species-specific recombinant cell lines as bioassay systems for the detection of 2,3,7,8-tetrachlorodibenzo-p-dioxin-like chemicals. Fundam Appl Toxicol. 1996;30(2):194–203.
Breznan D, Das D, MacKinnon-Roy C, Simard B, Kumarathasan P, Vincent R. Non-specific interaction of carbon nanotubes with the resazurin assay reagent: Impact on in vitro assessment of nanoparticle cytotoxicity. Toxicol In Vitro. 2015;29(1):142–7.
Reich M, Liefeld T, Gould J, Lerner J, Tamayo P, Mesirov JP. GenePattern 2.0. Nat Genet. 2006;38(5):500–1.
Saldanha AJ. Java Treeview--extensible visualization of microarray data. Bioinformatics. 2004;20(17):3246–8.
Hunter CA, Jones SA. IL-6 as a keystone cytokine in health and disease. Nat Immunol. 2015;16(5):448–57.
Scheller J, Chalaris A, Schmidt-Arras D, Rose-John S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim Biophys Acta. 2011;1813(5):878–88.
Frank JA. Claudins and alveolar epithelial barrier function in the lung. Ann N Y Acad Sci. 2012;1257:175–83.
Whitsett JA, Alenghat T. Respiratory epithelial cells orchestrate pulmonary innate immunity. Nat Immunol. 2015;16(1):27–35.
Chuquimia OD, Petursdottir DH, Periolo N, Fernández C. Alveolar epithelial cells are critical in protection of the respiratory tract by secretion of factors able to modulate the activity of pulmonary macrophages and directly control bacterial growth. Infect Immun. 2013;81(1):381–9.
Ralph P, Prichard J, Cohn M. Reticulum cell sarcoma: an effector cell in antibody-dependent cell-mediated immunity. J Immunol. 1975;114(2 pt 2):898–905.
Alfaro-Moreno E, Martínez L, Garcia-Cuellar C, Bonner JC, Murray JC, Rosas I, Rosales SP, Osornio-Vargas AR. Biologic effects induced in vitro by PM10 from three different zones of Mexico City. Environ Health Perspect. 2002;110:715–20.
Kumarathasan P, Breznan D, Das D, Salam MA, Siddiqui Y, MacKinnon-Roy C, Guan J, de Silva N, Simard B, Vincent R. Cytotoxicity of carbon nanotube variants: a comparative in vitro exposure study with A549 epithelial and J774 macrophage cells. Nanotoxicology. 2015;9:148–61.
Breznan D, Goegan P, Chauhan V, Karthikeyan S, Kumarathasan P, Cakmak S, Nadeau D, Brook JR, Vincent R. Respiratory burst in alveolar macrophages exposed to urban particles is not a predictor of cytotoxicity. Toxicol In Vitro. 2013;27(4):1287–97.
Donaldson K, Beswick PH, Gilmour PS. Free radical activity associated with the surface of particles: a unifying factor in determining biological activity? Toxicol Lett. 1996;88:293–8.
Beamer CA, Shepherd DM. Role of the aryl hydrocarbon receptor (AhR) in lung inflammation. Semin Immunopathol. 2013;35(6):693–704.
Kobayashi S, Okamoto H, Iwamoto T, Toyama Y, Tomatsu T, Yamanaka H, Momohara S. A role for the aryl hydrocarbon receptor and the dioxin TCDD in rheumatoid arthritis. Rheumatology (Oxford). 2008;47(9):1317–22.
Beamer CA, Seaver BP, Shepherd DM. Aryl hydrocarbon receptor (AhR) regulates silica-induced inflammation but not fibrosis. Toxicol Sci. 2012;126(2):554–68.
Dhasmana A, Sajid Jamal QM, Mir SS, Bhatt ML, Rahman Q, Gupta R, Siddiqui MH, Lohani M. Titanium dioxide nanoparticles as guardian against environmental carcinogen benzo[alpha]pyrene. PLoS One. 2014;9(9), e107068.
Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282(5396):2085–8.
Dong W, Lewtas J, Luster MI. Role of endotoxin in tumor necrosis factor alpha expression from alveolar macrophages treated with urban air particles. Exp Lung Res. 1996;22:577–92.
Fujii T, Hayashi S, Hogg JC, Vincent R, van Eeden SF. Particulate matter induces cytokine expression in human bronchial epithelial cells. Am J Respir Cell Mol Biol. 2001;25:265–71.
Mukae H, Hogg JC, English D, Vincent R, van Eeden SF. Phagocytosis of particulate air pollutants by human alveolar macrophages stimulates the bone marrow. Am J Physiol Lung Cell Mol Physiol. 2000;279:L924–31.
Øvrevik J, Låg M, Holme JA, Schwarze PE, Refsnes M. Cytokine and chemokine expression patterns in lung epithelial cells exposed to components characteristic of particulate air pollution. Toxicology. 2009;259(1–2):46–53.
Dick CA, Singh P, Daniels M, Evansky P, Becker S, Gilmour MI. Murine pulmonary inflammatory responses following instillation of size-fractionated ambient particulate matter. J Toxicol Environ Health A. 2003;66:2193–207.
Yuen IS, Hartsky MA, Snajdr SI, Warheit DB. Time course of chemotactic factor generation and neutrophil recruitment in the lungs of dust-exposed rats. Am J Respir Cell Mol Biol. 1996;15(2):268–74.
Happo MS, Salonen RO, Halinen AI, Jalava PI, Pennanen AS, Kosma VM, Sillanpaa M, Hillamo R, Brunekreef B, Katsouyanni K, et al. Dose and time dependency of inflammatory responses in the mouse lung to urban air coarse, fine, and ultrafine particles from six European cities. Inhal Toxicol. 2007;19:227–46.
Levinger I, Ventura Y, Vago R. Life is three dimensional-as in vitro cancer cultures should be. Adv Cancer Res. 2014;121:383–414.
Sambale F, Lavrentieva A, Stahl F, Blume C, Stiesch M, Kasper C, Bahnemann D, Scheper T. Three dimensional spheroid cell culture for nanoparticle safety testing. J Biotechnol. 2015;205:120–9.
Acknowledgements
The authors would like to acknowledge and thank Dr Azam Tayabali, Dr Guillaume Pelletier and Dr Marianne Ariganello for their manuscript review and valuable feedback. We would like to thank Dr. Denis Bérubé for ICP-MS analysis of elemental composition of EHC urban particles and Dr. Nimal DeSilva for conducting additional ICP-MS/AES analysis for CRI identity confirmation in archived, experimental working stock dilutions.
Funding
This work was supported by the Border Air Quality Strategy and the Clean Air Regulatory Agenda at Health Canada.
Availability of data and material
Additional data can be made available upon request.
Authors’ contributions
DB participated in the design of the study, carried out the cell culture experiments, coordinated, participated in the animal study, analyzed the data and drafted the manuscript. SK contributed to the statistical analyses performed in the study. MP assisted with the cell cultures, particle stock preparations, Bio-Plex analyses and the animal study. PK participated in the study design and provided expert advice on the study. SC contributed to and provided expert advice on the statistical analyses. MSD provided the H1L1.1c2 cells and advised on the AhR analysis. JRB provided the Trans-Canada Collection PM2.5 particles (DWR1). RV devised and oversaw the entire study. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
The animal treatment protocol was reviewed and approved by the Animal Care Committee of Health Canada.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1: Table S1.
Elemental composition and size of the urban particulate matter and mineral particles. Table S2. Pearson correlations for cytotoxic potency and cytokine inductions in cell lines versus BALB/c mice exposed to particles. Table S3. Pearson correlations for the combined average in vitro (cell lines) and in vivo (BALB/c mice) particle potency estimates. (DOC 77 kb)
Additional file 2: Figure S1.
Resazurin reduction by J774A.1 (A) and A549 (B) cells exposed to particles. Average fold-effect (FE) over control ± standard error values are shown (n = 5). ATP content of J774A.1 (C) and A549 (D) cells exposed to particles. (n = 3). Proliferation in J774A.1 (E) and A549 (F) cells exposed to particles. (n = 3). Two way ANOVA; Resazurin assay, J774A.1, PM main effect, p < 0.001, SRM-1648 vs. DWR1, EHC-98 or EHC-2000 (†), SRM-1649 vs. DWR1, EHC-93, EHC-98, EHC-2000, TiO2 or CRI (‡), Tukey test, p < 0.05, Dose main effect, p < 0.001, Doses 0, 10 or 20 vs. 40, 80 or 160 (#), Dose 40 vs. 80 or 160 (not shown), Dose 80 vs. 160 (not shown), Tukey test, p < 0.05; Resazurin assay, A549, PM × Dose interaction, p = 0.001, asterisks (*) represent effects significantly different from control, Tukey test, p < 0.05. ATP assay, J774A.1, PM main effect, p < 0.001, EHC-98 vs. SRM-1648, SRM-1649 or EHC-2000 (†), SRM-1649 vs. DWR1, TiO2 or EHC-93 (‡), Tukey test, p < 0.05, Dose main effect, p < 0.001, Dose 160 vs. all doses (#), Tukey test, p < 0.001; ATP assay, A549, PM main effect, p < 0.001, DWR1 vs. all particles (†), EHC-93 vs. SRM-1649, CRI or TiO2 (‡), Tukey test, p < 0.05, Dose main effect, p < 0.001, Dose 10 vs. 0, 40, 80 or 160 (not shown), Dose 20 vs. 80 (not shown), Dose 160 vs. 0, 20, 40 or 80 (#), Tukey test, p < 0.05. BrdU assay, J774A.1, PM main effect, p < 0.001, DWR1 or CRI vs. EHC-93, EHC-98, EHC-2000, SRM-1648, SRM-1649 or TiO2 (†), Tukey test, p < 0.05, Dose main effect, p < 0.001, Doses 0 or 10 vs. 20, 40, 80 or 160 (‡), Doses 20 or 40 vs. 80 or 160 (not shown), Dose 80 vs. 160 (not shown), Tukey test, p < 0.05; BrdU assay, A549, Dose main effect, p < 0.001, Dose 10 vs. 0, 20, 80 or 160 (†), Dose 40 vs. 80 or 160 (‡), Tukey test, p < 0.05. (DOCX 80 kb)
Additional file 3: Figure S2.
Granulocyte-Macrophage Colony-Stimulating Factor (GM-CSF) (A), interleukin (IL)-1α (B), IL-β (C), IL-10 (D), Regulated upon Activation Normal T cell Expressed and Secreted (RANTES) (E) and Tumor Necrosis Factor (TNF)-α (F) levels in cell culture supernatants of J774A.1 cells exposed to particles for 24 h. Values are presented as mean fold-effect (FE) ± standard error (n = 3). Two way ANOVA; GM-CSF, PM × Dose interaction, p = 0.001, asterisks (*) represent effects significantly different from Dose 0 control, Tukey test, p < 0.05; IL-1α, PM × Dose interaction, p < 0.001, asterisks (*) represent effects significantly different from Dose 0 control, Tukey test, p < 0.05; IL-β, PM × Dose interaction, p < 0.001, asterisks (*) represent effects significantly different from Dose 0 control, Tukey test, p < 0.05; IL-10, PM × Dose interaction, p = 0.026, asterisks (*) represent effects significantly different from Dose 0 control, Tukey test, p < 0.05; RANTES, PM × Dose interaction, p < 0.001, asterisks (*) represent effects significantly different from Dose 0 control, Tukey test, p < 0.05; TNF-α, PM × Dose interaction, p < 0.001, asterisks (*) represent effects significantly different from Dose 0 control, Tukey test, p < 0.05. (DOCX 73 kb)
Additional file 4: Figure S3.
Granulocyte-Macrophage Colony-Stimulating Factor (GM-CSF) (A), interleukin (IL)-1β (B), IL-8 (C), IL)-10 (D), Monocyte Chemoattractant Protein (MCP)-1 (E), Macrophage Inflammatory Protein (MIP)-1β (F) and Tumor Necrosis Factor (TNF)-α (G) levels in cell culture supernatants of A549 cells exposed to particles for 24 h. Values are presented as mean fold-effect (FE) ± standard error (n = 3). Two way ANOVA; GM-CSF, Dose main effect, p < 0.001, Dose 160 vs. 0, 10 or 40 (†), Dose 10 vs. 40 (not shown), Tukey test, p < 0.05; IL-1β, PM main effect, p = 0.004, TiO2 vs. DWR1 or EHC-93 (†), Tukey test, p < 0.05, Dose main effect, p < 0.001, Doses 40 or 160 vs. 0 or 10 (‡), Tukey test, p < 0.05; IL-8, PM × Dose interaction, p = 0.016, asterisks (*) represent effects significantly different from Dose 0 control, Tukey test, p < 0.05; IL-10, not statistically significant; MCP-1, PM × Dose interaction, p = 0.001, asterisks (*) represent effects significantly different from Dose 0 control, Tukey test, p < 0.05; MIP-1β, PM main effect, p = 0.014, TiO2 vs. EHC-93 (†), Tukey test, p < 0.05, Dose main effect, p = 0.023, Dose 160 vs. 10 (‡), Tukey test, p < 0.05; TNF-α, PM main effect, p < 0.001, TiO2 vs. DWR1, EHC-93 or EHC-2000 (†), CRI vs. DWR1 (‡), Tukey test, p < 0.05, Dose main effect, p < 0.001, Dose 0 vs. 10, 40 or 160 (#), Dose 10 vs. 40 (not shown), Dose 160 vs. 10 or 40 (not shown), Tukey test, p < 0.05. (DOCX 85 kb)
Additional file 5: Figure S4.
Lactate dehydrogenase (LDH) levels in bronchioalveolar lavage (BAL) fluid (A), 8-isoprostane levels in blood plasma (B), Macrophage (C), Band cell (D) and lymphocyte (E) counts in BAL fluid from BALB/c mice exposed to particles by intratracheal instillation, at 24 h post-exposure. Values represent mean fold-effect (FE) ± standard error of the mean (n = 5). Two way ANOVA; LDH, PM × Dose interaction, p = 0.035, asterisks (*) represent effects significantly different from Dose 0 control, Tukey test, p < 0.05; 8-isoprostane, Dose main effect, p = 0.006, Dose 0 vs. 100 (†), Tukey test, p = 0.004; Macrophages, PM main effect, p = 0.018, TiO2 vs. SRM-1649 (†), Tukey test, p = 0.049, Dose main effect, p < 0.001, Dose 0 vs. 50, 100 or 250 (‡), Dose 50 vs. 250 (#), Tukey test, p < 0.05; Band cells, Dose main effect, p < 0.001, Dose 0 vs. 50 or 100 (†), Tukey test, p < 0.001; Lymphocytes, Not statistically significant. (DOCX 63 kb)
Additional file 6: Figure S5.
Granulocyte Macrophage-Colony Stimulating Factor (GM-CSF) (A), interleukin (IL)-1α (B), IL-1β (C), IL-5 (D), IL-10 (E), Keratinocyte-derived Chemokine (KC) (F), Macrophage Inflammatory Protein (MIP)-1α (G), Regulated upon Activation Normal T cell Expressed and Secreted (RANTES) (H) and Tumor Necrosis Factor (TNF)-α (I) levels in bronchioalveolar lavage fluid of BALB/c mice exposed to particles by intratracheal instillation, at 24 h post-exposure. Values represent mean fold-effect (FE) ± standard error of the mean (n = 5). Two way ANOVA; GM-CSF, Dose main effect, p < 0.001, Dose 250 vs. 0 (†), Dose 250 vs. 50 (‡), Tukey test, p < 0.05; IL-1α, PM main effect, p < 0.001, CRI, EHC-2000 or SRM-1649 vs. DWR1 or TiO2 (†), Tukey test, p < 0.05, Dose main effect, p < 0.001, Dose 0 vs. 50, 100 or 250 (‡), Dose 50 vs. 100 (not shown), Dose 250 vs. 50 or 100 (not shown), Tukey test, p < 0.05; IL-β, PM × Dose interaction, p = 0.015, asterisks (*) represent effects significantly different from Dose 0 control, Tukey test, p < 0.05; IL-5, PM × Dose interaction, p = 0.007, asterisks (*) represent effects significantly different from Dose 0 control, Tukey test, p < 0.05; IL-10, not statistically significant; KC, PM × Dose interaction, p = 0.001, asterisks (*) represent effects significantly different from Dose 0 control, Tukey test, p < 0.05; MIP-1α, PM × Dose interaction, p < 0.001, asterisks (*) represent effects significantly different from Dose 0 control, Tukey test, p < 0.05; RANTES, Dose main effect, p < 0.001, Dose 0 vs. 100 or 250 (†), Dose 250 vs. 50 or 100 (‡), Tukey test, p < 0.05; TNF-α, PM main effect, p = 0.003, TiO2 vs. CRI, EHC-2000 or SRM-1649 (†), Tukey test, p < 0.05, Dose main effect, p < 0.001, Dose 0 vs. 50, 100 or 250 (‡), Dose 250 vs. 50 (#), Tukey test, p < 0.05. (DOCX 100 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Breznan, D., Karthikeyan, S., Phaneuf, M. et al. Development of an integrated approach for comparison of in vitro and in vivo responses to particulate matter. Part Fibre Toxicol 13, 41 (2015). https://doi.org/10.1186/s12989-016-0152-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12989-016-0152-6