
Abstract
HIV associated neurocognitive disorders (HAND) are the spectrum of cognitive impairments present in patients infected with human immunodeficiency virus type 1 (HIV-1). The number of patients affected with HAND ranges from 30 to 50% of HIV infected individuals and although the development of combinational antiretroviral therapy (cART) has improved longevity, HAND continues to pose a significant clinical problem as the current standard of care does not alleviate or prevent HAND symptoms. At present, the pathological mechanisms contributing to HAND remain unclear, but evidence suggests that it stems from neuronal injury due to chronic release of neurotoxins, chemokines, viral proteins, and proinflammatory cytokines secreted by HIV-1 activated microglia, macrophages and astrocytes in the central nervous system (CNS). Furthermore, the blood–brain barrier (BBB) not only serves as a route for HIV-1 entry into the brain but also prevents cART therapy from reaching HIV-1 brain reservoirs, and therefore could play an important role in HAND. The goal of this review is to discuss the current data on the epidemiology, pathology and research models of HAND as well as address the potential pharmacological treatment approaches that are being investigated.
Similar content being viewed by others

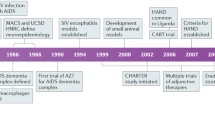
Epidemiology of HAND
Prevalence of HAND in the era of cART
Human immunodeficiency virus type 1 (HIV-1) is the enveloped retrovirus responsible for the development of Acquired immunodeficiency syndrome (AIDS) in patients. The Joint United Nations Program on HIV/AIDS (UNAIDS) estimated that about 36.9 million people worldwide were living with HIV as of 2017, with approximately 21.7 million of that population receiving combinational antiretroviral therapy (cART) [1]. In addition to the development of AIDS, patients with HIV have been documented to have neurological complications as early as the late 80’s [2]. In their 1986 paper, Navia and Price first described the observed motor and behavioural deficits associated with AIDS as the “AIDS dementia complex”(ADC) [3]. Since then, three types of disorders have been recognized to define the observed neurocognitive deficits. In order of increasing severity, the terms are Asymptomatic Neurocognitive Impairment (ANI), Minor Neurocognitive Disorder (MND), and HIV-associated Dementia (HAD) [4]. In 2007, HIV Associated Neurocognitive Disorders (HAND) was proposed by the US National Institute of Mental Health panel to be used to define the spectrum of the neurological disease associated with HIV infection [4].
Determining the prevalence of HAND continues to be challenging as reported numbers are variable within the literature. In a study using patients from the Multicentre AIDS Cohort Study (MACS), Sacktor et al. observed a frequency of HAND amongst 364 HIV+ gay/bisexual men within the 3 year period of 2007–2008 of 33% (14% classified as ANI, 14% classified as MND and 5% as HAD) [5]. Other groups have reported the prevalence of HAND to be between 15 and 55% of HIV+ individuals [6,7,8,9,10]. Since the development of cART over two decades ago, the prevalence of HAND has not changed significantly but the severity of neurocognitive impairment has noticeably dropped. In the pre-cART era, approximately 16% of HIV+ individuals were also suffering from HAD [11] but more recent data has shown that after the introduction of cART, prevalence rates of HAD are within the range of 2–8% [6]. Although cART appears to be beneficial for attenuating the presentation of HAD, it does not yield positive results in the prevention of milder forms of HAND, as ANI prevalence has dramatically increased since the dawn of cART, and it is now the most common form of HAND [6, 11, 12].
Despite, the asymptomatic nature of ANI, it remains a concern as mild cognitive deficiency can quickly progress to more severe forms of HAND. In the MACS study the research team enrolled a group of 197 HIV+ participants on cART and assessed them every 2 years over a 6 year time period and observed an increase in HAND prevalence from 25 to 31% [5]. Within the study, 15% of the subjects experienced further cognitive decline (ANI to MND or HAD) over the 6 years, while 14% saw improvements in their HAND stage [5]. In 2014, another study demonstrated that an ANI diagnosis was associated with a higher risk of developing MND or HAD when compared to HIV+ patients with no signs of diminished cognition [13].
Clinical aspects of HAND
Diagnosis and biomarker identification
A clinical diagnosis of HAND is reached based on the patient’s results on time-consuming neuropsychological tests assessing their abilities in memory, information processing speed, verbal language, attention and working memory, sensory perception, motor skills and executive functioning, where scoring at least one standard deviation below the age-appropriate mean in two categories or more is indicative of impairment [4]. The severity of impairment via neuropsychological testing cannot solely be taken into account when diagnosing patients. Healthcare professionals must also consider the degree of impact that these deficits have on a patient’s ability to function and the absence of any confounding factors that can otherwise explain the observed clinical symptoms [4, 14]. The typical diagnosis of ANI requires a below average performance on diagnostic tests, no negative impact on daily living, cognitive impairment not meeting the criteria for delirium or dementia, and the absence of other conditions that may cause cognitive impairment [4]. The classification of MND is similar except that the impairment must mildly impact the patient’s daily functioning through either self reported or observed deficiencies in work, homemaking, social interaction and mental acuity and typically results in a score between 0.5 and 1 on the Memorial Sloan Kettering scale. HAD patients typically score 2 or greater on the Memorial Sloan Kettering scale and have their daily functioning significantly hindered by their cognitive insufficiency [4].
At present, there are no validated biomarkers to diagnose HAND. Several potential markers have been identified in HIV+ individuals with neurological impairment, however most of them are typically associated with HAD rather than ANI and MND, the more prevalent forms of HAND [6]. The biomarkers can be divided into four general categories: (1) structural changes observed with neuroimaging, (2) markers of cellular or metabolic stress, (3) humoral markers of immune activation and (4) markers of neuronal injury.
In regards to brain structural changes, a multitude of studies conducting brain volumetric analysis with the use of magnetic resonance imaging (MRI) in the context of HAND are summarized in a review by Masters and Ances [15]. One of the more recent publications relates to a 2013 study including patients from the ANRS CO3 Aquitaine Cohort and reports that MND and HAD patients had lower gray matter and white matter volumes when compared to patients diagnosed with ANI [16]. Additionally, in a separate study, HIV+ individuals with quantifiable peripheral viral load displayed decreased subcortical and cerebellar gray matter volumes when compared to HIV+ subjects with undetectable viral load in the periphery [17]. The same study reported that HIV+ participants had enlarged ventricles and reduced putamina, hippocampi, nucleus accumbens, caudate nuclei, brainstems, thalami, total cortical gray matter and cerebral white matter compared to the HIV- control group [17].
The second type of biomarkers are classified as markers of cellular or metabolic stress. Elevated levels of Krebs Cycle substrates (acetate and citrate) in the cerebrospinal fluid (CSF) were found to be linked with worsening cognitive status in HIV+ patients [18]. A separate study published results indicating that reduced CSF concentrations of esterified cholesterols and sphingolipids in HIV+ patients increased the risk of cognitive decline [19]. Additionally, proton magnetic resonance spectroscopy was used to detect elevated choline compounds in the white matter of those diagnosed with ADC when compared to neuro-asymptomatic HIV subjects [20]. Other markers of cellular or metabolic stress include CSF heme oxygenase-1, CSF protein carbonyls, CSF 3-nitrotyrosine and brain inducible nitric oxide synthase (iNOS) [6].
Markers of immune activation are part of another class of potential biomarkers that include molecules such as neopterin. Neopterin is a metabolite of the guanosine triphosphate pathway in monocytes and macrophages and high levels have been reported in the CSF of HIV+ individuals [21]. Increased CSF neopterin is indicative of immune activation during HIV infection and thus significantly elevated concentrations suggests infection of the immune cells within the central nervous system (CNS). Neopterin levels are also correlated with the release of reactive oxygen species (ROS) by macrophages which is hypothesised to be a mechanism of neuronal cell damage leading to the symptoms of HAND in patients [21]. Another example of a marker of immune activation in the context of HAND is elevated C-C chemokine receptor type 2 (CCR2) on CD14+ and CD16+ monocytes [22]. CD14+CD16+ monocytes have a high vulnerability to HIV [23] and are considered to act as peripheral reservoirs for the virus. A 2014 study demonstrated that CCR2 was markedly increased in those suffering from HAND when compared to HIV+ patients with normal cognition and found that it did not change with viral load, CD4+ cell counts or with the use of cART [22]. CSF fractalkine, plasma sCD14 and sCD163, CSF osteopontin, CSF C-C motif chemokine ligand 2 (CCL2), brain interleukin-1β (IL-1β), and brain interleukin-10 (IL-10) are just a few of the numerous other potential immunological biomarkers for HAND under investigation [6, 24].
Finally, the last category of biomarkers are markers of neuronal damage. Neurofilament light chain (NFL) is well documented to be positively correlated with HAND [25, 26]. NFL is a surrogate marker for neuronal damage and elevated levels have been detected in the CSF of those with neurodegenerative disorders like subcortical vascular dementia and Alzheimer’s disease [27]. A recent study measured CSF NFL concentrations in 48 untreated HIV+ subjects not on ART and reported that NFL was significantly higher in patients with HAD compared to subjects with mild to no cognitive impairments [28]. It was also discovered that CSF NFL was positively correlated with plasma HIV-1 viral load and negatively correlated with peripheral CD4+ T cell count [28]. A separate study by Nitkiewicz et al. investigated the increased expression of complement proteins, such as complement 3 (C3), in human fetal astrocytes after exposure to HIV-1 [29]. The complement cascade is a critical factor in the pathogenesis of diseases in the CNS and C3 upregulation is indicative of neuronal injury and chronic neurodegenerative disorders. The upregulation of C3 is likely facilitated by the induction of interleukin 6 (IL-6) mediated by nuclear factor kappa-light-chain-enhancer of activated B cells NF-κB during HIV infection, as demonstrated in vitro [29]. Additional CSF markers for neuronal injury currently being investigated in the context of HAND are quinolinic acid, CSF total Tau concentrations, CSF soluble beta amyloid precursor protein and brain N-acetyl aspartate [6].
Risk factors
Risk factors for HAND have been well-documented and often overlap with observed comorbidities [6]. For example, one report documented that diabetes, high carotid intima media thickness, smoking, hypertension and dyslipidemia were highly prevalent in asymptomatic HIV+ subjects and subsequently were associated with lower cognitive performance [30]. Old age is also positively correlated with an increased risk of HAND. HIV+ individuals over the age of 50 years were twice as likely to develop HAD when compared to younger seropositive comparators, according to a study involving the Hawaiian Aging Cohort [31]. A more recent Japanese study also found that increased age was associated with a higher chance of developing MND and HAD [32]. Other risk factors reported in the literature include, sleep disorders such as sleep apnea, and co-infection with the Hepatitis C virus [6]. Substance abuse is another common risk factor, with the abuse of opioids, cocaine, marijuana, alcohol and methamphetamine being correlated with poor cognitive performance in HIV+ patients [33, 34]. Mental illnesses such as depression, schizophrenia and bipolar disorder are often observed in HIV+ patients and are strongly associated with medical nonadherence [35]. As a result, HAND patients may be at high risk of improperly using their antiretroviral (ARV) medications which could potentially lead to the exacerbation of their condition.
Neuropathogenesis
HIV infection and the CNS: cellular targets
The principal peripheral targets of the HIV-1 virion are circulating CD4+ T-lymphocytes and macrophages [7, 36]. The virus gains entry through interactions with the host’s CD4 surface protein and C-C chemokine receptor type 5 (CCR5) via its envelope glycoprotein [37]. Certain strains of HIV-1 can also infect cells using the C-X-C motif chemokine receptor 4 (CXCR4) as a coreceptor [38]. Due to the variable entry mechanisms used by the virus, it is suitable to distinguish HIV-1 into three groups. R5-tropic variants which make up the majority of HIV-1 and uses CCR5 as a co-receptor; X4-tropic variants which instead use CXCR4 to gain access to cells [39]; and finally dual-tropic HIV-1 strains which have the capability to use both co-receptors, yet the exact mechanisms on how it switches from one molecule to the other is not fully understood [40].
Within the first few weeks of infection, HIV-1 can enter the CNS [36]; viral RNA has been measured in the CSF of patients as early as 8 days after initial infection [41]. It is proposed that HIV-1 enters the CNS by crossing the blood–brain barrier (BBB) through the penetration of HIV-infected monocytes across the brain vascular endothelial cells or as cell free virions [42,43,44]. Approximately 5-10% of circulating monocytes are CD14 and CD16+, but that proportion increases during HIV infection [45, 46]. These monocytes have also been shown to be preferentially vulnerable to HIV-1 and thus are important to its associated neuropathogenesis [23]. Once infected, CD14+CD16+ cells present upregulated surface expression of the junctional proteins ALCAM and JAM-A and the receptor CCR2, which are required for CNS entry [43, 44]. Although baseline transport of normal human CD14+CD16+ cells was revealed to not be statistically different from infected cells, HIV-containing monocytes were shown to have a significantly higher transmigration across an in vitro human BBB model when compared to uninfected cells through a C-C chemokine ligand 2 (CCL2) mediated mechanism involving JAM-A and ALCAM surface proteins. Migration increased in a dose-dependent manner in the presence of CCL2, and antibody specific blocking of JAM-A and ALCAM fully inhibited BBB transport [44]. Blocking JAM-A, ALCAM or CCR2 may prove to be an effective prophylactic measure in the prevention of HAND in HIV+ individuals [43, 47].
In contrast, two studies have proposed that macrophages are not directly targeted by HIV-1 [48, 49] and that they do not contribute to virus production in vivo [48]. Instead, it was proposed that macrophages target HIV-infected CD4+ lymphocytes for phagocytosis, explaining the reason for viral DNA and proteins being detected in macrophages. This controversy led to a 2016 study by Honeycutt et al. which aimed to further identify the mechanism behind HIV-1 infection of the monocyte lineage. The study found no evidence of viral DNA in monocytes from HIV+ individuals [50]. The group then used a myeloid-only mouse model and observed that HIV-1 infection was observed to be sustained in macrophages in the brain and de novo HIV-1 synthesis in the absence of T-cells in vivo was demonstrated [50]. These results support the idea that HIV-1 spreading in the CNS can be facilitated by mature macrophages but suggests that initial CNS penetration by HIV-1 infected monocytes is not an entry mechanism of the virus, directly arguing against the predominant school of thought. This knowledge discrepancy highlights our need for further understanding of HAND pathogenesis in order to identify effective treatment approaches.
Cells of the monocyte–macrophage lineage are considered as a potential HIV-1 reservoir and play a role in virus dissemination. HIV-1 infected monocytes can circulate in the blood up to 3 days before migrating to various tissues such as the brain, where they differentiate into macrophages [51,52,53], and may infect microglial cells. Alternatively, but still not well understood, the cell-to-cell transfer of the virus from infected migrating CD4+ T cells into brain macrophages and microglia, and virus spreading between infected macrophages has been suggested [54, 55]. It has been shown that infected T-lymphocytes come in contact with macrophages leading to cell fusion and transfer of viruses to macrophages. The lymphocyte-macrophage cells further fuse with noninfected macrophages and produce highly virus-productive multinucleated giant cells, observed in lymphoid organs and CNS of HIV-1- infected individuals and SIV-infected macaques [54, 56,57,58,59,60,61,62]. Multinucleated giant cells, along with myelin pallor, activated microglia, reactive astrocytosis (proliferation and activation of astrocytes), and presence of microglial nodules are a hallmark of chronic HIV-1 infection [11, 63, 64].
HIV-1 is also thought to be able to cross the BBB and enter the CNS as cell free virions [65]. Firstly, it may cross paracellularly into the CNS through a “leaky” BBB whose tight junctions have been compromised due to HIV-1 exposure [66,67,68,69,70,71]. Alternatively, a series of experiments involving in vivo and in vitro mouse models identified that the mannose-6-phosphate receptor expressed on brain microvessel endothelial cells can also mediate the transport of HIV-1 [65]. Ultrastructural studies revealed HIV-1 transport by the mannose-6-phosphate receptor in a transcytotic manner and transport was inhibited in the presence of mannose-6-phosphate, mannan (a plant polysaccharide), and mannose in the in vitro models. Transport was further reduced after exposure with endoglycosidase, an enzyme that cleaves high mannose oligosaccharide residues [65] (Fig. 1).

(Figure adapted from Saylor et al. [6])
Neuropathogenesis of HAND. HIV-1 can enter the brain as a cell free virion or encased within infected monocytes or macrophages. Once in the CNS HIV-1 targets microglia, and to a lesser extent, astrocytes. Upon activation, these cells release numerous inflammatory markers (IL-1β, TNFα, CCL2 etc.) and can shed HIV-1 viral proteins (e.g. gp120, Tat). Chronic secretion of such factors which can exacerbate viral replication and pathogenic immune signalling ultimately leading to neuronal injury. HIV-1 infection in the brain may cause disruption of glutamate homeostasis leading to excitotoxicity
It should be noted that in addition to the BBB, the choroid plexus and meninges are also investigated as possible sites for virus dissemination to the CNS [72,73,74] as well as critical players in neuroinflammation [75, 76]. The CSF flow dynamics have also been recognized as a possible contribution of the CSF to disease pathogenesis. The potential for immune regulation in the CNS by the glymphatic system, an effector of neuroinflammation has also been suggested [77]. However, to keep the length of the manuscript concise, we primarily focused on the role of the BBB in HAND.
Following CNS infiltration, HIV-1 can infect the non-neuronal cells of the nervous system, namely microglia [78,79,80] and to lesser extent astrocytes [81,82,83]. Microglial cells initiate the brain’s innate immune response through their ability to congregate onto pathogens and destroy them. They express recognition receptors to detect pathogen-associated molecular signals and are capable of antigen presentation as well secreting cytokines and chemokines during an inflammatory response [79]. As microglia express both CD4 and CCR5 [78, 80], the mechanism by which they become infected with HIV is similar to that of leukocytes [80]. In the case of astrocytes however, this mechanism of HIV cellular entry cannot be applied. Astrocytes lack expression of the critical CD4 molecule that facilitates the infection of lymphocytes, microglia and macrophages but yet they have been shown to be susceptible to HIV and viral DNA and viral protein has been detected in post-mortem brain tissue from HIV+ patients [84,85,86,87]. Uptake of HIV-1 by astrocytes has been demonstrated to be facilitated by the human mannose receptor in a CD4-independent manner [88]. Expression of human mannose receptor rendered human astrocytes vulnerable to HIV-1 and siRNA silencing of the receptor blocked HIV-1 infection [88]. Despite the evidence in support of HIV-1 infection, controversial data exists that suggests that HIV-1 may not directly infect astrocytes after all. A 2001 in vitro study showed that there is a lack of intracellular mechanisms to restrict HIV-1 replication in primary human astrocytes [89], indicating that there was no evolutionary pressure for those cells in primates to defend themselves against HIV and related viruses and suggested that they are not main targets. Furthermore, Russel et al. suggested that the presence of HIV-1 DNA in cultured human astrocytes may be due to cell–cell interactions with infected macrophages [90]. Most recently, viral RNA and DNA were undetectable using single copy sensitive RNAscope and DNAscope on the astrocytes of aviremic HIV+ individuals on cART [91]. However, published data showed HIV-1 infection of astrocytes in HAND patients [92]. Together these conflicting reports highlight our limited knowledge in the understanding of HAND pathogenesis and present another barrier to effective treatment.
An additional group of cells susceptible to HIV-1 infection are the pericytes. Pericytes are known to wrap around the endothelial cells of the BBB with their cytoplasmic processes and facilitate interactions between endothelial cells. These cells are important for the formation, stabilization, and maintenance of the BBB [93]. Pericytes are known to express high levels of the CCR5 and CXCR4 receptors, as well as low-levels of the CD4 receptor [94], suggesting that pericytes can be infected by both X4 and R5 tropic HIV-1 strains. Recently, Bertrand et al. showed that pericytes can be infected with EcoHIV in vivo in mice, suggesting that pericytes could also be an HIV-1 target in the brain, however, the mechanism of HIV-1 entry into these cells remains unknown.
HIV-1 toxicity
The direct neurotoxic effects of HIV-1 are demonstrated to be mediated solely by its viral proteins. There is little to no published research investigating if HIV-1 RNA or DNA is neurotoxic and directly contributes to neurodegeneration. At present, the evidence of HIV-1 induced neurotoxicity does not implicate direct contribution to toxicity by viral nucleic acids in the pathogenesis of HAND.
Available data generally proposes two main mechanisms of HIV-1-mediated CNS pathology leading to the development of HAND: (1) viral proteins from the HIV genome directly causing neurotoxicity, and (2) activation of microglia, brain macrophages and astrocytes in response to HIV-1 and secretion of a range of proinflammatory cytokines and neurotoxins [tumor necrosis factor-α (TNFα), interleukin-1β (IL-1β), interleukin-6 (IL-6), interleukin-8 (IL-8), arachidonic and quinolinic acids, platelet-activating factor, neurotoxic amines, ROS, nitric oxide (NO), glutamate, macrophage inflammatory protein 1α (MIP-1α), monocyte chemoattractant protein 1 (MCP-1) and growth-related oncogene α (GRO-α)], resulting in neuronal injury and death [95, 96].
HIV-1 tat
Trans-activator for transcription (Tat) is encoded by the tat gene. As one of HIV-1’s regulatory proteins, it is a key regulator for the transcription of proviral DNA to mRNA [97, 98]. Tat DNA sequences have been detected in the brain tissue of AIDS patients who suffered from dementia [99] and Tat mRNA along with protein were preferentially detected in brain tissues of patients suffering from HIV encephalitis (HIVE) [100]. The viral protein binds to the trans-activation response elements located at the 5′ ends of HIV-1 transcripts where it increases the activity of RNA polymerase II and thus greatly increases viral transcription [101]. In terms of its contribution to neurotoxicity during HIV-1 infection, Tat has been demonstrated to stimulate tumour necrosis factor alpha (TNFα) released by infected astrocytes resulting in neuronal death [102]. Other groups have also demonstrated Tat-mediated secretion of TNFα in glial cells [102,103,104,105,106,107] as well as increased release of IL-1β [103, 108, 109] and CCL2 [106, 110]. Studies have shown that Tat causes an upregulation of glial fibrillary acidic protein (GFAP) [111, 112] as well as decreased expression of two subtypes of excitatory amino acid transporters (EAAT1 and EAAT2) in mouse astrocytes [112]. EAAT1/2 are glutamate uptake transporters and their reduced expression may lead to an increase in glutamate concentrations in the CNS microenvironment, resulting in excitotoxicity [113]. Tat released by infected astrocytes has also been demonstrated to alter gap junction protein expression on the endothelial cells of the BBB leading to increased permeability [66] and Tat directly induces neuronal necrosis by disrupting mitochondrial function [104]. Recently, Tat was discovered to form a rigid multifibrillar structure by interacting with amyloid beta proteins in a mouse model that forms aggregates and mechanically disrupts the cell membranes of neurons and lead to the formation of pore [98].
Nef
Negative replication factor (Nef) is another regulatory protein of HIV-1 and its primary role is to decrease the transcription of various surface molecules and receptors in infected cells (such as MHC-I, MHC-II, CD3, CD4, CXCR4, CCR5, etc.) in order to avoid detection by the host’s immune system [114]. Nef has also been detected in astrocytes of patients with HIV encephalitis [85]. Data is limited in regards to its direct toxic effects, although Saribas et al. found that Nef-containing extracellular vesicles released by astrocytes induced oxidative stress in neurons and that Nef expression through adenoviral transduction in neurons led to the degeneration of axons [115]. It was also demonstrated that extracellular vesicles containing Nef were capable of suppressing action potentials in neurons, suggesting a role for Nef in HIV neurotoxicity. The mechanisms underlying these observations are still unclear.
Altered behaviour in rodents exposed to Nef has also been reported [116, 117]. For example, transgenic mice expressing Nef under the control of the c-fms promoter exhibited enhanced mania-like behaviour as demonstrated through enhanced locomotor activity, increased exploration times in an open field test, shorter periods of immobility in a forced swim test, and increased exploration in an elevated plus maze when compared to wild type mice [116]. These results support surrogate measures for manic behaviour and gives further insight into the role of Nef in behavioural deficits in those experiencing HAND. In parallel, these animals showed increased CCL2, decreased interferon alpha (IFNα) and disrupted dopamine levels in the striatum. A separate study investigating rats engrafted with Nef-expressing hippocampal astrocytes showed that these rodents had impairments in spatial and recognition memory as demonstrated by their failure in both novel location and novel object recognition tests [117].
Gp120
Glycoprotein 120 (gp120) is the HIV envelope protein that interacts with CD4 and CCR5/CXCR4 receptors to gain access into target cells [37]. Previous studies from our group have shown that gp120 stimulates the immune response causing the release of inflammatory, immune and oxidative stress markers both in vitro, in human and rodent astrocytes, in mixed glia, and in vivo [64, 118,119,120], in addition, it has also been demonstrated to be directly toxic to neurons of the CNS [121]. gp120 has been detected in macrophages and microglia of patients with HIV encephalitis [122, 123]. Studies examining the substantia nigra and caudate-putamina of rats showed that gp120 caused a loss in dopaminergic neurons [124]. The involvement of ROS in gp120-mediated neurotoxicity was further demonstrated in vivo and in vitro where gp120 preferentially induced apoptosis in dopaminergic neurons over non-dopaminergic neurons [125]. The gp120-induced apoptotic pathway is believed to use ROS intermediates as secondary messengers to increase intracellular Ca2+ [125], which may affect the Ca2+ balance in mitochondria, leading to programmed cell death in neurons through release of mitochondrial components such as cytochrome C and apoptotic protease activating factor 1 [126, 127]. Direct neurotoxicity by gp120 has also been reported by Chen et al. where rat cortical neurons were treated with gp120 and found that A-type transient outward K+ currents were enhanced in a dose-dependent manner [128]. A proposed mechanism of direct gp120 neurotoxicity includes mitochondrial dysfunction in exposed neurons [121].
Multiple groups have also shown that gp120 can reduce glutamate uptake in microglia and astrocytes by inhibiting glutamate transporter-1 (GLT-1) otherwise known as EAAT2, leading to excessive stimulation of neuronal N-methyl D Aspartate receptor (NMDAR) which results in an influx of Na+ and Ca2+ and subsequent cellular excitotoxicity [129, 130]. Gp120 has also been studied in the context of drug transporter and tight junction protein regulation at the level of the BBB [71, 131, 132] and in glial cells [119]. Data from these studies suggests that gp120 may have an impact on the integrity of the BBB and ultimately the disposition of certain pharmacological agents and endogenous molecules.
In parallel with gp120 neurotoxicity, behavioral dysfunction has also been reported. Morris Water Maze tests have shown that the V3 loop polypeptide of the gp120 protein can impair spatial memory in rats [133, 134] and electrophysiology tests revealed that exposure reduced long term potentiation in the CA1 region [133]. Deficits in locomotion have also been established in gp120 exposed rats, with exploration times observed to be significantly reduced when compared to controls [135, 136].
Vpr
Viral protein R (Vpr) is another accessory protein of HIV-1 and is important for the infection of cells of the monocytes-macrophages lineage and the nuclear localization of the pre-integration complex [137]. Using immunohistochemistry, Vpr has been detected in the basal ganglia and frontal cortices of patients with HIV encephalitis [138]. Vpr has also been reported to cause neuronal degeneration in vivo as well as to induce apoptosis in cultured human neuroblastoma cells [139]. Jones et al. demonstrated Vpr-induced neuronal loss which was mediated by p53 induction, cytochrome C release and activation of caspase 9 [139]. Microglial cells activated by Vpr were also found to release neurotoxins in a dose dependent pattern [139]. In a recent study, Vpr was shown to disrupt mitochondrial transport [140]. Vpr-treated neurons showed signs of accelerated aging through their increased expression of markers such as peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1α), X-gal, and p21WAF−1 [140]. HIV-1 infected microglia have also been known to express Vpr and release for subsequent uptake by proximal neurons [141]. A study by Rom et al. showed that human neuronal exposure to recombinant Vpr resulted in a sustained increase in intracellular Ca2+ which could impair glutamate signaling in neural cells as well as lead to the production of ROS [141]. Lastly, monocytes and macrophages infected with an HIV-1 variant that was incapable of producing Vpr released significantly less IL-1β, interleukin-8 (IL-8) and TNF-α when compared to HIV-1 wildtypes [142]. The observed reduction in apoptosis suggest a Vpr-dependent necrotic pathway mediated by proinflammatory molecules. An extensive 2016 review on the neuropathogenesis of Vpr conducted by James et al. further sheds light on the neurotoxicity of the viral protein [137].
ARV treatment
HIV+ individuals have been treated with a combination of ARVs since their development in 1996 [143]. Patients on cART are required to take multiple therapeutic agents of different pharmacological classes designed to inhibit viral replication and entry. These classes include nucleoside reverse transcriptase inhibitors, non-nucleoside reverse transcriptase inhibitors, protease inhibitors, fusion inhibitors, entry inhibitors and integrase inhibitors [143]. Despite the complex pharmacotherapeutic intervention, HAND still prevails as a chronic condition that negatively impact patients’ quality of life, drug adherence and survival. cART is effective at decreasing viral load in the periphery of patients but the persistence of ANI and MND suggests that it could fall short on adequately controlling viral load in the CNS. A 2008 study followed 467 HIV+ individuals on cART (number of drugs included in their regimens ranged from 1 to 3) and assigned a CNS Penetration Effectiveness (CPE) score to each of the drugs within their regimen [144]. Scores from 0 (low CNS penetration) to 4 (high penetration) were given based on their chemical properties and observed concentrations in the CSF. The median CPE amongst the cohort was 1.5 and scores less than 2 were associated with an 88% increase in the odds of detectable CSF viral load [144]. Additionally, neurological impairment was negatively correlated to CPE scores in a cohort study involving 417 HIV+ participants on cART [145].
Recently, using a mouse model of HIV infection (EcoHIV) and the middle cerebral artery occlusion model, Bertrand et al. reported that HIV infection significantly increases the severity of ischemic stroke by affecting the BBB integrity and enhancing inflammatory response. Treatment of this mouse model with high CPE ART was more beneficial than low CPE ART in limiting tissue injury and accelerating post-stroke recovery, [146], thus establishing an additional beneficial effect of AVRs with a high CNS penetrance.
Improving CNS penetration of new ARV therapies may lead to a decrease in CNS viral load, but it poses an increased risk of neurotoxicity to patients [147]. The neurotoxicity of various ARVs was demonstrated in primary cultures of rat forebrain neurons where treatment with abacavir, efavirenz, etravirine, nevirapine, and atazanavir induced neuronal loss and damage. These compounds were highly toxic at levels above their current therapeutic concentrations [147], suggesting that increased CNS permeability of ARVs could be detrimental to patients. Clinical data on the use of efavirenz, for example, has shown that its use is associated with lower working memory, global functioning, processing speed, motor functioning and other signs of neurological decline [148, 149].
The lack of CNS penetration of ARVs may be due to the expression of membrane-associated drug efflux transporters at the BBB. Transporters such as P-gp are expressed on the luminal side of the vascular endothelial cells comprising the BBB and are responsible for pumping substances back into the blood to protect the brain from potentially harmful molecular entities [150]. Our group previously demonstrated that the ARVs abacavir, efavirenz, and nevirapine can activate the nuclear receptor Human Constitutive Androstane Receptor while the ARVs amprenavir, atazanavir, darunavir, efavirenz, ritonavir, and lopinavir can activate the Human Pregnane X Receptor in human brain microvessel endothelial cells [151]. These two nuclear receptors act as transcription factors and can regulate efflux transporters such as P-gp. For further details, please refer to our previous reviews [53, 152].
Approaches to eliminating the HIV-1 brain reservoir
Although antiretroviral therapy (ART) has significantly decreased the HIV-1 associated mortality and morbidity, the prevalence of HAND is continuing to increase, and conventional ARV regimens are insufficient to improve this condition. This is partly because many ARVs exhibit poor permeability across the BBB and blood-cerebrospinal fluid barrier (BCSFB), which results in low tissue bioavailability and subtherapeutic ARVs concentrations in the brain. The BBB and BCSFB are protected by brain microvessel endothelial cells and epithelial cells, respectively, and are known to physically and metabolically restrict ARV delivery into the brain [53, 153,154,155]. In addition to the presence of tight junctions, the permeability of ARVs into the CNS can also be highly regulated by the expression of drug efflux transporters such as P-gp, expressed at the BBB and BCSFB [53, 64]. As a result, the brain constitutes a reservoir for HIV-1 and presents a significant challenge to treating HAND. To bypass the effect of efflux transporters expressed at the BBB and BCSFB, and to increase the delivery of ARVs into the brain, multiple alternative approaches have been recently tested.
In particular, nanoparticle-based delivery of ARV drugs has recently been investigated. This system is known to protect ARVs from the effect of efflux transporters as well as from enzymatic and hydrolytic degradation, and can be used for a sustained release of therapeutics, in particular the novel integrase inhibitor, elvitegravir [156]. Although the data demonstrated that the encapsulated elvitegravir nanoparticle formulation improved the ability of the antiviral drug to cross the BBB model in vitro, in vivo data are needed in order to conclude that this strategy has a potential for therapeutic interventions in reducing HAND [156]. Agrawal et al. investigated the feasibility of developing a trojan horse prodrug that could simultaneously inhibit P-gp and have anti-HIV properties [157]. This could be a very promising approach which will need further investigation. In addition, Kaushik et al. developed a magnetic nanoformulation consisting of genome editing Cas9/gRNA bound with magneto-electric nanoparticles with the aim of targeting HIV-1 long terminal repeat, thereby stopping viral transcription and eradicating latent HIV infection. This is a very innovative approach deserving further investigation and that could potentially have clinical utility in the management of HIV infection of the brain [158].
Animal models of HAND
Since the discovery of HIV-1, laboratory and animal models were rapidly implemented and developed to recapitulate the human disease. Animal models have helped to facilitate an in-depth understanding of HIV-1 while avoiding the use of human brain tissue which is challenging. Various animal models for investigating HIV-1 neuropathogenesis have been developed in order to investigate how systemic infection, immune activation and nervous system infection drive neuronal cell damage and death. A summary of various implemented models are outlined (Table 1). Several factors should be taken into account when aiming to reproduce HAND in animals. For example, the model should contain virus susceptible target cells, including CD4+ T lymphocytes, dendritic cells, monocytes and macrophages that display receptors and co-receptors for viral infection and possess the host cell machinery to complete the viral life cycle [37, 159]. Ideally animals should be infected through recognized sites of viral entry (e.g. blood, mucosal layers). Additionally, infection occurring for prolonged periods of time should be achieved in order to reflect the chronic nature of the disease [160]. Finally, viral infection should result in BBB impairment so that leukocyte transmigration can occur [161].
Rodent models have been an instrumental tool used to study neuropathogenesis of HIV-1, as they offer several advantages such as: convenient handling, housing, and well characterized methods for manipulating their genome and affordability. Although HIV-1 does not naturally infect rodent cells, many approaches have been developed to circumvent this problem. For a more thorough review on animal models of HIV-1 please refer to the following publications [161, 162].
Transgenic animals
One of the earliest approaches for modeling CNS infection was the generation of transgenic rodent models expressing human proteins necessary for HIV-1 replication. Initial attempts included transgenic rodents expressing human viral receptors CD4, CCR5 and CXCR4, however, all these were unsuccessful and with limited use [163,164,165]. The next approach was the implementation of transgenic animals expressing HIV-1 viral proteins in the brain. As mentioned earlier, several reports have demonstrated neurotoxicity associated with HIV-1 viral proteins. One of the original models investigating the role of viral proteins in the brain was the gp120 transgenic mouse model, where CXCR4 tropic gp120 was primarily expressed in astrocytes [166]. These mice developed age-specific memory deficits and the model helped to delineate cellular pathways involved in gp120 mediated neurotoxicity [167, 168]. Similarly, a Tat transgenic mouse model was also developed where Tat is expressed under the control of a doxycycline-dependent GFAP promoter, allowing for these mice to develop Tat-dependent brain pathologies such as astrogliosis, infiltration of monocytes/T-cells and premature death [169]. One of the more recent transgenic models is the Vpr transgenic mouse model, where Vpr is specifically expressed in myeloid cells in both the central and peripheral nervous systems [139, 170]. These mice acquire CNS abnormalities, and signs of peripheral neuropathy that are linked to mitochondrial dysfunction [171]. Other groups, including our own, have also demonstrated that a faster and more acute approach for investigating the role of these viral proteins in neuropathogenesis is direct injection of recombinant proteins into the brain [103, 118]. An alternative approach for viral protein expression in rodents through transgenic technology was the development of the transgenic rat model where gag-pol are deleted from the HIV-1 genome to render the virus non-infectious [172]. These rats develop behavioral and motor deficits [173].
Chimeric viruses
Another innovative strategy for generating a small animal model of HIV-1 infection was demonstrated by Potash and colleagues who reengineered the virus in order to circumvent the obstacle of viral entry into murine cells [174]. This chimeric virus replaces gp120 with the murine leukemia virus gp80, facilitating entry into mouse cells [174]. Several studies have shown that mice infected with this chimeric strain display stable pro-virus in T-cells and macrophages, mucosal transmission of virus, normal CD4:CD8 ratios, a partially functional immune system and neurocognitive impairment [175, 176]. Our group along with others has demonstrated that intracranial (IC) administration of EcoHIV at a dose of 1x106 pg p24 directly into the caudate putamen results in increased levels of several inflammatory genes [177,178,179]. Moreover, this dose leads to hippocampal dysfunction shown by defective long-term potentiation in hippocampal slices ex vivo and significant reduction in MAP-2 and synapsin II staining. Recently, several groups have investigated behavioural deficits and potential adjuvant therapies in this mouse model [179,180,181,182]. In one report, Kelschenbach and colleagues administered EcoHIV at a dose of 1 × 106 pg p24 through intracranial (IC) injection directly into the caudate putamen of adult mice. The infected mice were subjected to radial arm water maze and cued-fear conditioning tests; deficits in both behavioural tests were observed starting at 19 days post infection [179]. Others have shown that inoculation with EcoHIV at a dose of 1 × 106 pg p24 through intraperitoneal (IP) injection leads to the same behavioural impairments as early as 1 month post infection [180,181,182].
Humanized mouse models
Humanized mouse models are becoming increasing popular for investigating interactions between the virus and host. Several variations of humanized mice have been developed through the use of severe combined immunodeficiency (SCID) genetic backgrounds. These mice have a mutant DNA-dependent protein kinase catalytic subunit, which causes the mice to lack functional B cells and T cells [183].
HIVE model
One of the earliest humanized models used for investigating neuroAIDS was the HIVE mouse model, where HIV-1 infected human monocyte-derived macrophages (MDMs) were injected directly into the basal ganglia of immunodeficient mice. Histopathological changes observed in these mouse brains included: formation of multinucleated giant cells, astrogliosis, microglial activation, and neuronal cell death. These mice also exhibited behavioural deficits [184,185,186,187].
huPBL-HIVE model
The next development of humanized mouse models aimed to examine the peripheral immunity in HIVE mice in order to get an in depth understanding of the adaptive immune system during HIV neuropathogenesis. Non-obese diabetic (NOD) mice were crossed with SCID background in order to enhance reconstitution with human peripheral lymphocytes (huPBLs). huPBL mice were synergistically injected in the brain with HIV-1-infected human macrophages [188, 189]. This model demonstrated the transmission of HIV-1 infected cells in the brain to human lymphocytes, the dissemination of virus throughout the blood, and the detection of HIV-1-specific cellular immune responses in the periphery [188]. A major limitation of this model is that these animals die within 4–5 weeks of engraftments due to human PBLs inducing graft-versus-host disease, where the human immune cells recognize the host mouse cells as foreign and attack them [188].
Human CD34+ cells and mouse lymphoid tissue re-population
The following series of models developed used NOD SCID mice crossed with interleukin-2 receptor g-chain (IL2Rg−/−) mice. The two versions of these mice were either engineered with partial deletion of IL2Rg−/− (NOG) [190] or complete deletion of IL2Rg−/− (NSG) [191]. IL2R is an essential protein for immune cell growth and maturation, and this mutation prevents the development of lymphomas. Therefore, the life spans of these mice are longer when compared to the standard NOD SCID mice. This model offered several other advantages compared to the HIVE model, for example, the ability to investigate peripheral and brain infection. Engraftment of NOD or NSG mice is performed by injecting human hematopoietic CD34+ stem cells derived from either cord blood, fetal liver or adult blood [191,192,193]. Human immune cells are present in numerous sites including the peripheral blood, liver, lung, vagina and rectum of these mice. NSG mice have been used as a useful model for investigating HIV-1-induced neuropathogenesis [194, 195].
BLT mice
The most recent humanized mouse model available is the bone liver thymus (BLT) mouse model. The approach for generating this model involves transplanting NOD SCID or NSG mice with human fetal thymus and liver cells following irradiation with human CD34+ cells from the same donor [196, 197]. The major advantage of these mice is that human T-cells develop within a human thymus, reflecting the clinical situation. Additionally, transmission of the virus can occur through the mucosal route. Studies have shown that BLT mice inoculated with HIV-1 have detectable levels of viral RNA and DNA in the brain [198, 199] and therefore, this could constitute another tool for investigating mechanisms and treatments for HAND.
It is important to note some of the limitations that exist in using humanized murine systems. For example, inconsistency in the amounts of grafted human cells, different mouse-to-human and human-to-mouse receptor-ligand interactions, varied populations of human and mouse macrophages and altered levels of infection dependent on human cell reconstitution and lack of significant mouse microglia. For more detail on brain pathologies of humanized mice please refer to [200].
Non-rodent animal models
SIV
Simian immunodeficiency virus (SIV) infection is another closely related lentivirus which can be used as a tool to study HIV-1 pathogenesis. Over 40 strains of SIV have been discovered that naturally infect African non-human primate species [201]. The natural SIV infection of hosts does not typically lead to disease due to thousands of years of virus-to-host co-evolution, therefore these infected primates are not useful pathogenic models [162]. On the other hand, infection of Asian macaques with specific strains of SIV recapitulates many aspects of the disease in humans therefore, these have become the most widely accepted models for HIV/AIDS research. The macaque models have allowed for several advances in our knowledge of viral transmission, pathogenesis and latency, as explained in a review by Clements and colleagues [202]. The search for an effective vaccine and microbicides for prevention of HIV-1 has been extensively studied in this model. The most commonly used macaque species for AIDS are the rhesus macaque, the pig-tailed macaque and the cynomolgus macaque [162]. The use of SIV infected Asian macaques has undoubtedly provided tremendous insight into the pathogenesis of HIV/AIDS, however, it is important to note that there are fundamental differences between SIV and HIV-1 that limit the use of SIV-macaque models for the investigation of specific research questions. SIV is non-responsive to several drugs targeting HIV-1 protease, reverse transcriptase and integrase enzymes. Viral entry between the two strains can also differ as HIV-1 can in some cases utilize CXCR4, whereas SIV rarely uses this co-receptor but is capable of binding to other co-receptors that are not used by HIV-1 [203]. In order to circumvent this limitation, efforts have geared towards the developments of SHIVs, chimeric viruses which contain recombinants generated by replacing SIV viral genes such as rev, tat and env with corresponding HIV-1 genes.
Macaques have several advantages over small-animal models. SIV or SHIV infection of macaques reflects human infection in regard to cell tropism of viral infection, progressive depletion of CD4+ T cells and development of opportunistic infections typical of AIDS. Additionally, macaques and humans have a close phylogenetic relationship where many of the human genes controlling immune responses to HIV are similar to those in macaques. However, the use of this model as a research tool for many laboratories is limited due to high maintenance costs and genetic variability which can complicate studies. Nonetheless, despite these limitations, the SIV/SHIV model remains a precious tool to drive the HIV research field forward and ultimately bring us closer to a vaccine or cure.
FIV
Feline immunodeficiency virus (FIV) infection is a model which offers a natural approach to studying lentiviral-associated neuropathology. Similar to HIV-1, FIV infection results in an acute phase with minor symptoms, an inconstant latent phase and detrimental CD4+ T-cell depletion [204, 205]. Specific strains of FIV can lead to infection in the CNS and subsequent neuropathological changes similar to those evident in HIV+ patients [206, 207]. This model has served as a great tool for the development of several ARVs [208, 209].
Potential pharmacological therapies for HAND
As the implementation of cART appears to do little to alleviate HAND, recent research has focussed on identifying new therapeutics to prevent cognitive decline in HIV-1+ individuals. Below is a summary of recent investigations. A chart displaying the disease models on which these compounds were tested can also be found (Table 2).
Natural compounds
Studies investigating natural compounds as potential anti-inflammatory agents for the treatment of HAND are limited. Curcumin is the most extensively studied anti-inflammatory natural product in the context of HAND to date but there are numerous other compounds that may be viable candidates. A review by Kurapati et al. summarizes a number of plant derived compounds showing antiviral activity against HIV, however, none of these compounds have shown to target inflammation [210]. Additionally, a review by Shal et al. discusses the neuroprotective potential of several natural products in the context of Alzheimer’s disease [211]. The compounds discussed in both reviews with exception of resveratrol and curcumin have not been examined as treatment options for HAND and may be considered as candidates in the future.
Resveratrol
Resveratrol is a stilbene derivative that is synthesized in grape skin and is found in wine, with a significantly higher concentration present in red wines [212]. To the best of our knowledge, there is only one report investigating the anti-inflammatory effects of this compound in the context of HAND [105]. This study used an ex vivo system of rat hippocampal slices exposed to HIV-1 Tat. Treatment with resveratrol reversed the Tat induced expression of CCL2 and TNFα pro-inflammatory molecules. Western blot analysis showed an elevation of phosphorylated ERK2 in Tat-exposed slices, which was also reversed with treatment of resveratrol, suggesting that resveratrol attenuates inflammation by inhibiting the ERK1/2 pathway [105]. It is difficult to fully evaluate resveratrol’s potential in HAND therapy based on a single ex vivo study. A prior study showed its capability to inhibit TNFα and iNOS production in lipopolysaccharide-activated rat microglia [213] however, extensive investigation is required before considering resveratrol as a candidate therapeutic agent for HAND.
Curcumin
Curcumin is a bright yellow organic compound derived from turmeric, a member of the ginger root family [214]. Outside of its uses as a herbal supplement, cosmetic ingredient, spice, and food coloring agent, curcumin has also been investigated as a potential treatment for HIV-associated neuropathologies. A 2013 in vitro study examined the effects of curcumin on gp120 V3 loop-treated mouse microglia cultures and primary cultures of rat neurons [215]. The results showed that curcumin treatment led to an inhibition of gp120 V3-loop induced ROS production as well as TNFα and CCL2 mRNA upregulation [215]. Furthermore, the authors found curcumin to be protective in rat neurons by reducing apoptosis in gp120 V3 loop exposed cells [215]. Additionally, this study also demonstrated that curcumin treatment in rat neurons attenuated the HIV-1 gp120 V3 loop-mediated increased K+ current, and this was likely a mechanism mediating curcumin‘s anti-apoptotic effects [215]. In a separate study, an ex vivo system of rat hippocampal slices exposed to gp120 V3-loop curcumin treatment improved synaptic plasticity, as demonstrated by a decrease in Ca2+ concentrations in the hippocampal synaptosomes [216]. These results are consistent with older studies which reported that treatment of curcumin to gp120 V3 loop exposed rat hippocampal neurons resulted in attenuated neuronal injury, decreased caspase-3 expression and improved mitochondrial function and synaptic growth [217, 218]. Recently, in murine microglial cells exposed to gp120 in vitro, curcumin treatment was able to inhibit autophagy and inflammatory responses (e.g. CCL2, IL-17) [219]. The authors demonstrated that the anti-inflammatory actions of curcumin were mediated through the PI3K/AKT/IKK/NF-κB autophagic pathway [219].
Together, the present studies on curcumin’s neuroprotective properties in the context of HAND appear promising, however in vivo studies investigating this potential therapy are scarce and further research is required in order to move forward with this natural product. In addition to its anti-inflammatory effects, studies have also demonstrated curcumin’s anti-viral properties. A review by Prasad and Tyagi summarizes curcumin’s inhibition of HIV-1 proteins and enzymes such as HIV protease, HIV integrase and Tat [220].
Anti-diabetic agents
PPARγ agonists
Peroxisome proliferator-activated receptor-gamma (PPARγ) is a ligand activated transcription factor that belongs to the nuclear receptors for steroid, thyroid hormones and retinoids and plays a major role in lipid and glucose regulation [221]. PPARγ agonists rosiglitazone and pioglitazone have been clinically proven as effective treatments for type 2 diabetes [221]. There is ample evidence suggesting that targeting the PPAR family is neuroprotective in several animal models of neurological disorders [222,223,224]. In the context of HIV-1, ample data from pre-clinical studies also supports PPARγ as an effective anti-inflammatory target in the brain [118, 225,226,227,228]. Studies have also demonstrated protective effects of PPARγ activation in reducing HIV-1 or Tat induced dysfunction in brain microvessel endothelial cells [226, 228]. Overexpression of PPARγ in a human brain microvessel endothelial cell line (hCMEC/D3) inhibited HIV-1 or Tat-mediated increases in IL-1β, TNF-α, CCL2 and E-selectin, which was partly mediated through inhibition of NF-кB transcriptional activity [228]. In this study, activation of PPARγ by an exogenous agonists (i.e., rosiglitazone) in hCMEC/D3 also protected against these responses, whereas, the antagonist of PPARγ reversed the protective effects [228]. The same group followed up with subsequent studies where, overexpression of PPARγ protected against increased matrix metalloproteases and proteasome activity, downregulation of tight junction proteins and increased monocyte migration in a co-culture model of hCMEC/D3 and astrocytes exposed to HIV-1 infected monocytes [226]. These results were further corroborated in an in vivo mouse model of Tat exposure through injection into internal carotid artery. Treatment with PPARγ agonist, rosiglitazone reduced Tat-induced BBB impairments, astrogliosis, and neuronal loss [227]. Our group has also demonstrated that both PPARγ agonists, rosiglitazone and pioglitazone, are protective against gp120 mediated inflammatory responses (IL-1β, TNF-α) in primary cultures of rat mixed glial cell cultures as well as in vivo in a rat model of gp120 exposure through intracerebroventricular injection [118]. Although the precise mechanism of how PPARγ agonists activate an anti-inflammatory response to HIV-1 remains unclear, our group has recently shown evidence of PPARγ agonists inhibiting NF-κB [118].Furthermore, targeting PPARγ has also proven to be anti-viral. Potula et al. have demonstrated that PPARγ activation by rosiglitazone resulted in suppression of HIV-1 LTR promoter activity and HIV-1 replication in MDMs through transrepression of NF-кB [225]. These results were corroborated in the HIVE model which demonstrated rosiglitazone-mediated suppression of viral replication in macrophages in brain tissues and 50% reduction in viremia in vivo [225]. More recently, our group has showed that treatment with PPARγ agonists rosiglitazone and/or pioglitazone in an EcoHIV mouse model effectively reduced HIV viral p24 protein burden in mice brains [178].
Insulin
Insulin is a hormone produced by the beta cells of the pancreas. The primary role of insulin is to help regulate blood sugar. Insulin is administered to diabetic patients who are unable to produce sufficient levels of insulin. Recently, the use of insulin as a therapeutic target for HAND has been explored. Mamik et al. demonstrated the anti-inflammatory effect of insulin in vitro and in vivo. In primary cultures of HIV-1 infected human microglia, insulin treatment reduced inflammatory genes CXCL10 and IL-6 as well as HIV-1 p24 levels in the supernatant [229]. In primary cultures of human neurons, insulin exposure prevented Vpr-mediated cell death [229]. In vivo, intranasal insulin treatment in FIV infected cats reduced the same markers (CXCL10, IL-6) and FIV RNA in the brain. Immunohistochemical analysis revealed diminished levels of glial activation and protection of cortical neurons. The neuroprotective mechanism behind insulin is not yet understood, but the authors hypothesize that insulin treatment regulates PPARγ expression in microglia and astrocytes, which may explain the reported observations. The molecular results were accompanied by functional improvement of neurobehavioral performance, including both motor and memory [229]. Furthermore, in an EcoHIV mouse model intranasal insulin beginning 23 days or 3 months post infection reversed neurocognitive impairments in mice. Insulin treatment also reduced HIV DNA in the brain, however, this was only achieved when treatment was initiated at earlier time points post infection (e.g. 23 days post infection) [182]. At present, there are at least two clinical trials that have been initiated for intranasal insulin for HAND treatment (Clinical trial IDs: NCT03081117 and NCT03277222) [230].
The prevalence of comorbid metabolic disorders is increasing as the HIV/AIDS populations ages. The risk of type 2 diabetes, stroke, hypertension and hyperlipidemia can be further exacerbated by cART [231,232,233]. Type 2 diabetes has been reported to be an established risk factor for HAND and other neurological disorders such as Alzheimer’s. Therefore, these compounds seem to be the most promising HAND treatments, as they could be used to treat not only the neurocognitive aspect but also the metabolic comorbidities. Additionally, as they are already in clinical use this could allow for relatively quick translation for use as therapy for HAND.
Glutamate modulators
Glutamate is an abundant excitatory neurotransmitter present in the brain which plays a critical role in synaptic plasticity. Under homeostatic conditions, glutamate is important for cognitive functions such as learning and memory. Glutamate is cleared from the extracellular space by specific uptake transporters expressed in neurons and glial cells. EAAT2 in humans or GLT-1 in rodents is mainly expressed in astrocytes and is the primary transporter responsible for glutamate clearance [234]. Once glutamate enters astrocytes it is converted into glutamine and released to the extracellular space where it is subsequently taken up by neurons which convert it back into glutamate which plays a role in neurotransmission. Impaired glutamate homeostasis can lead to glutamate excitotoxicity which is defined as excessive activation of receptors such as NMDA, leading to increased intracellular Ca2+ levels and subsequent activation of proteases and endonucleases that can damage cellular components. Excitotoxicity has been proposed to contribute to several neurological diseases including HAND [235]. Studies have shown that HIV-1 infected individuals have five-fold greater levels of glutamate in the CSF when compared to healthy controls [236]. Another report investigating patients receiving combinational ARV therapy, observed that those diagnosed with HAND had increases in CSF glutamate levels compared to the individuals without neurological impairments [237].
As ample evidence has demonstrated the importance of glutamate regulation in the context of neurological disorders including HAND, several groups have employed different strategies to modulate glutamate excitotoxicity. The use of memantine, an uncompetitive NMDAR antagonist which has been clinically validated as a treatment for moderate to severe Alzheimer’s Disease, is one such example. Numerous pre-clinical studies have demonstrated neuroprotective effects of memantine in various HAND models; gp120 mouse models [132, 238], HIVE SCID mice [239] and SIV-infected macaques [240]. Unfortunately, the benefits of memantine failed to translate in clinical trials in patients with HAND [241, 242]. More recently, Nakanishi and colleagues developed a series of improved derivatives of memantine, termed NitroMemantines, which allosterically inhibit NMDAR activity through an adducted nitro group that reacts with redox modulatory sites on the receptor. Treatment with NitroMemantine protected against gp120 induced neuronal damage and synaptic loss in the hippocampus of gp120 transgenic mice [243]. Although memantine was unsuccessful in improving neurocognitive function in HAND patients, it is possible that NitroMemantine could result in a better outcome for these patients, as it preferentially inhibits extra synaptic NMDARs, which are relevant to glutamate mediated excitotoxicity [244].
Other strategies that have been investigated for modulating glutamate homeostasis include regulation of enzymes that are responsible for generating glutamate [245] or regulation of the glutamate transporters themselves [118, 246]. Erdmann et al. showed that inhibition of glutaminase by glutaminase specific small molecule inhibitors or glutaminase specific siRNA were successful in preventing increased glutamate production in vitro by HIV-1 infected macrophages [245]. Unfortunately, there are no clinically available glutaminase inhibitors that penetrate the CNS. More recently, Nedelcovych et al. 2017 used glutamine antagonist 6-diazo-5-oxo-l-norleucine and successfully showed that this compound attenuates glutamate synthesis in HIV-infected microglia/macrophages and prevents spatial memory deficits in EcoHIV infected mice [180]. Although these results are exciting, this compound cannot be used clinically due to associated peripheral toxicities. The same group synthesized several prodrugs of 6-diazo-5-oxo-l-norleucine which aimed to enhance brain delivery while limiting peripheral exposure [180]. Given the efficacy of glutamine antagonists in neuroprotective effects, these novel compounds should be considered for clinical testing in patients with HAND. Another strategy that has been investigated is to specifically target the transporters for glutamate uptake, such as EAAT2/GLT-1. A series of reports have demonstrated that this transporter is dysregulated in the context of HIV-1 associated neurological complications, likely mediated by HIV-1 viral proteins such as gp120 [118, 129, 130]. Our group has shown that treatment with PPARγ agonists (rosiglitazone or pioglitazone) reverses the gp120 mediated downregulation of GLT-1 in primary cultures of rat astrocytes [118]. Previous bioinformatic analyses revealed that there are at least 6 putative consensus PPAR response element sites in the promoter region of the EAAT2 gene and in vitro treatment with rosiglitazone increased promoter activity, therefore PPARγ may be regulating GLT-1/EAAT2 at the transcriptional level [247].
The importance of glutamate involvement in HAND has been made abundantly clear, establishing it as an important component that warrants further study. Attempts to modulate glutamate response to alleviate HAND symptoms and pathogenesis have thus far been unsuccessful in the few existing clinical studies. However, with newly developed promising compounds, such as NitroMemantine or PPARγ agonists, research can now offer new candidates for human studies.
Statins
Statins make up a class of drugs capable of reducing cholesterol production in the liver by inhibiting the enzyme HMG-CoA reductase [248]. They are primarily used for the management of hypercholesteremia and the reduction of cardiovascular risk in patients. Aside from their role in limiting blood cholesterol, statins have also been shown to exhibit anti-inflammatory properties although the investigation of their effects on HIV-1 associated brain inflammation is limited. In a study previously performed by our group examining the anti-inflammatory properties in a rat model of gp120 induced neuroinflammation, simvastatin reduced enhanced brain expression of iNOS and IL-1β [64]. Yadav and colleagues demonstrated the abilities of atorvastatin and simvastatin in modulating the function and phenotype of human peripheral blood mononuclear cells. Treatment with a combination of atorvastatin and simvastatin reduced the proportion of CD16+ monocytes in PBMCs and purified monocyte cultures [249]. This is relevant as CD16+ cells are highly susceptible to HIV-1 and their migration to the CNS is vital to HIV-1 neuropathogenesis [250]. Treatment with simvastatin alone was also observed to reduce monocyte chemotaxis through inhibition of CCL2 secretion [249].
Despite the encouraging data from in vitro studies, the efficacy of statins has not been as promising in clinical studies. Bandaru and colleagues found no association with worsening or improved cognitive status in 19 HIV+ subjects actively on statin therapy [19]. This aligns with a previous pilot study in HIV+, ARV naïve males where treatment with atorvastatin failed to reduce HIV-1 RNA levels in the CSF and there were no changes in white blood cell counts or neopterin CSF concentrations [251]. However, there is a need to properly evaluate their therapeutic potential in the context of HAND, as a comprehensive review by van der Most and colleagues described the neuroprotective mechanism of statins in Alzheimer’s, Parkinson’s, Multiple Sclerosis and strokes [252] which may translate into HAND treatment.
ARVs
Maraviroc
Maraviroc (MVC) is an approved ARV that acts as a CCR5 receptor blocker to prevent the entry of HIV into target cells [253]. Its relatively high CNS penetration and low neurotoxicity [147, 254, 255] has made MVC an attractive compound for the treatment of HAND. Early in vitro studies have shown MVC to inhibit the migratory response of macrophages to CCL2 [256], suggesting anti-inflammatory actions of this CCR5 antagonist. SIV infected monkeys treated with MVC monotherapy for 5 months had decreased viral loads in the CNS as proven by an observed reduction in viral RNA and DNA in the basal ganglia [257]. Additionally, treated macaques had lower expression of TNFα, CCL2, and reduced macrophage activation in the brain [257]. Amyloid precursor protein levels were also reduced further supporting the concept that MVC is neuroprotective in an SIV macaque model [257].
The encouraging results from in vitro and in vivo studies combined with the fact that MVC is already approved for human use allowed for pilot clinical trials to be performed relatively quickly. A small, single arm, open label study intensified the cART regimen of 12 stable HIV+ patients with undetectable plasma viral RNA but detectable monocyte HIV DNA. These patients were subjected to MVC treatment for 24 weeks and their monocyte HIV DNA, circulating CD16+ monocytes levels and neuropsychological performance were monitored [258]. Flow cytometry and RT-PCR results demonstrated a reduction in both monocyte HIV DNA and plasma CD16+ monocytes content in study participants. MVC treatment improved neuropsychological test scores in half of the participants who had previously showed evidence of mild to moderate cognitive impairment [258]. Further pilot studies with MVC intensified cART therapy in patients with HAND show similar results. For example, in a separate single arm trial, HIV+ participants with associated cognitive deficits and plasma viral suppression were switched from their cART regimen comprised of tenofovir, emtricitabine, and efavirenz to one containing abacavir, lamivudine and MVC for 48 weeks. These patients underwent neuropsychological testing as well as had their blood and CSF analysed. There was no significant difference in improved global deficit scores or CSF inflammatory markers with the exception of reduced CSF TNFα [259]. More recently, a randomized controlled clinical study performed in HIV+ patients diagnosed with HAND compared the efficacy of MVC intensified cART on cognition to participants’ existing cART treatment for 12 months [260]. Results from this study showed an improved global neurocognitive performance in the MVC group over the control but the authors could not detect metabolic differences in the brains of subjects between the two groups nor could they detect significant treatment-related changes in neopterin or β2-microglobulin levels in the CSF, two molecules associated with neurocognitive impairment in HAND [21, 261].
The evidence in support of MVC’s beneficial effects on cognition in HAND patients is a positive first step into investigating its use as a therapeutic option, but the current published studies are plagued by poor sample sizes. Also, to the best of our knowledge, there is only one study conducted that was randomized and contained a control group, which is clearly insufficient to fully evaluate the potential of MVC. The studies also seem to be lacking in information on the effects of MVC on chemokine and cytokine levels in those with HAND in order to evaluate the efficacy of MVC in the context of HAND pathogenesis. Further clinical studies with larger sample sizes and more sensitive assays to detect changes in inflammatory markers must be implemented in order to make an informed evaluation of the therapeutic potential of MVC.
Type I interferon modulation
Type I interferons (IFN) are protein cytokines released by host immune cells in the presence of pathogens such as bacteria, viruses and tumor cells. Specifically, type I IFNs (IFNα and IFNβ) are secreted during viral infections where they bind to Interferon-alpha–beta receptors (INFARs) to upregulate antigen presentation and to express proteins to inhibit viral replication. Studies have shown that type I IFNs are implicated in both attenuating and exacerbating neuroinflammation triggered by HIV-1 infection. As a result, modulation of type I IFNs may be a key factor in treating HAND. A review on type I IFNs and their responses by Donlin and Ivashkiv provides great detail on this topic [262].
Interferonβ
Cocchi and colleagues showed that INFβ exposure in human microglia cell cultures induces the secretion of β-chemokines CCL3, CCL4 and CCL5 [263]. These endogenous CCR5 receptor ligands have been shown to inhibit HIV-1 infection and the progression to disease [264] and therefore make IFNβ a potential therapeutic option. IFNβ has already been shown to facilitate anti-inflammatory responses by down-regulating cytokine expression in CD4+ lymphocytes (IL-2, IFNγ, and IL-12), suppressing IFNγ-triggered iNOS expression by glial cells, and inhibiting the induction of MHC antigens [265,266,267]. Specifically, evidence supporting the neuroprotective roles of CCL4 and CCL5 against gp120-mediated toxicity in rat and murine neuronal cultures have been previously reported [268, 269]. Furthermore, INFβ inhibits HIV-1 infection in primary cultures of human fetal microglia [270]. Infection was enhanced when the cells were co-administered with anti-CCL4/5 antibodies, suggesting that INFβ indirectly supresses HIV-1 infection by upregulating β-chemokines [270].
Recently, the protective effects of IFNβ in HIV-1 related neuronal injury was investigated. In vitro and in vivo experiments with IFNβ were performed in rat cerebrocortical cultures containing astrocytes, neurons and microglia and in gp120-expressing transgenic mice [271]. In their in vitro experiments, recombinant murine IFNβ (mIFNβ) prevented gp120-mediated neuronal death in rat mixed cultures. Interferon-stimulated gene and protein expression (CCL5, CCL4, CCL3, CXCL10) were increased following IFNβ treatment. Neutralizing antibodies against each interferon-stimulated chemokine revealed that CCL4 is responsible for mediating the neuroprotective effects of IFNβ [271]. In vivo, gp120 transgenic mice treated with intranasal mIFNβ had increased CCL4 mRNA expression, higher levels of MAP-2 and synaptophysin and lower levels of Iba1 when compared to vehicle treated mice, demonstrating that IFNβ mediated neuroprotection [271]. This supports the results of an earlier in vivo study where type I IFN receptor knock out mice injected with EcoHIV experienced worse progression to disease when compared to wildtype, suggesting the involvement of type I IFN signalling in restricting HIV-1 infection and pathogenesis in the mice brains [177].
IFNβ is proposed to inhibit HIV-associated neuronal damage by binding to INFAR1/2 and increasing the expression of interferon-stimulated genes, particularly CCL4, to inhibit dendritic and synaptic injury. Current available studies with IFNβ in the context of HAND illustrate the signalling protein as a promising therapeutic option however there is a lack of behavioural studies. IFNβ is already FDA-approved for the management of multiple sclerosis [272] and therefore remains one of the most promising options for treating HAND.
B18r
Interferon alpha (IFNα) has been implicated in contributing to the neuropathology observed in HAND patients as high CSF concentrations of this cytokine have been reported to be positively correlated with cognitive impairment in HIV+ individuals [187, 273,274,275]. It is thought that IFNα leads to dendritic simplification through a mechanism mediated by INFAR and NMDA receptors, with the 2A subunit of the NMDA receptor playing a vital role in IFNα-mediated neurotoxicity [276]. In the endeavour to prevent IFNα related neurocognitive decline in HIV+ patients, research has shifted towards controlling IFNα with B18R, a recombinant protein originally expressed in vaccinia virus that is a type I IFN receptor capable of inhibiting type I IFNs in a wide variety of species [277]. In a HIVE/SCID mouse model of HAND, B18R was investigated for its BBB permeability, ability to neutralize IFNα and subsequently attenuate histopathological complications. HIVE/SCID mice administered IP with B18R three times daily for a total of 10 days [278]. Immunohistochemistry staining of brain tissue detected B18R, confirming its ability to cross the BBB. RT-PCR analysis also demonstrated the inhibitory effect of B18R on IFNα signalling by showing a downregulation of IFNα stimulated genes such as ISG15, IFNA4, and Ifrng15 [278]. More recently, Koneru et al., 2018 also investigated the neuroprotective effects of B18R in the HIVE/SCID mouse model. In this study, subcutaneous treatment twice daily with B18R in combination with a common cART regimen (atazanavir, tenofovir, and emtricitabine) prevented astrogliosis, the presence of mononuclear phagocytes and preserved staining of MAP2. Behavioural deficits in memory were investigated and results showed that treatment with B18R alone improved HIV-1 induced decrease in discrimination indexes [279]. Although the behavioural studies are limited, the results are promising and the authors plan to move forward with a Phase I clinical trial for B18R [279].
Others
Dimethyl fumarate
Dimethyl fumarate (DMF) is a methyl ester of fumaric acid and is hydrolysed to its active form monomethyl fumarate. Currently, it is approved for the treatment of psoriasis and multiple sclerosis in adults [280]. Although its precise mechanism of action is unknown, DMF has shown to be immunomodulatory and anti-inflammatory, making it a potential agent for targeting the neuroinflammation observed in HAND. A 2011 study found that direct treatment with DMF was able to reduce HIV-1 reverse transcriptase activity in human monocytes infected with HIV-1 [281]. In addition, when primary cultures of rat neurons were exposed to HIV infected human macrophages, DMF treatment increased neuronal survival rates, likely due to their role in suppressing HIV replication and neurotoxin release. CCL2 release by infected monocytes was attenuated by DMF and its metabolite, and DMF was also shown to inhibit TNFα production and NF-κB signalling, suggesting that DMF’s effects may be due to their modulation of inflammatory pathways [281].
One of the most recent studies investigating DMF and its possible role in treating HAND looks at lysosomal dysfunction and the release of neurotoxic cathepsin B from infected macrophages. Increased cathepsin B release has been evidenced in HIV-infected macrophages and promotes neuronal apoptosis [282,283,284] and infected macrophages treated with DMF showed decreases in HIV-1 replication, cathepsin B secretion and ROS/RNS production [285]. DMF has consistently shown evidence of reducing HIV-1 replication in macrophages and inhibiting the release of neurotoxic compounds in vitro, but conflicting results on neuronal cell viability prevents a definitive conclusion of DMF’s role in HAND therapy. The discrepancy may be due to species differences between human and rats or to the differences in cell types used (primary neurons vs neuroblastoma cells). To the best of our knowledge, in vivo studies using fumaric acid derivatives for HAND treatment do not exist and further work is needed to confirm its therapeutic potential.
Minocycline
In the search for alternative treatments for the associated neuroinflammation in HAND, the potential efficacy of minocycline has been examined. As a second generation tetracycline antibiotic, minocycline has shown promise as an inhibitor of microglia activation; in vitro models of brain inflammation demonstrated its neuroprotective effects through reducing expression of pro-inflammatory cytokines [286, 287], inhibition of apoptotic cell death [288,289,290], nitric oxide synthesis [291]. This, coupled with its highly lipophilic profile that allows it to easily cross the BBB [292], renders minocycline as a promising candidate for HAND therapy. Early work investigating the neuroprotective impact of minocycline on HIV-related neuroinflammation used an SIV macaque model of neuroAIDS [293]. Daily oral administration of minocycline to SIV infected macaques prevented neuronal injury, decreased astrogliosis and microglial activation [293]. Additionally, further evidence to support the anti-inflammatory effects of minocycline in the context of HIV-1 was reported by our group, where intracerebroventricularly administered gp120 rats treated with minocycline had reduced protein and mRNA levels of inflammatory markers [64].
Results from the in vitro and in vivo studies encouraged the investigation of minocycline in clinical trials for HAND therapy. A randomized double blind, placebo-controlled study treated HIV+ participants with cognitive impairment with oral minocycline every 12 h for 24 weeks where its safety and efficacy was assessed through frequency of adverse events and changes in neuropsychological test composite z scores(NPZ-8) [294]. The authors found no change in NPZ-8 over 24 weeks between the treatment group and the control group. Similar results were observed in a later clinical trial in 2013 where HIV+ participants with low CD4+ cell counts were given oral minocycline every 12 h and had their cognitive function measured using the Uganda Neuropsychological Test Battery Summary Measures and the MSK scale [295]. Minocycline was shown not to be effective for improving cognitive function. A year later, minocycline’s effects on CSF markers of neuronal injury, inflammation, neurotransmitter levels and oxidative stress were tested using the same participants from the previous 2013 clinical study. CSF concentrations of numerous markers such as TNFα, IL-6, ceramides, quinolinic acid and glutamate was measured after 24 weeks while participants were receiving oral minocycline [296]. Minocycline was only found to significantly reduce CSF ceramide, a marker of oxidative stress. This study further demonstrated minocycline’s ineffectiveness as a therapy for HAND clinically, despite previous animal studies suggesting the opposite.
The discrepancy between the effects of minocycline in SIV encephalitis models and HIV+ patients has yet to be explained. A recent study reports that viral load and neuronal damage were reduced with minocycline in a SIV neuroAIDS model using macaque monkeys [297]. Perhaps the interspecies differences between humans and macaques are great enough to demonstrate different responses to minocycline, which may be the explanation for the contradictory data. Furthermore, the macaques studied in the SIV research models were studied from a maximum of 24 weeks after being inoculated with SIV, whereas participants in clinical trials may have been living with their infection for years prior to their involvement in studies. Based on the data from the clinical studies, minocycline does not appear as a therapeutic option for HAND.
Meloxicam
Meloxicam is a nonsteroidal anti-inflammatory drug, meloxicam reduces prostaglandin production, and thus inflammation, by selectively inhibiting COX-2 enzymes. Used in veterinary and human medicine, it has been FDA approved for the treatment of osteoarthritis. To the best of our knowledge, only one study investigated the anti-inflammatory effects of meloxicam in the context of HIV-1 associated brain pathology. Transgenic HIV-1 female rats were administered meloxicam daily and underwent a battery of behavioural tests. The purpose of the study was to verify if HIV-1 proteins can cause depressive-like behaviours in rats via alterations in neuroinflammation and cell proliferation, and if the treatment with meloxicam can reverse the observed behavioural patterns. The authors found that transgenic mice had increased CCL2 gene expression and displayed depressive like behaviours [298]. Treatment with meloxicam reduced CCL2 expression but was not able to attenuate depressive behaviours. From this study alone, meloxicam’s anti-inflammatory profile is somewhat promising but it is unclear if meloxicam will be a viable treatment option, continued research will provide further insight on the role of meloxicam in HAND therapy.
Didehydro-cortistatin A
Didehydro-cortistatin A (dCA) is a steroidal alkaloid analog isolated from Corticium simplex, a marine sponge [299]. Initially discovered to have anti-proliferative effects [300], dCA was also found to inhibit Tat-mediated HIV-1 replication in CD4+ T-cells from infected patients [301]. A study published in 2015 investigated the therapeutic potential of dCA in a tat-transfected human astrocytic cell line as well as its effect on cocaine reward in Tat transgenic mice [302]. In the Tat-expressing astrocytic cell line, qPCR analysis revealed an increase in TNFα and IL-1β gene expression and surprisingly, a decrease in CCL2 gene expression, which were all returned to baseline upon dCA treatment [302]. Additionally, extracellular uptake of Tat into human glial cell lines was reduced upon dCA treatment. In vivo, dCA was able to efficiently cross the BBB and its treatment reversed Tat mediated potentiation of cocaine reward in Tat transgenic mice [302]. This is thought to be of clinical importance as substance abusers make up a significant population amongst HIV+ individuals and previous studies have shown that Tat and cocaine can collectively augment neuronal damage [303,304,305].
Other studies examining dCA as an inhibitor of neuroinflammation in HAND are scarce. Investigations primarily look at dCA as a potential therapy to manage HIV-1 infection in patients due to the evidence of dCA being a potent inhibitor of Tat, and by extension, HIV-1 replication [306,307,308]. It is proposed that dCA inhibits Tat by binding to its basic domain and blocking its interaction with viral RNA [301]. With a lack of available studies on the topic, it is unclear how likely dCA will function as a therapeutic agent. Although it was shown to reduce Tat-mediated inflammation in vitro, there is no in vivo data to corroborate this. Additionally, there are several other HIV-1 proteins other than Tat that can initiate an immune response in the brain. With that being said, there is still much to learn before an informed assessment of dCA’s potential can be made. However, with its ability to cross the BBB, its lack of toxicity in astrocytic cell lines, and its modulation of inflammatory cytokines further testing of dCA is needed.
Galectin-1
Galectin-1 is part of a large family of β-galactoside-binding proteins. Evidence suggests that it has a role in modulating cell to cell interactions and its expression is correlated with neuro-regeneration [309, 310]. Galectin-1 is thought to be a key modulator of CNS homeostasis, with specific effects attenuating proinflammatory cytokines and oxidative stress markers while enhancing expression of anti-inflammatory cytokines such as TGF-β and IL-10 in microglia [311]. In the context of HIV-1 associated brain pathology, a recent study showed that galectin-1 treatment in HIV-1 Tat transfected primary human microglia exposed to TNFα was able to inhibit nitric oxide production and ROS/RNS activity, likely through limiting the availability of l-arginine in the iNOS-mediated nitric oxide production pathway, and by inhibiting the expression of iNOS [312]. Furthermore, galectin-1 promoted the protective M2 phenotype in the transfected microglia [312]. To the best of our knowledge, this is the only published study investigating the effect of galactin-1 treatment in the context of HIV-1. Further in vitro and in vivo studies are necessary for a more complete understanding of galectin-1’s potential as a HAND therapeutic.
Cannabinoids
The endocannabinoid system is a complex signalling system comprised of lipid molecules, enzymes and G-protein coupled receptors responsible for mediating a wide range of effects in organisms such as modifying neurotransmitter release, modulating learning and memory, regulating food intake and modulating inflammation and pain reception [313]. These effects are mediated through the activation of cannabinoid receptor 1 (CB1) and cannabinoid receptor 2 (CB2), the latter of which is primarily expressed in the cells of the immune system, including microglia [314, 315]. Synthetic cannabinoid agonists have been evidenced to yield beneficial effects in animal models of numerous neurodegenerative diseases such as Parkinson’s disease, multiple sclerosis and Huntington’s disease [316,317,318], thus cannabinoids may have similar effects in models of HAND.
Numerous in vitro studies have investigated the potential of cannabinoids in HIV-1 induced neuropathology. For example, using an in vitro co-culture composed of human brain microvascular endothelial cells and human astrocytes, synthetic cannabinoids ACEA and CP55,940 were investigated for their protective effects against gp120 [319]. ACEA and CP55,940 preserved the permeability of the endothelial cells, protected dysregulation of the tight junctions, maintained Ca2+ intracellular concentrations, and reduced monocytes transmigration, all of which were affected by gp120 exposure [319]. In another in vitro study, the synthetic CB1/CB2 agonist WIN55,212-2 protected human neurons that were exposed to gp120. Using dopamine transporter suppression as a measure of cellular injury, treatment with WIN55,212-2 blocked the downregulation of dopamine transporter activity and prevented neuronal apoptosis [320]. These effects appear to be primarily mediated by CB2 as the protective effects of WIN55,212-2 were abolished by CB2 antagonist co-treatment, whereas, co-treatment with CB1 antagonist was not sufficient in abolishing the protective effects [320]. WIN55,212-2 and the endocannabinoid anandamide displayed evidence of preventing Tat-induced decreases in GABAergic postsynaptic currents (frequency and amplitude) in murine prefrontal cortices [321]. Interestingly enough, this outcome was implied to be primarily through interaction of CB1 and not CB2, as additional treatment with a CB1 antagonist prevented the occlusion of Tat-mediated GABAergic signalling but the addition of a CB2 antagonist did not [321]. Furthermore, an in vitro study using primary human and murine neural progenitor cells exposed to gp120 showed that treatment with CB2 agonist AM1241 protected against toxicity and apoptosis [322].
In vivo studies investigating cannabinoids in the context of HAND treatment exist but are limited. Gorantla and colleagues tested the immunoregulatory effects of the synthetic CB2 agonist Gp1a in the HIVE mouse model [323]. Treatment with the CB2 agonist resulted in decreases in TNFα expression and microglia activation in the brain as well as a reduction of CCR5 expression in CD4+ cells in the spleen [323]. Another study which used gp120 transgenic mice showed that administration with the CB2 agonist AM1241 increased the number of neurons, neuroblasts and cells positive in markers of proliferation (BrdU, PCNA) in the hippocampi of these mice [322]. Cannabinoid treated gp120 transgenic mice also had decreased instances of astrogliosis when compared to vehicle treated mice [322].
Cannabinoids may have a place in treating HAND. As their exact mechanism in immunomodulation is still not yet fully understood, the details of their role cannot be fully described. There seems to be some inconsistency over which isoform of the cannabinoid receptors (CB1 or CB2) is critical to the immunomodulation of cannabinoids in HAND. Evidence does seem to favour CB2 as being the more important receptor as Purohit and colleagues have written a review focusing on CB2 and how its activation affects HAND [324]. Cannabinoids have also been reported to have antiviral activity and a study demonstrated their ability to inhibit HIV-1 replication in macrophages via activation of CB2 [325]. Studies have demonstrated the beneficial effect that cannabinoids have on GABAergic and dopaminergic signalling, two pathways whose dysregulation is associated with HAND [326,327,328,329,330], which further implicates their therapeutic potential. Continued study of this class of compounds is warranted as the current available data is encouraging.
Paroxetine and fluconazole combination
Fluconazole is an approved oral anti-fungal medication whereas paroxetine is a selective serotonin reuptake inhibitor. In 2014, Meulendyke and colleagues screened 2000 FDA approved drugs and natural compounds for neuroprotection against gp120 and Tat neurotoxicity and found the molecules fluconazole and paroxetine to be the most promising candidates [331]. This group then demonstrated that once daily oral treatment with combined fluconazole and paroxetine (FluPar) in SIV-infected rhesus macaques reduced CSF NFL concentrations, accumulation of amyloid precursor proteins and prevented CaMKIIα loss in the frontal cortex [331]. Despite the favourable neuroprotective effects, FluPar did not inhibit viral replication in the brain, CSF or periphery and had little effect on inflammatory markers such as CCL2 and IL-6 in the CNS [331]. Although this study did not show FluPar’s control of SIV infection, it did establish proof of concept of FluPar’s beneficial neuroprotective properties.
As both compounds are already approved for use in humans, a double-blind placebo controlled clinical study testing the efficacy FluPar in attenuating HAND was published shortly after the initial pre-clinical work. The parameters for this clinical trial were as follows: HIV+ patients with cognitive impairment were randomly assigned to placebo, fluconazole, paroxetine, or combination fluconazole and paroxetine treatment for 24 weeks and underwent neurological tests (NPZ8 and the Global Deficit Scale [332]. Participants receiving paroxetine had improved scores in tests measuring reaction time, verbal fluency, visual attention and task switching, with an overall improvement in NPZ8 summary scores but had lower performances in letter number sequencing tests when compared to participants not receiving paroxetine [332]. Paroxetine also had little effect on cellular stress, inflammatory and neuronal damage biomarkers. In contrast, fluconazole treatment, did not show any improvements on cellular stress markers or cognition [332].
Very much like meloxicam, fluconazole showed promise in treating HAND in an SIV macaque model but failed to prove efficacy in human trials. However, paroxetine still may have a significant role in achieving a viable HAND therapeutic as it is well tolerated in humans and showed encouraging data on improving cognition in HIV+ patients [332].
Chloroquine
Chloroquine is an FDA approved antiparasitic drug for the treatment of malaria. It disrupts the ability of the malarial parasite to metabolize toxic heme proteins, thus leading to an internal build up of the molecule and ultimately resulting in death [333]. Chloroquine also has mild immunosuppressive effects by inhibiting thiamine uptake via the SLC19A3 transporter [334]. In its treatment for rheumatoid arthritis, its activity reduces lymphocyte proliferation, antigen presentation in dendritic cells, ROS production by macrophages, enzyme release by lysozymes and inhibits phospholipase A2. It is these demonstrated effects that calls into question chloroquine’s potential in treating HIV-associated brain inflammation.
In a 2014 animal model of HIV-induced brain inflammation, rats were intracerebroventricularly injected with gp120 while being pretreated with interparietal minocycline, simvastatin or chloroquine. Upregulation of IL-1β and iNOS was attenuated in the frontal cortex, striatum and hippocampus of the animals pretreated with chloroquine while secretions of IL-1β in the CSF were reduce [64].
Although animal studies with chloroquine and HIV induced brain inflammation appear to be promising, clinical trial assessments have demonstrated no benefit in HIV replication, as well as insignificant immune-dampening properties. HIV+ patients undergoing chloroquine treatment for 24 weeks experienced no change in T-cell activation, absolute counts of CD8+ and CD4+ cells and no reductions in plasma concentrations of cytokines including IFN-γ, TNF-α, IL-1β, IL-6, IL-7, IL-8, IL-12 and IL-13 [335]. A separate randomized trial not only established chloroquine’s inability to reduce CD8+ cell activation in HIV+ patients not on ART, but also found that it exacerbated CD4+ cell count decline and increased HIV replication [336].
Conclusion
The advent of cART has helped millions of people with HIV-1 and allowed for the management of their infection. Despite cART efficacy, neurological disorders still persist in up to half of those living with HIV-1. Diagnosis of HAND requires exhaustive efforts, and the lack of viable biomarkers increases the challenge of properly identifying affected individuals. Several factors can be associated with the persistence of HAND such as continuous brain inflammation caused by low level HIV-1 replication in cellular reservoirs such as microglia, secretion/shedding of viral proteins and poor penetration of ARVs across the BBB. In order to overcome this pressing issue in such a vulnerable subset of the general population, researchers have employed the use of various animal models in order to further understand the pathology and test pharmacological agents in the pursuit of achieving an effective treatment for HAND. Animal models such as SIV infected macaques and mice treated with chimeric viruses have been useful for evaluating drug candidates in a preclinical setting. The ongoing research has led to promising discoveries such as the efficacy of exogenous IFNβ, maraviroc, and PPARγ agonists. Further studies are still required for such pharmacological entities in order to reach definitive conclusions on their possible use in the clinic.
Availability of data and materials
Confirmed.
Abbreviations
- AIDS:
-
Acquired immunodeficiency syndrome
- ADC:
-
AIDS dementia complex
- ALCAM:
-
Activated leukocyte cell adhesion molecule
- AMPA:
-
α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- ARV:
-
Antiretroviral
- ANI:
-
Asymptomatic neurocognitive impairment
- BBB:
-
Blood–brain barrier
- BCSFB:
-
Blood–cerebrospinal barrier
- BLT:
-
Bone liver thymus
- CCR(n):
-
C-C chemokine receptor type(n)
- CCL(n):
-
C-C motif chemokine ligand(n)
- CNS:
-
Central nervous system
- CSF:
-
Cerebrospinal fluid
- CD(n):
-
Cluster of differentiation(n)
- cART:
-
Combined antiretroviral therapy
- C3:
-
Complement component 3
- CXCL10:
-
C-X-C motif chemokine ligand 10
- CPE:
-
CNS penetration effectiveness
- CXCR4:
-
C-X-C motif chemokine receptor 4
- COX-2:
-
Cyclooxygenase-2
- DNA:
-
Deoxyribose nucleic acid
- dCA:
-
Didehydro-cortistatin A
- DMF:
-
Dimethyl fumarate
- EAAT(n):
-
Excitatory amino acid transporter(n)
- FIV:
-
Feline immunodeficiency virus
- GFAP:
-
Glial fibrillary acidic protein
- gp120:
-
Glycoprotein 120
- HAD:
-
HIV-associated dementia
- HAND:
-
HIV associated neurocognitive disorders
- HIVE:
-
HIV encephalitis
- HIV:
-
Human immunodeficiency virus
- iNOS:
-
Inducible nitric oxide synthase
- INFAR(n):
-
Interferon alpha–beta receptor
- IFNα:
-
Interferon α
- IFNβ:
-
Interferon β
- IL-(n):
-
Interleukin-(n)
- IL-1β:
-
Interleukin-1β
- IL2R:
-
Interleukin-2 receptor
- UNAIDS:
-
Joint United Nations program on HIV/AIDS
- JAM-A:
-
Junctional adhesion molecule A
- MAP-2:
-
Microtubule-associated protein
- MND:
-
Minor neurocognitive disorder
- MDM:
-
Monocyte derived macrophages
- NFL:
-
Neurofilament light chain
- NMDA:
-
N-methyl-d-aspartate
- NF-κB:
-
Nuclear factor kappa-light-chain-enhancer of activated B cells
- PPARgamma:
-
Peroxisome proliferator-activated receptor gamma
- qPCR:
-
Quantitative polymerase chain reaction
- SCID:
-
Severe combined immunodeficiency
- SIV:
-
Simian immunodeficiency virus
- Tat:
-
Trans-activator for transcription
- TNFα:
-
Tumor necrosis factor α
- Vpr:
-
Viral protein R
References
Unaids. UNAIDS DATA 2018; 2018. http://www.unaids.org/sites/default/files/media_asset/unaids-data-2018_en.pdf. Accessed 7 Aug 2018.
Cecilia RE, Ovidiu R, Mihaela S, Dana CR. HIV-associated neurocognitive disorders. Neurologist. 2012;18(2):64–7.
Navia BA, Jordan BD, Price RW. The AIDS dementia complex: I. Clinical features. Ann Neurol. 1986;19(6):517–24.
Antinori A, Becker N. Updated research nosology for HIV-associated neuro cognitive disorders. Neurology. 2007;69:1789–99.
Sacktor N, Skolasky RL, Seaberg E, Munro C, Becker JT, Martin E, et al. Prevalence of HIV-associated neurocognitive disorders in the Multicenter AIDS Cohort Study. Neurology. 2015;86:334–40.
Saylor D, Dickens AM, Sacktor N, Haughey N, Slusher B, Pletnikov M, et al. HIV-associated neurocognitive disorder—pathogenesis and prospects for treatment. Nat Rev Neurol. 2016;12(4):234–48.
Fox H, Purnell P. HIV-associated neurocognitive disorders. Neuroimmune Pharmacology. Cham: Springer; 2016. p. 407–20.
Goodkin K, Hardy DJ, Singh D, Lopez E. Diagnostic utility of the international HIV dementia scale for HIV-associated neurocognitive impairment and disorder in South Africa. J Neuropsychiatry Clin Neurosci. 2014;26(4):352–8.
Jika Yusuf A, Hassan A, Indo Mamman A, Mohammed Muktar H, Maude Suleiman A, Baiyewu O. HIV clinical management prevalence of HIV-associated neurocognitive disorder (HAND) among patients attending a tertiary health facility in northern nigeria. J Int Assoc Provid AIDS Care. 2017;16(1):48–55.
Sacktor N. Changing clinical phenotypes of HIV-associated neurocognitive disorders. J Neurovirol. 2018;24:141–5.
Heaton RK, Franklin DR, Ellis RJ, McCutchan JA, Letendre SL, LeBlanc S, et al. HIV-associated neurocognitive disorders before and during the era of combination antiretroviral therapy: differences in rates, nature, and predictors. J Neurovirol. 2011;17(1):3–16.
Heaton RK, Clifford DB, Franklin DR, Woods SP, Ake C, Vaida F, et al. HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy: charter Study. Neurology. 2010;75(23):2087–96.
Grant I, Franklin DR, Deutsch R, Woods SP, Vaida F, Ellis RJ, et al. Asymptomatic HIV-associated neurocognitive impairment increases risk for symptomatic decline. Neurology. 2014;82(23):2055–62.
Robertson K, Yosief S. Neurocognitive assessment in the diagnosis of HIV-associated neurocognitive disorders. Semin Neurol. 2014;34(01):021–6.
Masters MC, Ances BM. Role of neuroimaging in HIV-associated neurocognitive disorders. Semin Neurol. 2014;34(1):89–102.
Bonnet F, Amieva H, Marquant F, Bernard C, Bruyand M, Dauchy FA, et al. Cognitive disorders in HIV-infected patients: are they HIV-related? Aids. 2013;27(3):391–400.
Kallianpur KJ, Shikuma C, Kirk GR, Shiramizu B, Valcour V, Chow D, et al. Peripheral blood HIV DNA is associated with atrophy of cerebellar and subcortical gray matter. Neurology. 2013;80(19):1792–9.
Dickens AM, Anthony DC, Deutsch R, Mielke MM, Claridge TDW, Grant I, et al. Cerebrospinal fluid metabolomics implicate bioenergetic adaptation as a neural mechanism regulating shifts in cognitive states of HIV-infected patients. AIDS. 2015;29(5):1.
Bandaru VVR, Mielke MM, Sacktor N, Mcarthur JC, Grant I, Letendre S, et al. A lipid storage—like disorder contributes to cognitive decline in HIV-infected subjects. Neurology. 2013;81:1492–9.
Chang L, Lee PL, Yiannoutsos CT, Ernst T, Marra CM, Richards T, et al. A multicenter in vivo proton-MRS study of HIV-associated dementia and its relationship to age. Neuroimage. 2004;23(4):1336–47.
Hagberg L, Cinque P, Gisslen M, Brew BJ, Spudich S, Bestetti A, et al. Cerebrospinal fluid neopterin: an informative biomarker of central nervous system immune activation in HIV-1 infection. AIDS Res Ther. 2010;7(1):1–12.
Williams DW, Byrd D, Rubin LH, Anastos K, Morgello S, Berman JW. CCR2 on CD14+CD16+monocytes is a biomarker of HIV-associated neurocognitive disorders. Neurol Neuroimmunol Neuroinflamm. 2014;1(3):1–9.
Ellery PJ, Tippett E, Chiu Y-L, Paukovics G, Cameron PU, Solomon A, et al. The CD16+ monocyte subset is more permissive to infection and preferentially harbors HIV-1 in vivo. J Immunol. 2007;178(10):6581–9.
Cassol E, Misra V, Morgello S, Gabuzda D. Applications and limitations of inflammatory biomarkers for studies on neurocognitive impairment in HIV infection. J Neuroimmune Pharmacol. 2013;8(5):1087–97.
Peterson J, Gisslen M, Zetterberg H, Fuchs D, Shacklett BL, Hagberg L, et al. Cerebrospinal fluid (CSF) neuronal biomarkers across the spectrum of HIV infection: hierarchy of injury and detection. PLoS ONE. 2014;9(12):e116081.
Abdulle S, Mellgren Å, Brew BJ, Cinque P, Hagberg L, Price RW, et al. CSF neurofilament protein (NFL)—a marker of active HIV-related neurodegeneration. J Neurol. 2007;254(8):1026–32.
Norgren N, Rosengren L, Stigbrand T. Elevated neurofilament levels in neurological diseases. Brain Res. 2003;987(1):25–31.
McGuire JL, Gill AJ, Douglas SD, Kolson DL. Central and peripheral markers of neurodegeneration and monocyte activation in HIV-associated neurocognitive disorders. J Neurovirol. 2015;21(4):439–48.
Nitkiewicz J, Borjabad A, Morgello S, Murray J, Chao W, Emdad L, et al. HIV induces expression of complement component C3 in astrocytes by NF-κB-dependent activation of interleukin-6 synthesis. J Neuroinflamm. 2017;14(1):23.
Fabbiani M, Ciccarelli N, Tana M, Farina S, Baldonero E, Di Cristo V, et al. Cardiovascular risk factors and carotid intima-media thickness are associated with lower cognitive performance in HIV-infected patients. HIV Med. 2013;14(3):136–44.
Valcour V, Shikuma C, Shiramizu B, Watters M, Poff P, Selnes O, et al. Higher frequency of dementia in older HIV-1 individuals: the Hawaii Aging with HIV-1 Cohort. Neurology. 2004;63(5):822–7.
Kinai E, Komatsu K, Sakamoto M, Taniguchi T, Nakao A, Igari H, et al. Association of age and time of disease with HIV-associated neurocognitive disorders: a Japanese nationwide multicenter study. J Neurovirol. 2017;23(6):864–74.
Gonzalez R, Schuster. Substance abuse, hepatitis C, and aging in HIV: common cofactors that contribute to neurobehavioral disturbances. Neurobehav HIV Med. 2012;33(7):15.
Gonzalez R, Schuster RM, Vassileva J, Martin EM. Impact of HIV and a history of marijuana dependence on procedural learning among individuals with a history of substance dependence. J Clin Exp Neuropsychol. 2011;33(7):735–52.
Tedaldi EM, Minniti NL, Fischer T. HIV-associated neurocognitive disorders: the relationship of HIV infection with physical and social comorbidities. Biomed Res Int. 2015;2015:1–13.
Spudich S, González-Scarano F. HIV-1-related central nervous system disease: current issues in pathogenesis, diagnosis, and treatment. Cold Spring Harb Perspect Med. 2012;2(6):1–17.
Wilen CB, Tilton JC, Doms RW. HIV: cell binding and entry. Cold Spring Harb Perspect Med. 2012;2(8):a006866.
Shepherd JC, Jacobson LP, Qiao W, Jamieson BD, Phair JP, Piazza P, et al. Emergence and persistence of CXCR4-tropic HIV-1 in a population of men from the multicenter AIDS cohort study; 2008. http://www.statepi. Accessed 6 Sept 2018.
Joseph SB, Arrildt KT, Sturdevant CB, Swanstrom R. HIV-1 target cells in the CNS. J Neurovirol. 2015;21(3):276–89.
Toma J, Whitcomb JM, Petropoulos CJ, Huang W. Dual-tropic HIV type 1 isolates vary dramatically in their utilization of CCR5 and CXCR4 coreceptors. Aids. 2010;24(14):2181–6.
Valcour V, Chalermchai T, Sailasuta N, Marovich M, Lerdlum S, Suttichom D, et al. Central nervous system viral invasion and inflammation during acute HIV infection. J Infect Dis. 2012;206(2):275–82.
Liu NQ, Lossinsky AS, Popik W, Li X, Gujuluva C, Kriederman B, et al. Human immunodeficiency virus type 1 enters brain microvascular endothelia by macropinocytosis dependent on lipid rafts and the mitogen-activated protein kinase signaling pathway. J Virol. 2002;76(13):6689–700.
Veenstra M, León-Rivera R, Li M, Gama L, Clements JE, Berman JW. Mechanisms of CNS viral seeding by HIV+ CD14+CD16+ monocytes: establishment and reseeding of viral reservoirs contributing to HIV-associated neurocognitive disorders. MBio. 2017;8(5):e01280-17.
Williams DW, Calderon TM, Lopez L, Carvallo-Torres L, Gaskill PJ, Eugenin EA, et al. Mechanisms of HIV Entry into the CNS: increased sensitivity of HIV infected CD14+CD16+ monocytes to CCL2 and key roles of CCR2, JAM-A, and ALCAM in diapedesis. Public Libr Sci ONE. 2013;8(6):e69270.
Pulliam L, Gascon R, Stubblebine M, McGuire D, McGrath MS. Unique monocyte subset in patients with AIDS dementia. Lancet. 1997;349(9053):692–5.
Thieblemont N, Weiss L, Sadeghi HM, Estcourt C, Haeffner-Cavaillon N. CD1410wCD16high: a cytokine-producing monocyte subset which expands during human immunodeficiency virus infection. Eur J Immunol. 1995;25:3418–24.
Williams DW, Anastos K, Morgello S, Berman JW. JAM-A and ALCAM are therapeutic targets to inhibit diapedesis across the BBB of CD14+CD16+ monocytes in HIV-infected individuals. J Leukoc Biol. 2015;97(2):401–12.
Calantone N, Wu F, Klase Z, Deleage C, Perkins M, Matsuda K, et al. Tissue myeloid cells in SIV-infected primates acquire viral DNA through phagocytosis of infected T cells. Immunity. 2014;41(3):493–502.
Baxter AE, Russell RA, Duncan CJA, Moore MD, Willberg CB, Pablos JL, et al. Macrophage infection via selective capture of HIV-1-infected CD4+ T cells. Cell Host Microbe. 2014;16(6):711–21.
Honeycutt JB, Wahl A, Baker C, Spagnuolo RA, Foster J, Zakharova O, et al. Macrophages sustain HIV replication in vivo independently of T cells. J Clin Invest. 2016;126(4):1353–66.
Ho DD, Rota TR, Hirsch MS. Infection of monocyte/macrophages by human T lymphotropic virus type III. J Clin Invest. 1986;77(5):1712–5.
Swingler S, Mann AM, Zhou J, Swingler C, Stevenson M. Apoptotic killing of HIV-1-infected macrophages is subverted by the viral envelope glycoprotein. PLoS Pathog. 2007;3(9):e134.
Alam C, Whyte-Allman S-K, Omeragic A, Bendayan R. Role and modulation of drug transporters in HIV-1 therapy. Adv Drug Deliv Rev. 2016;103:121–43.
Bracq L, Xie M, Lambelé M, Vu L-T, Matz J, Schmitt A, et al. T Cell-macrophage fusion triggers multinucleated giant cell formation for HIV-1 spreading. J Virol. 2017;91(24):1–19.
Wallet C, De Rovere M, Van Assche J, Daouad F, De Wit S, Gautier V, et al. Microglial cells: the main HIV-1 reservoir in the brain. Front Cell Infect Microbiol. 2019;9:1–18.
Geny C, Gherardi R, Boudes P, Lionnet F, Cesaro P, Gray F. Multifocal multinucleated giant cell myelitis in an AIDS patient. Neuropathol Appl Neurobiol. 1991;17(2):157–62.
Dargent J-L, Lespagnard L, Kornreich A, Hermans P, Clumeck N, Verhest A. HIV-associated multinucleated giant cells in lymphoid tissue of the Waldeyer’s ring: a detailed study. Mod Pathol. 2000;13(12):1293–9.
Lewin-Smith M, Wahl SM, Orenstein JM. Human immunodeficiency virus-rich multinucleated giant cells in the colon: a case report with transmission electron microscopy, immunohistochemistry, and in situ hybridization. Mod Pathol. 1999;12(1):75–81.
Ryzhova EV, Crino P, Shawver L, Westmoreland SV, Lackner AA, González-Scarano F. Simian immunodeficiency virus encephalitis: analysis of envelope sequences from individual brain multinucleated giant cells and tissue samples. Virology. 2002;297(1):57–67.
Soulas C, Conerly C, Kim W-K, Burdo TH, Alvarez X, Lackner AA, et al. Recently infiltrating MAC387+ monocytes/macrophages. Am J Pathol. 2011;178(5):2121–35.
Teo I, Veryard C, Barnes H, An SF, Jones M, Lantos PL, et al. Circular forms of unintegrated human immunodeficiency virus type 1 DNA and high levels of viral protein expression: association with dementia and multinucleated giant cells in the brains of patients with AIDS. J Virol. 1997;71(4):2928–33.
Vicandi B, Jiménez-Heffernan JA, López-Ferrer P, Patrón M, Gamallo C, Colmenero C, et al. HIV-1 (p24)-positive multinucleated giant cells in HIV-associated lymphoepithelial lesion of the parotid gland. Acta Cytol. 1999;43(2):247–51.
Warriner EM, Rourke SB, Rourke BP, Rubenstein S, Millikin C, Buchanan L, et al. Immune activation and neuropsychiatric symptoms in HIV infection. J Neuropsychiatry Clin Neurosci. 2010;22(3):321–8.
Ashraf T, Jiang W, Hoque M, Henderson J, Wu C, Bendayan R. Role of anti-inflammatory compounds in human immunodeficiency virus-1 glycoprotein120-mediated brain inflammation. J Neuroinflamm. 2014;11(1):91.
Dohgu S, Ryerse JS, Robinson SM, Banks WA. Human immunodeficiency virus-1 uses the mannose-6-phosphate receptor to cross the blood-brain barrier. PLoS ONE. 2012;7(6):1–12.
Toborek M, Lee YW, Flora G, Pu H, András IE, Wylegala E, et al. Mechanisms of the blood-brain barrier disruption in HIV-1 infection. Cell Mol Neurobiol. 2005;25(1):181–99.
Xiong H, Ton H. Astrocyte dysfunctions and HIV-1 neurotoxicity. J AIDS Clin Res. 2013;04(01):255.
András IE, Pu H, Tian J, Deli MA, Nath A, Hennig B, et al. Signaling mechanisms of HIV-1 Tat-induced alterations of Claudin-5 expression in brain endothelial cells. J Cereb Blood Flow Metab. 2005;25(9):1159–70.
Pu H, Tian J, Andras IE, Hayashi K, Flora G, Hennig B, et al. HIV-1 Tat protein-induced alterations of ZO-1 expression are mediated by redox-regulated ERK1/2 activation. J Cereb Blood Flow Metab. 2005;25(10):1325–35.
Johnsen AR, Jacobsen OS, Gudmundsson L, Albers CN. Erratum to: Chloroform emissions from arctic and subarctic ecosystems in Greenland and Northern Scandinavia (Biogeochemistry, https://doi.org/10.1007/s10533-016-0241-5). Biogeochemistry. 2016;130(1–2):67.
Kanmogne GD, Primeaux C, Grammas P. HIV-1 gp120 proteins alter tight junction protein expression and brain endothelial cell permeability: implications for the pathogenesis of HIV-associated dementia. J Neuropathol Exp Neurol. 2005;64(6):498–505.
Chen H, Wood C, Petito CK. Comparisons of HIV-1 viral sequences in brain, choroid plexus and spleen: potential role of choroid plexus in the pathogenesis of HIV encephalitis. J Neurovirol. 2000;6(6):498–506.
Burkala EJ, He J, West JT, Wood C, Petito CK. Compartmentalization of HIV-1 in the central nervous system: role of the choroid plexus. Aids. 2005;19(7):675–84.
Lamers SL, Gray RR, Salemi M, Huysentruyt LC, McGrath MS. HIV-1 phylogenetic analysis shows HIV-1 transits through the meninges to brain and peripheral tissues. Infect Genet Evol. 2011;11(1):31–7.
Pal S, Rao S, Louveau A. Meningeal lymphatic network: the middleman of neuroinflammation. Clin Exp Neuroimmunol. 2020;11(1):21–5.
Schwartz M, Baruch K. The resolution of neuroinflammation in neurodegeneration: leukocyte recruitment via the choroid plexus. EMBO J. 2014;33(1):7–22.
Jessen NA, Munk ASF, Lundgaard I, Nedergaard M. The glymphatic system: a beginner’s guide. Neurochem Res. 2015;40(12):2583–99.
Jordan CA, Watkins BA, Kufta C, Dubois-Dalcq M. Infection of brain microglial cells by human immunodeficiency virus type 1 is CD4 dependent. J Virol. 1991;65(2):736–42.
Chen NC, Partridge AT, Sell C, Torres C, Martín-García J. Fate of microglia during HIV-1 infection: from activation to senescence? Glia. 2017;65(3):431–46.
Castellano P, Prevedel L, Eugenin EA. HIV-infected macrophages and microglia that survive acute infection become viral reservoirs by a mechanism involving Bim. Sci Rep. 2017;7(1):12866.
Gray LR, Turville SG, HItchen TL, Cheng W-J, Ellett AM, Salimi H, et al. HIV-1 Entry and trans-infection of astrocytes involves CD81 vesicles. PLoS ONE. 2014;9(2):e90620.
Luo X, He JJ. Cell–cell contact viral transfer contributes to HIV infection and persistence in astrocytes. J Neurovirol. 2015;21(1):66–80.
Eugenin EA, Berman JW. Cytochrome c dysregulation induced by HIV infection of astrocytes results in bystander apoptosis of uninfected astrocytes by an IP3 and calcium-dependent mechanism. J Neurochem. 2013;127(5):644–51.
An SF, Groves M, Gray F, Scaravilli F. Early entry and widespread cellular involvement of HIV-1 DNA in brains of HIV-1 positive asymptomatic individuals. J Neuropathol Exp Neurol. 1999;58(11):1156–62.
Anderson CE, Tomlinson GS, Pauly B, Brannan FW, Chiswick A, Brack-Werner R, et al. Relationship of Nef-positive and GFAP-reactive astrocytes to drug use in early and late HIV infection. Neuropathol Appl Neurobiol. 2003;29(4):378–88.
Trillo-Pazos G, Diamanturos A, Rislove L, Menza T, Chao W, Belem P, et al. Detection of HIV-1 DNA in microglia/macrophages, astrocytes and neurons isolated from brain tissue with HIV-1 encephalitis by laser capture microdissection. Brain Pathol. 2003;13(2):144–54.
Churchill MJ, Wesselingh SL, Cowley D, Pardo CA, McArthur JC, Brew BJ, et al. Extensive astrocyte infection is prominent in human immunodeficiency virus—associated dementia. Ann Neurol. 2009;66:253–8.
Liu Y, Liu H, Kim BO, Gattone VH, Li J, Nath A, et al. CD4-independent infection of astrocytes by human immunodeficiency virus type 1: requirement for the human mannose receptor. J Virol. 2004;78(8):4120–33.
Canki M, Thai JNF, Chao W, Ghorpade A, Potash MJ, Volsky DJ. Highly productive infection with pseudotyped human immunodeficiency virus type 1 (HIV-1) indicates no intracellular restrictions to HIV-1 replication in primary human astrocytes. J Virol. 2001;75(17):7925–33.
Russell RA, Chojnacki J, Jones DM, Johnson E, Do T, Eggeling C, et al. Astrocytes resist HIV-1 fusion but engulf infected macrophage material. Cell Rep. 2017;18(6):1473–83.
Ko A, Kang G, Hattler JB, Galadima HI, Zhang J, Li Q, et al. Macrophages but not astrocytes harbor HIV DNA in the brains of HIV-1-infected aviremic individuals on suppressive antiretroviral therapy. J Neuroimmune Pharmacol. 2018;14:110–9.
Churchill MJ, Wesselingh SL, Cowley D, Pardo CA, McArthur JC, Brew BJ, et al. Extensive astrocyte infection is prominent in human immunodeficiency virus—associated dementia. Ann Neurol. 2009;66(2):253–8.
Bertrand L, Cho HJ, Toborek M. Blood–brain barrier pericytes as a target for HIV-1 infection. Brain. 2019;142(3):502–11.
Nakagawa S, Castro V, Toborek M. Infection of human pericytes by HIV-1 disrupts the integrity of the blood-brain barrier. J Cell Mol Med. 2012;16(12):2950–7.
Sillman B, Woldstad C, Mcmillan J, Gendelman HE. Neuropathogenesis of human immunodeficiency virus infection. Handbook of clinical neurology. Amsterdam: Elsevier; 2018. p. 21–40.
Lindl KA, Marks DR, Kolson DL, Jordan-Sciutto KL. HIV-associated neurocognitive disorder: pathogenesis and therapeutic opportunities. J Neuroimmune Pharmacol. 2010;5(3):294–309.
Mujeeb A, Bishop K, Peterlintt BMM, Turck C, Parslowtllli TGG, James TLL. NMR structure of a biologically active peptide containing the RNA-binding domain of human immunodeficiency virus type 1 Tat (protein structure/RNA-binding proteins/gene regulation/AIDS). Proc Natl Acad Sci USA. 1994;91:8248–52.
Hategan A, Bianchet MA, Steiner J, Karnaukhova E, Masliah E, Fields A, et al. HIV Tat protein and amyloid-β peptide form multifibrillar structures that cause neurotoxicity. Nat Struct Mol Biol. 2017;24(4):379–86.
Bratanich AC, Liu C, Mcarthur JC, Fudyk T, Glass JD, Mittoo S, et al. Brain-derived HIV-1 tat sequences from AIDS patients with dementia show increased molecular heterogeneity. J Neurovirol. 1998;4(4):387–93.
Hudson L, Liu J, Nath A, Jones M, Raghavan R, Narayan O, et al. Detection of the human immunodeficiency virus regulatory protein tat in CNS tissues. J Neurovirol. 2000;6(2):145–55.
Marciniak RA, Sharp PA. HIV-1 Tat protein promotes formation of more-processive elongation complexes. EMBO J. 1991;10(13):4189–96.
Chen P, Mayne M, Power C, Nath A. The Tat protein of HIV-1 induces tumor necrosis factor-α production. J Biol Chem. 1997;272(36):22385–8.
Li ZH, Pu SS, Gao WH, Chi YY, Wen HL, Wang ZY, Song YY, Yu XJ, et al. Effects of HIV-1 tat on secretion of TNF-(alpha) and IL-1(beta) by U87 cells in AIDS patients with or without AIDS dementia complex. Biomed Environ Sci. 2014;27(2):111–7.
Chauhan A, Turchan J, Pocernich C, Bruce-Keller A, Roth S, Butterfield DA, et al. Intracellular human immunodeficiency virus Tat expression in astrocytes promotes astrocyte survival but induces potent neurotoxicity at distant sites via axonal transport. J Biol Chem. 2003;278(15):13512–9.
Lee EO, Kim SE, Park HK, Kang JL, Chong YH. Extracellular HIV-1 Tat upregulates TNF-α dependent MCP-1/CCL2 production via activation of ERK1/2 pathway in rat hippocampal slice cultures: inhibition by resveratrol, a polyphenolic phytostilbene. Exp Neurol. 2011;229(2):399–408.
Kim SE, Lee EO, Yang JH, Kang JHL, Suh YH, Chong YH. 15-deoxy-Δ12,14-prostaglandin J2inhibits human immunodeficiency virus-1 tat-induced monocyte chemoattractant protein-1/CCL2 production by blocking the extracellular signal-regulated kinase-1/2 signaling pathway independently of peroxisome proliferator-act. J Neurosci Res. 2012;90(9):1732–42.
Sawaya BE, Thatikunta P, Denisova L, Brady J, Khalili K, Amini S. Regulation of TNFα and TGFβ-1 gene transcription by HIV-1 Tat in CNS cells. J Neuroimmunol. 1998;87(1–2):33–42.
King JE, Eugenin EA, Buckner CM, Berman JW. HIV tat and neurotoxicity. Microbes Infect. 2006;8(5):1347–57.
Nath A, Conant K, Chen P, Scott C, Major EO. Transient exposure to HIV-1 Tat protein results in cytokine production in macrophages and astrocytes. J Biol Chem. 1999;274(24):17098–102.
Conant K, Garzino-Demo A, Nath A, McArthur JC, Halliday W, Power C, et al. Induction of monocyte chemoattractant protein-1 in HIV-1 Tat-stimulated astrocytes and elevation in AIDS dementia. Med Sci. 1998;95(6):3117–21.
Fan Y, He JJ. HIV-1 tat promotes lysosomal exocytosis in astrocytes and contributes to astrocyte-mediated Tat neurotoxicity. J Biol Chem. 2016;291(43):22830–40.
Zhou BY, Liu Y, Oh Kim B, Xiao Y, He J. Astrocyte activation and dysfunction and neuron death by HIV-1 Tat expression in astrocytes. Mol Cell Neurosci. 2004;27(3):296–305.
Kim K, Lee S-G, Kegelman TP, Su Z-Z, Das SK, Dash R, et al. Role of Excitatory Amino Acid Transporter-2 (EAAT2) and glutamate in neurodegeneration: opportunities for developing novel therapeutics. J Cell Physiol. 2011;226(10):2484–93.
Landi A, Iannucci V, Van Nuffel A, Meuwissen P, Verhasselt B. One protein to rule them all: modulation of cell surface receptors and molecules by HIV Nef. Curr HIV Res. 2011;9(7):496–504.
Sami Saribas A, Cicalese S, Ahooyi TM, Khalili K, Amini S, Sariyer IK. HIV-1 Nef is released in extracellular vesicles derived from astrocytes: evidence for Nef-mediated neurotoxicity. Cell Death Dis. 2017;8(1):e2542.
Acharjee S, Branton WG, Vivithanaporn P, Maingat F, Paul AM, Dickie P, et al. HIV-1 Nef expression in microglia disrupts dopaminergic and immune functions with associated mania-like behaviors. Brain Behav Immun. 2014;40:74–84.
Chompre G, Cruz E, Maldonado L, Rivera-Amill V, Porter JT, Noel RJ. Astrocytic expression of HIV-1 Nef impairs spatial and recognition memory. Neurobiol Dis. 2013;49(11):128–36.
Omeragic A, Hoque MT, Choi U, Bendayan R. Peroxisome proliferator-activated receptor-gamma: potential molecular therapeutic target for HIV-1-associated brain inflammation. J Neuroinflamm. 2017;14(1):183.
Ronaldson PT, Bendayan R. HIV-1 viral envelope glycoprotein gp120 triggers an inflammatory response in cultured rat astrocytes and regulates the functional expression of P-glycoprotein. Mol Pharmacol. 2006;70(3):1087–98.
Ashraf T, Ronaldson PT, Persidsky Y, Bendayan R. Regulation of P-glycoprotein by human immunodeficiency virus-1 in primary cultures of human fetal astrocytes. J Neurosci Res. 2011;89(11):1773–82.
Avdoshina V, Fields JA, Castellano P, Dedoni S, Palchik G, Trejo M, et al. The HIV protein gp120 alters mitochondrial dynamics in neurons. Neurotox Res. 2016;29(4):583–93.
Nath A, Haughey NJ, Jones M, Anderson C, Bell JE, Geiger JD. Synergistic neurotoxicity by human immunodeficiency virus proteins Tat and gp120: protection by memantine. Ann Neurol. 2000;47(2):186–94.
Jones MV, Bell JE, Nath A. Immunolocalization of HIV envelope gp120 in HIV encephalitis with dementia. AIDS. 2000;14(17):2709–13.
Louboutin J-P, Agrawal L, Reyes BAS, Van Bockstaele EJ, Strayer DS. HIV-1 gp120 neurotoxicity proximally and at a distance from the point of exposure: protection by rSV40 delivery of antioxidant enzymes. Neurobiol Dis. 2009;34(3):462–76.
Agrawal L, Louboutin J-P, Marusich E, Reyes BAS, Van Bockstaele EJ, Strayer DS. Dopaminergic neurotoxicity of HIV-1 gp120: reactive oxygen species as signaling intermediates. Brain Res. 2010;1306:116–30.
Gorman AM. Neuronal cell death in neurodegenerative diseases: recurring themes around protein handling: Apoptosis Review Series. J Cell Mol Med. 2008;12(6A):2263–80.
Pinton P, Giorgi C, Siviero R, Zecchini E, Rizzuto R. Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene. 2008;27(50):6407–18.
Chen L, Liu J, Xu C, Keblesh J, Zang W, Xiong H. HIV-1gp120 induces neuronal apoptosis through enhancement of 4-aminopyridine-senstive outward K + currents. PLoS ONE. 2011;6(10):e25994.
Wang Z, Pekarskaya O, Bencheikh M, Chao W, Gelbard HA, Ghorpade A, et al. Reduced expression of glutamate transporter EAAT2 and impaired glutamate transport in human primary astrocytes exposed to HIV-1 or gp120. Virology. 2003;312(1):60–73.
Melendez RII, Roman C, Capo-Velez CMM, Lasalde-Dominicci JAA. Decreased glial and synaptic glutamate uptake in the striatum of HIV-1 gp120 transgenic mice. J Neurovirol. 2016;22(3):358–65.
Kanmogne GD, Schall K, Leibhart J, Knipe B, Gendelman HE, Persidsky Y. HIV-1 gp120 compromises blood-brain barrier integrity and enhance monocyte migration across blood-brain barrier: implication for viral neuropathogenesis. J Cereb Blood Flow Metab. 2007;27(1):123–34.
Louboutin J-P, Strayer DS. Blood-brain barrier abnormalities caused by HIV-1 gp120: mechanistic and therapeutic implications. Sci World J. 2012;2012:1–15.
Tang H, Lu D, Pan R, Qin X, Xiong H, Dong J. Curcumin improves spatial memory impairment induced by human immunodeficiency virus type 1 glycoprotein 120 V3 loop peptide in rats. Life Sci. 2009;85(1–2):1–10.
Glowa JR, Panlilio LV, Brenneman DE, Gozes I, Fridkin M, Hill JM. Learning impairment following intracerebral administration of the HIV envelope protein gp120 or a VIP antagonist. Brain Res. 1992;570(1–2):49–53.
Barak O, Weidenfeld J, Goshen I, Ben-Hur T, Taylor AN, Yirmiya R. Intracerebral HIV-1 glycoprotein 120 produces sickness behavior and pituitary–adrenal activation in rats: role of prostaglandins. Brain Behav Immun. 2002;16(6):720–35.
Barak O, Goshen I, Ben-Hur T, Weidenfeld J, Taylor AN, Yirmiya R. Involvement of brain cytokines in the neurobehavioral disturbances induced by HIV-1 glycoprotein120. Brain Res. 2002;933(2):98–108.
James T, Nonnemacher MR, Wigdahl B, Krebs FC. Defining the roles for Vpr in HIV-1-associated neuropathogenesis. J Neurovirol. 2016;22(4):403–15.
DA Wheeler E, Achim CL, Ayyavoo V. Immunodetection of human immunodeficiency virus type 1 (HIV-1) Vpr in brain tissue of HIV-1 encephalitic patients. J Neurovirol. 2006;12(3):200–10.
Jones GJ, Barsby NL, Cohen EA, Holden J, Harris K, Dickie P, et al. HIV-1 Vpr causes neuronal apoptosis and in vivo neurodegeneration. J Neurosci. 2007;27(14):3703–11.
Wang Y, Santerre M, Tempera I, Martin K, Mukerjee R, Sawaya BE. HIV-1 Vpr disrupts mitochondria axonal transport and accelerates neuronal aging. Neuropharmacology. 2017;117:364–75.
Rom I, Deshmane SL, Mukerjee R, Khalili K, Amini S, Sawaya BE. HIV-1 Vpr deregulates calcium secretion in neural cells. Brain Res. 2009;1275:81–6.
Guha D, Nagilla P, Redinger C, Srinivasan A, Schatten GP, Ayyavoo V. Neuronal apoptosis by HIV-1 Vpr: contribution of proinflammatory molecular networks from infected target cells. J Neuroinflammation. 2012;9(1):664.
Vella S, Schwartländer B, Sow SP, Eholie SP, Murphy RL. The history of antiretroviral therapy and of its implementation in resource-limited areas of the world. AIDS. 2012;26(10):1231–41.
Letendre S, Marquie-Beck J, Capparelli E, Best B, Clifford D, Collier AC, et al. Validation of the CNS penetration-effectiveness rank for quantifying antiretroviral penetration into the central nervous system. Arch Neurol. 2008;65(1):65–70.
Carvalhal A, Gill MJ, Letendre SL, Rachlis A, Bekele T, Raboud J, et al. Central nervous system penetration effectiveness of antiretroviral drugs and neuropsychological impairment in the Ontario HIV Treatment Network Cohort Study. J Neurovirol. 2016;22(3):349–57.
Bertrand L, Méroth F, Tournebize M, Leda AR, Sun E, Toborek M. Targeting the HIV-infected brain to improve ischemic stroke outcome. Nat Commun. 2019;10(1):2009.
Robertson K, Liner J, Meeker RB. Antiretroviral neurotoxicity. J Neurovirol. 2012;18(5):388–99.
Ciccarelli N, Fabbiani M, Baldonero E. Efavirenz associated with cognitive disorders in otherwise asymptomatic HIV- infected patients. Neurology. 2011;76:1403–9.
Ma Q, Vaida F, Wong J, Sanders CA, Kao Y-T, Croteau D, et al. Long-term efavirenz use is associated with worse neurocognitive functioning in HIV-infected patients. J Neurovirol. 2016;22(2):170–8.
Staud F, Ceckova M, Micuda S, Pavek P. Expression and function of P-glycoprotein in normal tissues: effect on pharmacokinetics. Totowa: Humana Press; 2010. p. 199–222.
Chan GNY, Patel R, Cummins CL, Bendayan R. Induction of P-glycoprotein by antiretroviral drugs in human brain microvessel endothelial cells. Antimicrob Agents Chemother. 2013;57(9):4481–8.
Chan GNY, Hoque MT, Bendayan R. Role of nuclear receptors in the regulation of drug transporters in the brain. Trends Pharmacol Sci. 2013;34:361–72.
Best BM, Letendre SL, Koopmans P, Rossi SS, Clifford DB, Collier AC, et al. Low cerebrospinal fluid concentrations of the nucleotide HIV reverse transcriptase inhibitor, tenofovir. JAIDS J Acquir Immune Defic Syndr. 2012;59(4):376–81.
Best BM, Letendre SL, Brigid E, Clifford DB, Collier AC, Gelman BB, et al. Low atazanavir concentrations in cerebrospinal fluid. AIDS. 2009;23(1):83–7.
Gisolf EH, Enting RH, Jurriaans S, de Wolf F, van der Ende ME, Hoetelmans RMW, et al. Cerebrospinal fluid HIV-1 RNA during treatment with ritonavir/saquinavir or ritonavir/saquinavir/stavudine. AIDS. 2000;14(11):1583–9.
Gong Y, Chowdhury P, Nagesh PKB, Rahman MA, Zhi K, Yallapu MM, et al. Novel elvitegravir nanoformulation for drug delivery across the blood-brain barrier to achieve HIV-1 suppression in the CNS macrophages. Sci Rep. 2020;10(1):3835.
Agrawal N, Rowe J, Lan J, Yu Q, Hrycyna CA, Chmielewski J. Potential tools for eradicating HIV reservoirs in the brain: development of trojan horse prodrugs for the inhibition of P-glycoprotein with anti-HIV-1 activity. J Med Chem. 2020;63(5):2131–8.
Kaushik A, Yndart A, Atluri V, Tiwari S, Tomitaka A, Gupta P, et al. Magnetically guided non-invasive CRISPR-Cas9/gRNA delivery across blood-brain barrier to eradicate latent HIV-1 infection. Sci Rep. 2019;9(1):3928.
Friedrich BM, Dziuba N, Li G, Endsley MA, Murray JL, Ferguson MR. Host factors mediating HIV-1 replication. Virus Res. 2011;161(2):101–14.
Rawat P, Spector SA. Development and characterization of a human microglia cell model of HIV-1 infection. J Neurovirol. 2017;23(1):33–46.
Gorantla S, Poluektova L, Gendelman HE. Rodent models for HIV-associated neurocognitive disorders. Trends Neurosci. 2012;35(3):197–208.
Hatziioannou T, Evans DT. Animal models for HIV/AIDS research. Nat Rev Microbiol. 2012;10(12):852–67.
Browning J, Horner JW, Pettoello-Mantovani M, Raker C, Yurasov S, DePinho RA, et al. Mice transgenic for human CD4 and CCR5 are susceptible to HIV infection. Proc Natl Acad Sci. 1997;94(26):14637–41.
Dunn CS, Mehtali M, Houdebine LM, Gut J-P, Kirn A, Aubertin A-M. Human immunodeficiency virus type 1 infection of human CD4-transgenic rabbits. J Gen Virol. 1995;76(6):1327–36.
Keppler OT, Welte FJ, Ngo TA, Chin PS, Patton KS, Tsou C-L, et al. Progress toward a human CD4/CCR5 transgenic rat model for De Novo infection by human immunodeficiency virus type 1. J Exp Med. 2002;195(6):719–36.
Toggas SM, Masliah E, Rockenstein EM, Rail GF, Abraham CR, Mucke L. Central nervous system damage produced by expression of the HIV-1 coat protein gpl20 in transgenic mice. Nature. 1994;367(6459):188–93.
Garden GA, Budd SL, Tsai E, Hanson L, Kaul M, D’Emilia DM, et al. Caspase cascades in human immunodeficiency virus-associated neurodegeneration. J Neurosci. 2002;22(10):4015–24.
Toneatto S, Finco O, Van Der Putten H, Abrignani S, Annunziata P. Evidence of blood-brain barrier alteration and activation in HIV-1 gp120 transgenic mice. Aids. 1999;13(17):2343–8.
Kim BO, Liu Y, Ruan Y, Xu ZC, Schantz L, He JJ. Neuropathologies in transgenic mice expressing human immunodeficiency virus type 1 Tat protein under the regulation of the astrocyte-specific glial fibrillary acidic protein promoter and doxycycline. Am J Pathol. 2003;162(5):1693–707.
Acharjee S, Noorbakhsh F, Stemkowski PL, Olechowski C, Cohen ÉA, Ballanyi K, et al. HIV-1 viral protein R causes peripheral nervous system injury associated with in vivo neuropathic pain. FASEB J. 2010;24(11):4343–53.
Power C, Hui E, Vivithanaporn P, Acharjee S, Polyak M. Delineating HIV-associated neurocognitive disorders using transgenic models: the neuropathogenic actions of Vpr. J Neuroimmune Pharmacol. 2012;7(2):319–31.
Reid W, Sadowska M, Denaro F, Rao S, Foulke J, Hayes N, et al. An HIV-1 transgenic rat that develops HIV-related pathology and immunologic dysfunction. Proc Natl Acad Sci. 2001;98(16):9271–6.
Vigorito M, Connaghan KP, Chang SL. The HIV-1 transgenic rat model of neuroHIV. Brain Behav Immun. 2015;48:336–49.
Potash MJ, Chao W, Bentsman G, Paris N, Saini M, Nitkiewicz J, et al. A mouse model for study of systemic HIV-1 infection, antiviral immune responses, and neuroinvasiveness EcoHIV infection of mice forms a useful model of HIV-1 infection of human beings for convenient and safe investigation of HIV-1 therapy, vaccines; 2005.
Gu CJ, Borjabad A, Hadas E, Kelschenbach J, Kim BH, Chao W, et al. EcoHIV infection of mice establishes latent viral reservoirs in T cells and active viral reservoirs in macrophages that are sufficient for induction of neurocognitive impairment. PLoS Pathog. 2018;14:1–33.
Geraghty P, Hadas E, Kim B-H, Dabo AJ, Volsky DJ, Foronjy R. HIV infection model of chronic obstructive pulmonary disease in mice. Am J Physiol Lung Cell Mol Physiol. 2017;312(4):L500–9.
He H, Sharer LR, Chao W, Gu CJJ, Borjabad A, Hadas E, et al. Enhanced human immunodeficiency virus Type 1 expression and neuropathogenesis in knockout mice lacking Type I interferon responses. J Neuropathol Exp Neurol. 2014;73(1):59–71.
Omeragic A, Kara-Yacoubian N, Kelschenbach J, Sahin C, Cummins CL, Volsky DJ, et al. Peroxisome proliferator-activated receptor-gamma agonists exhibit anti-inflammatory and antiviral effects in an EcoHIV mouse model. Sci Rep. 2019;9(1):9428.
Kelschenbach J, He H, Kim B, Borjabad A, Gu C, Chao W, et al. Efficient expression of HIV in immunocompetent mouse brain reveals a novel nonneurotoxic viral function in hippocampal synaptodendritic injury and memory impairment. Am Soc Microbiol. 2019;10(4):1–17.
Nedelcovych MT, Tenora L, Kim BH, Kelschenbach J, Chao W, Hadas E, et al. N-(Pivaloyloxy)alkoxy-carbonyl prodrugs of the glutamine antagonist 6-diazo-5-oxo-l-norleucine (DON) as a potential treatment for HIV associated neurocognitive disorders. J Med Chem. 2017;60(16):7136–98.
Nedelcovych MT, Kim B-H, Zhu X, Lovell LE, Manning AA, Kelschenbach J, et al. Glutamine antagonist JHU083 normalizes aberrant glutamate production and cognitive deficits in the EcoHIV murine model of HIV-associated neurocognitive disorders. J Neuroimmune Pharmacol. 2019;14:391–400.
Kim BH, Kelschenbach J, Borjabad A, Hadas E, He H, Potash MJ, et al. Intranasal insulin therapy reverses hippocampal dendritic injury and cognitive impairment in a model of HIV-associated neurocognitive disorders in EcoHIV-infected mice. AIDS. 2019;33(6):973–84.
Blunt T, Gell D, Fox M, Taccioli GE, Lehmann AR, Jackson SP, et al. Identification of a nonsense mutation in the carboxyl-terminal region of DNA-dependent protein kinase catalytic subunit in the scid mouse. Proc Natl Acad Sci. 1996;93(19):10285–90.
Avgeropoulos N, Kelley B, Middaugh L, Arrigo S, Persidsky Y, Gendelman HE, et al. SCID mice with HIV encephalitis develop behavioral abnormalities. J Acquir Immune Defic Syndr Hum Retrovirol. 1998;18(1):13–20.
Anderson E, Boyle J, Zink W, Persidsky Y, Gendelman H, Xiong H. Hippocampal synaptic dysfunction in a murine model of human immunodeficiency virus type 1 encephalitis. Neuroscience. 2003;118(2):359–69.
Xiong H, Zeng YC, Lewis T, Zheng J, Persidsky Y, Gendelman HE. HIV-1 infected mononuclear phagocyte secretory products affect neuronal physiology leading to cellular demise: relevance for HIV-1-associated dementia. J Neurovirol. 2000;6 Suppl 1(5):S14–23.
Sas AR, Bimonte-Nelson H, Smothers CT, Woodward J, Tyor WR. Interferon-α causes neuronal dysfunction in encephalitis. J Neurosci. 2009;29(12):3948–55.
Poluektova L, Gorantla S, Faraci J, Birusingh K, Dou H, Gendelman HE. Neuroregulatory events follow adaptive immune-mediated elimination of HIV-1-infected macrophages: studies in a murine model of viral encephalitis. J Immunol. 2004;172(12):7610–7.
Poluektova LY, Munn DH, Persidsky Y, Gendelman HE. Generation of cytotoxic T cells against virus-infected human brain macrophages in a murine model of HIV-1 encephalitis. J Immunol. 2002;168(8):3941–9.
Ito M, Hiramatsu H, Kobayashi K, Suzue K, Kawahata M, Hioki K, et al. NOD/SCID/gamma cnull mouse: an excellent recipient mouse model for engraftment of human cells. Blood. 2002;100(9):3175–82.
Ishikawa F, Yasukawa M, Lyons B, Yoshida S, Miyamoto T, Yoshimoto G, et al. Development of functional human blood and immune systems in NOD/SCID/IL2 receptor chainnull mice. Blood. 2005;106(5):1565–73.
Shultz LD, Lyons BL, Burzenski LM, Gott B, Chen X, Chaleff S, et al. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R null mice engrafted with mobilized human hemopoietic stem cells. J Immunol. 2005;174(10):6477–89.
Brehm MA, Cuthbert A, Yang C, Miller DM, DiIorio P, Laning J, et al. Parameters for establishing humanized mouse models to study human immunity: analysis of human hematopoietic stem cell engraftment in three immunodeficient strains of mice bearing the IL2rγnull mutation. Clin Immunol. 2010;135(1):84–98.
Dash PK, Gorantla S, Gendelman HE, Knibbe J, Casale GP, Makarov E, et al. Loss of neuronal integrity during progressive HIV-1 infection of humanized mice. J Neurosci. 2011;31(9):3148–57.
Gorantla S, Makarov E, Finke-Dwyer J, Castanedo A, Holguin A, Gebhart CL, et al. Links between progressive HIV-1 infection of humanized mice and viral neuropathogenesis. Am J Pathol. 2010;177(6):2938–49.
Melkus MW, Estes JD, Padgett-Thomas A, Gatlin J, Denton PW, Othieno FA, et al. Humanized mice mount specific adaptive and innate immune responses to EBV and TSST-1. Nat Med. 2006;12(11):1316–22.
Wege AK, Melkus MW, Denton PW, Estes JD, Garcia JV. Functional and phenotypic characterization of the humanized BLT mouse model. Current topics microbiology immunology. Berlin: Springer; 2008. p. 149–65.
Araínga M, Su H, Poluektova LY, Gorantla S, Gendelman HE. HIV-1 cellular and tissue replication patterns in infected humanized mice. Sci Rep. 2016;6(1):23513.
Asahchop EL, Meziane O, Mamik MK, Chan WF, Branton WG, Resch L, et al. Reduced antiretroviral drug efficacy and concentration in HIV-infected microglia contributes to viral persistence in brain. Retrovirology. 2017;14(1):47.
Honeycutt JB, Sheridan PA, Matsushima GK, Garcia JV. Humanized mouse models for HIV-1 infection of the CNS. J Neurovirol. 2015;21(3):301–9.
Chahroudi A, Bosinger SE, Vanderford TH, Paiardini M, Silvestri G. Natural SIV Hosts: Showing AIDS the Door. Science (80-). 2012;335(6073):1188–93.
Clements JE, Mankowski JL, Gama L, Zink MC. The accelerated simian immunodeficiency virus macaque model of human immunodeficiency virus–associated neurological disease: from mechanism to treatment. J Neurovirol. 2008;14(4):309–17.
Riddick NE, Hermann EA, Loftin LM, Elliott ST, Wey WC, Cervasi B, et al. A novel CCR5 mutation common in sooty mangabeys reveals sivsmm infection of CCR5-null natural hosts and efficient alternative coreceptor use in vivo. PLoS Pathog. 2010;6(8):71–2.
Pedersen N, Ho E, Brown M, Yamamoto J. Isolation of a T-lymphotropic virus from domestic cats with an immunodeficiency-like syndrome. Science (80-). 1987;235(4790):790–3.
Torten M, Franchini M, Barlough JE, George JW, Mozes E, Lutz H, et al. Progressive immune dysfunction in cats experimentally infected with feline immunodeficiency virus. J Virol. 1991;65(5):2225–30.
Dow SW, Poss ML, Hoover EA. Feline immunodeficiency virus: a neurotropic lentivirus. J Acquir Immune Defic Syndr. 1990;3:658–68.
Podell M, March PA, Buck WR, Mathes LE. The feline model of neuroAIDS: understanding the progression towards AIDS dementia. J Psychopharmacol. 2000;14(3):205–13.
Egberink H, Borst M, Niphuis H, Balzarini J, Neu H, Schellekens H, et al. Suppression of feline immunodeficiency virus infection in vivo by 9-(2-phosphonomethoxyethyl)adenine. Proc Natl Acad Sci U S A. 1990;87(8):3087–91.
Hartmann K, Donath A, Beer B, Egberink HF, Horzinek MC, Lutz H, et al. Use of two virustatica (AZT, PMEA) in the treatment of FIV and of FeLV seropositive cats with clinical symptoms. Vet Immunol Immunopathol. 1992;35(1–2):167–75.
Kurapati KRV, Atluri VS, Samikkannu T, Garcia G, Nair MPN. Natural products as Anti-HIV agents and role in HIV-associated neurocognitive disorders (HAND): a brief overview. Front Microbiol. 2016;6:1–14.
Shal B, Ding W, Ali H, Kim YS, Khan S. Anti-neuroinflammatory potential of natural products in attenuation of Alzheimer’s disease. Front Pharmacol. 2018;9:548.
Szekeres T, Fritzer-Szekeres M, Saiko P, Jäger W. Resveratrol and resveratrol analogues—structure—activity relationship. Pharm Res. 2010;27(6):1042–8.
Bi XL, Yang JY, Dong YX, Wang JM, Cui YH, Ikeshima T, et al. Resveratrol inhibits nitric oxide and TNF-α production by lipopolysaccharide-activated microglia. Int Immunopharmacol. 2005;5(1):185–93.
Hewlings S, Kalman D. Curcumin: a review of its’ effects on human health. Foods. 2017;6(10):92.
Guo L, Xing Y, Pan R, Jiang M, Gong Z, Lin L, et al. Curcumin protects microglia and primary rat cortical neurons against HIV-1 gp120-mediated inflammation and apoptosis. PLoS ONE. 2013;8(8):e70565.
Shen L-L, Jiang M-L, Liu S-S, Cai M-C, Hong Z-Q, Lin L-Q, et al. Curcumin improves synaptic plasticity impairment induced by HIV-1gp120 V3 loop. Neural Regen Res. 2015;10(6):925.
Gong Z, Yang L, Tang H, Pan R, Xie S, Guo L, et al. Protective effects of curcumin against human immunodeficiency virus 1 gp120 V3 loop-induced neuronal injury in rats. Neural Regen Res. 2012;7(3):171–5.
Tang H, Pan R, Fang W, Xing Y, Chen D, Chen X, et al. Curcumin ameliorates hippocampal neuron damage induced by human immunodeficiency virus-1. Neural Regen Res. 2013;8(15):1368–75.
Chen G, Liu S, Pan R, Li G, Tang H, Jiang M, et al. Curcumin attenuates gp120-induced microglial inflammation by inhibiting autophagy via the PI3K pathway. Cell Mol Neurobiol. 2018;38:1465–77.
Prasad S, Tyagi AK. Curcumin and its analogues: a potential natural compound against HIV infection and AIDS. Food Funct. 2015;6(11):3412–9.
Grygiel-Górniak B. Peroxisome proliferator-activated receptors and their ligands: nutritional and clinical implications–a review. Nutr J. 2014;13:17.
Polak PE, Kalinin S, Dello Russo C, Gavrilyuk V, Sharp A, Peters JM, et al. Protective effects of a peroxisome proliferator-activated receptor-β/δ agonist in experimental autoimmune encephalomyelitis. J Neuroimmunol. 2005;168(1–2):65–75.
Barbiero JK, Santiago R, Tonin FS, Boschen S, Da Silva LM, De Paula Werner MF, et al. PPAR-α agonist fenofibrate protects against the damaging effects of MPTP in a rat model of Parkinson’s disease. Prog Neuro-Psychopharmacol Biol Psychiatry. 2014;53:35–44.
Shao ZQ, Liu ZJ. Neuroinflammation and neuronal autophagic death were suppressed via Rosiglitazone treatment: new evidence on neuroprotection in a rat model of global cerebral ischemia. J Neurol Sci. 2015;349(1–2):65–71.
Potula R, Ramirez SH, Knipe B, Leibhart J, Schall K, Heilman D, et al. Peroxisome proliferator-activated receptor-g activation suppresses HIV-1 replication in an animal model of encephalitis. AIDS. 2008;22(13):1539–49.
Huang W, Eum SY, András IE, Hennig B, Toborek M. PPAR and PPAR attenuate HIV-induced dysregulation of tight junction proteins by modulations of matrix metalloproteinase and proteasome activities.
Huang W, Chen L, Zhang B, Park M, Toborek M. PPAR agonist-mediated protection against HIV Tat-induced cerebrovascular toxicity is enhanced in MMP-9-deficient mice. J Cereb Blood Flow Metab. 2014;34(4):646–53.
Huang W, Rha GB, Han MJJ, Eum SY, András IE, Zhong Y, et al. PPARalpha and PPARgamma effectively protect against HIV-induced inflammatory responses in brain endothelial cells. J Neurochem. 2008;107(2):497–509.
Mamik MK, Asahchop EL, Chan WF, Zhu Y, Branton WG, McKenzie BA, et al. Insulin treatment prevents neuroinflammation and neuronal injury with restored neurobehavioral function in models of HIV/AIDS neurodegeneration. J Neurosci. 2016;36(41):10683–95.
U.S. National Library of Medicine. Search of: intranasal insulin, HAND—list results—ClinicalTrials.gov.
Achhra AC, Mocroft A, Reiss P, Sabin C, Ryom L, de Wit S, et al. Short-term weight gain after antiretroviral therapy initiation and subsequent risk of cardiovascular disease and diabetes: the D: A: D study. HIV Med. 2016;17(4):255–68.
Silva-Pinto A, Costa A, Serrão R, Sarmento A, Abreu P. Ischaemic stroke in HIV-infected patients: a case–control study. HIV Med. 2017;18(3):214–9.
Divala OH, Amberbir A, Ismail Z, Beyene T, Garone D, Pfaff C, et al. The burden of hypertension, diabetes mellitus, and cardiovascular risk factors among adult Malawians in HIV care: consequences for integrated services. BMC Public Health. 2016;16(1):1–11.
Anderson CM, Swanson RA. Astrocyte glutamate transport: review of properties, regulation, and physiological functions. Glia. 2000;32(1):1–14.
Vázquez-Santiago FJ, Noel RJ, Porter JT, Rivera-Amill V. Glutamate metabolism and HIV-associated neurocognitive disorders. J Neurovirol. 2014;20(4):315–31.
Ferrarese C, Aliprandi A, Tremolizzo L, Stanzani L, De Micheli A, Dolara A, et al. Increased glutamate in CSF and plasma of patients with HIV dementia. Neurology. 2001;57(4):671–5.
Cassol E, Misra V, Dutta A, Morgello S, Gabuzda D. Cerebrospinal fluid metabolomics reveals altered waste clearance and accelerated aging in HIV patients with neurocognitive impairment. Aids. 2014;28(11):1579–91.
Raber J, Toggas SM, Lee S, Bloom FE, Epstein CJ, Mucke L. Central nervous system expression of HIV-1 gp120 activates the hypothalamic-pituitary-adrenal axis: evidence for involvement of NMDA receptors and nitric oxide synthase. Virology. 1996;226(2):362–73.
Anderson ER. Memantine protects hippocampal neuronal function in murine human immunodeficiency virus type 1 encephalitis. J Neurosci. 2004;24(32):7194–8.
Meisner F, Scheller C, Kneitz S, Sopper S, Neuen-Jacob E, Riederer P, et al. Memantine upregulates BDNF and prevents dopamine deficits in SIV-infected macaques: a novel pharmacological action of memantine. Neuropsychopharmacology. 2008;33(9):2228–36.
Zhao Y, Navia BA, Marra CM, Singer EJ, Chang L, Berger J, et al. Memantine for AIDS dementia complex: open-label report of ACTG 301. HIV Clin Trials. 2010;11(1):59–67.
Schifitto G, Yiannoutsos CT, Simpson DM, Marra CM, Singer EJ, Kolson DL, et al. A placebo-controlled study of memantine for the treatment of human immunodeficiency virus-associated sensory neuropathy. J Neurovirol. 2006;12(4):328–31.
Nakanishi N, Kang YJ, Tu S, McKercher SR, Masliah E, Lipton SA. Differential effects of pharmacologic and genetic modulation of NMDA receptor activity on HIV/gp120-induced neuronal damage in an in vivo mouse model. J Mol Neurosci. 2016;58(1):59–65.
Talantova M, Sanz-Blasco S, Zhang X, Xia P, Akhtar MW, Okamoto SI, et al. A induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc Natl Acad Sci. 2013;110(27):E2518–27.
Erdmann N, Zhao J, Lopez AL, Herek S, Curthoys N, Hexum TD, et al. Glutamate production by HIV-1 infected human macrophage is blocked by the inhibition of glutaminase. J Neurochem. 2007;102(2):539–49.
Thomas AG, Sattler R, Tendyke K, Loiacono KA, Hansen H, Sahni V, et al. High-throughput assay development for cystine-glutamate antiporter (xc-) highlights faster cystine uptake than glutamate release in glioma cells. PLoS ONE. 2015;10(8):e0127785.
Romera C, Hurtado O, Mallolas J, Pereira MP, Morales JR, Romera A, et al. Ischemic preconditioning reveals that GLT1/EAAT2 glutamate transporter is a novel PPARγ target gene involved in neuroprotection. J Cereb Blood Flow Metab. 2007;27(7):1327–38.
Stancu C, Sima A. Statins: mechanism of action and effects. J Cell Mol Med. 2001;5(4):378–87.
Yadav A, Betts MR, Collman RG. Statin modulation of monocyte phenotype and function: implications for HIV-1-associated neurocognitive disorders. J Neurovirol. 2016;22(5):584–96.
Williams DW, Eugenin EA, Calderon TM, Berman JW. Monocyte maturation, HIV susceptibility, and transmigration across the blood brain barrier are critical in HIV neuropathogenesis. J Leukoc Biol. 2012;91(3):401–15.
Probasco JC, Spudich SS, Critchfield J, Lee E, Lollo N, Deeks SG, et al. Failure of atorvastatin to modulate CSF HIV-1 infection: results of a pilot study. Neurology. 2008;71(7):521–4.
van der Most PJ, Dolga AM, Nijholt IM, Luiten PGM, Eisel ULM. Statins: mechanisms of neuroprotection. Prog Neurobiol. 2009;88(1):64–75.
Tan Q, Zhu Y, Li J, Chen Z, Han GW, Kufareva I, et al. Structure of the CCR5 chemokine receptor-HIV entry inhibitor maraviroc complex. Science (80-). 2013;341(6152):1387–90.
Smurzynski M, Wu K, Letendre S, Robertson K, Bosch RJ, Clifford DB, et al. Effects of central nervous system antiretroviral penetration on cognitive functioning in the ALLRT cohort. Aids. 2011;25(3):357–65.
Letendre S. Central nervous system complications in HIV disease: HIV-associated neurocognitive disorder. Top Antivir Med. 2011;19(4):137–42.
Rossi R, Lichtner M, de Rosa A, Sauzullo I, Mengoni F, Massetti AP, et al. In vitro effect of anti-human immunodeficiency virus CCR5 antagonist maraviroc on chemotactic activity of monocytes, macrophages and dendritic cells. Clin Exp Immunol. 2011;166(2):184–90.
Kelly KM, Beck SE, Pate KAM, Queen SE, Dorsey JL, Adams RJ, et al. Neuroprotective maraviroc monotherapy in simian immunodeficiency virus-infected macaques: reduced replicating and latent SIV in the brain. Aids. 2013;27(18):F21.
Ndhlovu LC, Umaki T, Chew GM, Chow DC, Agsalda M, Kallianpur KJ, et al. Treatment intensification with maraviroc (CCR5 antagonist) leads to declines in CD16-expressing monocytes in cART-suppressed chronic HIV-infected subjects and is associated with improvements in neurocognitive test performance: implications for HIV-associate. J Neurovirol. 2014;20(6):571–82.
Tiraboschi JM, Muñoz-Moreno JA, Puertas MC, Alonso-Villaverde C, Prats A, Ferrer E, et al. Viral and inflammatory markers in cerebrospinal fluid of patients with HIV-1-associated neurocognitive impairment during antiretroviral treatment switch. HIV Med. 2015;16(6):388–92.
Gates TM, Cysique LA, Siefried KJ, Chaganti J, Moffat KJ, Brew BJ. Maraviroc-intensified combined antiretroviral therapy improves cognition in virally suppressed HIV-associated neurocognitive disorder. Aids. 2016;30(4):591–600.
Brew BJ, Letendre SL. Biomarkers of HIV related central nervous system disease. Int Rev Psychiatry. 2008;20(1):73–88.
Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol. 2014;14(1):36–49.
McManus CM, Liu JS, Hahn MT, Hua LL, Brosnan CF, Berman JW, et al. Differential induction of chemokines in human microglia by type I and II interferons. Glia. 2000;29(3):273–80.
Cocchi F, DeVico AL, Garzino-Demo A, Arya SK, Gallo RC, Lusso P. Identification of RANTES, MIP-1alpha, and MIP-1beta as the major HIV-suppressive factors produced by CD8+ T cells. Science (80-). 1995;270(5243):1811–5.
Lu HT. Interferon (IFN) beta acts downstream of IFN-gamma-induced class II transactivator messenger RNA accumulation to block major histocompatibility complex class II gene expression and requires the 48- kD DNA-binding protein, ISGF3-gamma. J Exp Med. 1995;182(5):1517–25.
Hua LL, Liu JSH, Brosnan CF, Lee SC. Selective inhibition of human glial inducible nitric oxide synthase by interferon-α: implications for multiple sclerosis. Ann Neurol. 1998;43(3):384–7.
Hua LL, Lee SC. Distinct patterns of stimulus-inducible chemokine mRNA accumulation in human fetal astrocytes and microglia. Glia. 2000;30(1):74–81.
Kaul M, Ma Q, Medders KE, Desai MK, Lipton SA. HIV-1 coreceptors CCR5 and CXCR4 both mediate neuronal cell death but CCR5 paradoxically can also contribute to protection. Cell Death Differ. 2007;14(2):296–305.
Kaul M, Lipton SA. Chemokines and activated macrophages in HIV gp120-induced neuronal apoptosis. Proc Natl Acad Sci U S A. 1999;96(14):8212–6.
Kitai R, Zhao ML, Zhang N, Hua LL, Lee SC. Role of MIP-1β and RANTES in HIV-1 infection of microglia: inhibition of infection and induction by IFNβ. J Neuroimmunol. 2000;110(1–2):230–9.
Thaney VE, O’Neill AM, Hoefer MM, Maung R, Sanchez AB, Kaul M. IFNβ protects neurons from damage in a murine model of HIV-1 associated brain injury. Sci Rep. 2017;7(1):46514.
McGraw CA, Lublin FD. Interferon beta and glatiramer acetate therapy. Neurotherapeutics. 2013;10(1):2–18.
Anderson AM, Lennox JL, Mulligan MM, Loring DW, Zetterberg H, Blennow K, et al. Cerebrospinal fluid interferon alpha levels correlate with neurocognitive impairment in ambulatory HIV-Infected individuals. J Neurovirol. 2017;23(1):106–12.
Fritz-French C, Tyor W. Interferon-α (IFNα) neurotoxicity. Cytokine Growth Factor Rev. 2012;23(1–2):7–14.
Sas AR, Bimonte-Nelson HA, Tyor WR. Cognitive dysfunction in HIV encephalitic SCID mice correlates with levels of Interferon-α in the brain. Aids. 2007;21(16):2151–9.
Kessing CF, Tyor WR. Interferon-α induces neurotoxicity through activation of the type I receptor and the GluN2A subunit of the NMDA receptor. J Interf Cytokine Res. 2015;35(4):317–24.
Symons JA, Alcamí A, Smith GL. Vaccinia virus encodes a soluble type I interferon receptor of novel structure and broad species soecificity. Cell. 1995;81(4):551–60.
Fritz-French C, Shawahna R, Ward JE, Maroun LE, Tyor WR. The recombinant vaccinia virus gene product, B18R, neutralizes interferon alpha and alleviates histopathological complications in an HIV encephalitis mouse model. J Interf Cytokine Res. 2014;34(7):510–7.
Koneru R, Bimonte-Nelson H, Ciavatta V, Haile W, Elmore K, Ward J, et al. Reversing interferon-alpha neurotoxicity in a HIV-associated neurocognitive disorder mouse model. Aids. 2018;32(11):1403–11.
Food and Drug Administration. TECFIDERA product monograph. Washington: Food and Drug Administration; 2014.
Cross SA, Cook DR, Chi AWS, Vance PJ, Kolson LL, Wong BJ, et al. Dimethyl fumarate, an immune modulator and inducer of the antioxidant response, suppresses hiv replication and macrophage-mediated neurotoxicity: a novel candidate for HIV neuroprotection. J Immunol. 2011;187(10):5015–25.
Rodriguez-Franco EJ, Cantres-Rosario YM, Plaud-Valentin M, Romeu R, Rodríguez Y, Skolasky R, et al. Dysregulation of macrophage-secreted cathepsin B contributes to HIV-1-linked neuronal apoptosis. PLoS ONE. 2012;7(5):e36571.
Cantres-Rosario YM, Hernandez N, Negron K, Perez-Laspiur J, Leszyk J, Shaffer SA, et al. Interacting partners of macrophage-secreted cathepsin B contribute to HIV-induced neuronal apoptosis. Aids. 2015;29(16):2081–92.
Zenón F, Cantres-Rosario Y, Adiga R, Gonzalez M, Rodriguez-Franco E, Langford D, et al. HIV-infected microglia mediate cathepsin B-induced neurotoxicity. J Neurovirol. 2015;21(5):544–58.
Rosario-Rodríguez LJ, Colón K, Borges-Vélez G, Negrón K, Meléndez LM. Dimethyl fumarate prevents HIV-induced lysosomal dysfunction and cathepsin B release from macrophages. J Neuroimmune Pharmacol. 2018;13(3):345–54.
Nikodemova M, Duncan ID, Watters JJ. Minocycline exerts inhibitory effects on multiple mitogen-activated protein kinases and IκBα degradation in a stimulus-specific manner in microglia. J Neurochem. 2006;96(2):314–23.
Henry CJ, Huang Y, Wynne A, Hanke M, Himler J, Bailey MT, et al. Minocycline attenuates lipopolysaccharide (LPS)-induced neuroinflammation, sickness behavior, and anhedonia. J Neuroinflamm. 2008;5:1–14.
Du Y, Ma Z, Lin S, Dodel RC, Gao F, Bales KR, et al. Minocycline prevents nigrostriatal dopaminergic neurodegeneration in the MPTP model of Parkinson’s disease. Proc Natl Acad Sci. 2001;98(25):14669–74.
Lee PL, Yiannoutsos CT, Ernst T, Chang L, Marra CM, Jarvik JG, et al. A multi-center 1H MRS study of the AIDS dementia complex: validation and preliminary analysis. J Magn Reson Imaging. 2003;17(6):625–33.
Teng YD, Choi H, Onario RC, Zhu S, Desilets FC, Lan S, et al. Minocycline inhibits contusion-triggered mitochondrial cytochrome c release and mitigates functional deficits after spinal cord injury. Proc Natl Acad Sci. 2004;101(9):3071–6.
Amin AR, Attur MG, Thakker GD, Patel PD, Vyas PR, Patel RN, et al. A novel mechanism of action of tetracyclines: effects on nitric oxide synthases. Med Sci. 1996;93:14014–9.
Brogden RN, Speight TM, Avery GS. Minocycline: a review of its antibacterial and pharmacokinetic properties and therapeutic use. Drugs. 1975;9(4):251–91.
Ratai EM, Bombardier JP, Joo CG, Annamalai L, Burdo TH, Campbell J, et al. Proton magnetic resonance spectroscopy reveals neuroprotection by oral minocycline in a nonhuman primate model of accelerated NeuroAIDS. PLoS ONE. 2010;5(5):e10523.
Sacktor N, Miyahara S, Deng L, Evans S, Schifitto G, Cohen BA, et al. Minocycline treatment for HIV-associated cognitive impairment: results from a randomized trial. Neurology. 2011;77(12):1135–42.
Nakasujja N, Miyahara S, Evans S, Lee A, Musisi S, Katabira E, et al. Randomized trial of minocycline in the treatment of HIV-associated cognitive impairment. Neurology. 2013;80(2):196–202.
Sacktor N, Miyahara S, Evans S, Schifitto G, Cohen B, Haughey N, et al. Impact of minocycline on cerebrospinal fluid markers of oxidative stress, neuronal injury, and inflammation in HIV-seropositive individuals with cognitive impairment. J Neurovirol. 2014;20(6):620–6.
Gilberto González R, Fell R, He J, Campbell J, Burdo TH, Autissier P, et al. Temporal/compartmental changes in viral RNA and neuronal injury in a primate model of NeuroAIDS. PLoS ONE. 2018;13(5):1–21.
Nemeth CL, Glasper ER, Harrell CS, Malviya SA, Otis JS, Neigh GN. Meloxicam blocks neuroinflammation, but not depressive-like behaviors, in HIV-1 transgenic female rats. PLoS ONE. 2014;9(10):108399.
Aoki S, Watanabe Y, Sanagawa M, Setiawan A, Kotoku N, Kobayashi M, Cortistatins A. B, C, and D, anti-angiogenic steroidal alkaloids, from the marine sponge corticium simplex. J Am Chem Soc. 2006;128(10):3148–9.
Aoki S, Watanabe Y, Tanabe D, Arai M, Suna H, Miyamoto K, et al. Structure-activity relationship and biological property of cortistatins, anti-angiogenic spongean steroidal alkaloids. Bioorg Med Chem. 2007;15(21):6758–62.
Mousseau G, Clementz MA, Bakeman WN, Nagarsheth N, Cameron M, Shi J, et al. An analog of the natural steroidal alkaloid cortistatin A potently suppresses Tat-dependent HIV transcription. Cell Host Microbe. 2012;12(1):97–108.
Mediouni S, Jablonski J, Paris JJ, Clementz MA, Thenin-Houssier S, Mclaughlin JP, et al. Didehydro-cortistatin A inhibits HIV-1 Tat mediated neuroinflammation and prevents potentiation of cocaine reward in Tat transgenic mice. Curr HIV Res. 2015;13(1):64–79.
Bagashev A, Sawaya BE. Roles and functions of HIV-1 Tat protein in the CNS: an overview. Virol J. 2013;10(1):358.
Nath A, Anderson C, Jones M, Maragos W, Booze R, Mactutus C, et al. Neurotoxicity and dysfunction of dopaminergic systems associated with AIDS dementia. J Psychopharmacol. 2000;14(3):222–7.
Kousik SM, Napier TC, Carvey PM. The effects of psychostimulant drugs on blood brain barrier function and neuroinflammation. Front Pharmacol. 2012;3:1–12.
Kessing CF, Nixon CC, Li C, Tsai P, Takata H, Mousseau G, et al. In vivo suppression of HIV rebound by didehydro-cortistatin A, a “Block-and-Lock” strategy for HIV-1 treatment. Cell Rep. 2017;21(3):600–11.
Mousseau G, Kessing CF, Fromentin R, Trautmann L, Chomont N, Valente ST. The tat inhibitor didehydro-cortistatin a prevents HIV-1 reactivation from latency. MBio. 2015;6(4):e00465.
Mousseau G, Valente ST. Didehydro-Cortistatin A: a new player in HIV-therapy? Expert Rev Anti Infect Ther. 2016;14(2):145–8.
McGraw J, Gaudet AD, Oschipok LW, Kadoya T, Horie H, Steeves JD, et al. Regulation of neuronal and glial galectin-1 expression by peripheral and central axotomy of rat primary afferent neurons. Exp Neurol. 2005;195(1):103–14.
Wada M, Ono S, Kadoya T, Kawanami T, Kurita K, Kato T. Decreased galectin-1 immunoreactivity of the skin in amyotrophic lateral sclerosis. J Neurol Sci. 2003;208(1–2):67–70.
Almkvist J, Karlsson A. Galectins as inflammatory mediators. Glycoconj J. 2002;19(7–9):575–81.
Aalinkeel R, Mangum CS, Abou-Jaoude E, Reynolds JL, Liu M, Sundquist K, et al. Galectin-1 reduces neuroinflammation via modulation of nitric oxide-arginase signaling in HIV-1 transfected microglia: a gold nanoparticle-galectin-1 “Nanoplex” a possible neurotherapeutic? J Neuroimmune Pharmacol. 2017;12(1):133–51.
Hansen HS, Petersen G, Artmann A, Madsen AN. Endocannabinoids. Eur J Lipid Sci Technol. 2006;108(10):877–89.
Klein TW, Cabral GA. Cannabinoid-induced immune suppression and modulation of antigen-presenting cells. J Neuroimmune Pharmacol. 2006;1(1):50–64.
Raborn ES, Cabral GA. Cannabinoid inhibition of macrophage migration to the trans-activating (Tat) protein of HIV-1 Is linked to the CB2 cannabinoid receptor. J Pharmacol Exp Ther. 2010;333(1):319–27.
Price DA, Martinez AA, Seillier A, Koek W, Acosta Y, Fernandez E, et al. WIN55,212-2, a cannabinoid receptor agonist, protects against nigrostriatal cell loss in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. Eur J Neurosci. 2009;29(11):2177–86.
Croxford JL, Miller SD. Immunoregulation of a viral model of multiple sclerosis using the synthetic cannabinoid R(+)WIN55,212. J Clin Invest. 2003;111(8):1231–40.
Palazuelos J, Aguado T, Pazos MR, Julien B, Carrasco C, Resel E, et al. Microglial CB2 cannabinoid receptors are neuroprotective in Huntington’s disease excitotoxicity. Brain. 2009;132(11):3152–64.
Lu T-S, Avraham HK, Seng S, Tachado SD, Koziel H, Makriyannis A, et al. Cannabinoids inhibit HIV-1 Gp120-mediated insults in brain microvascular endothelial cells. J Immunol. 2008;181(9):6406–16.
Hu S, Sheng WS, Rock RB. CB2 receptor agonists protect human dopaminergic neurons against damage from HIV-1 gp120. PLoS ONE. 2013;8(10):77577.
Xu C, Hermes DJ, Mackie K, Lichtman AH, Ignatowska-Jankowska BM, Fitting S. Cannabinoids occlude the HIV-1 Tat-induced decrease in GABAergic neurotransmission in prefrontal cortex slices. J Neuroimmune Pharmacol. 2016;11(2):316–31.
Avraham HK, Jiang S, Fu Y, Rockenstein E, Makriyannis A, Zvonok A, et al. The cannabinoid CB2 receptor agonist AM1241 enhances neurogenesis in GFAP/Gp120 transgenic mice displaying deficits in neurogenesis. Br J Pharmacol. 2014;171(2):468–79.
Gorantla S, Makarov E, Roy D, Finke-Dwyer J, Murrin LC, Gendelman HE, et al. Immunoregulation of a CB2 receptor agonist in a murine model of NeuroAIDS. J Neuroimmune Pharmacol. 2010;5(3):456–68.
Purohit V, Rapaka RS, Rutter J. Cannabinoid receptor-2 and HIV-associated neurocognitive disorders. J Neuroimmune Pharmacol. 2014;9(4):447–53.
Ramirez SH, Reichenbach NL, Fan S, Rom S, Merkel SF, Wang X, et al. Attenuation of HIV-1 replication in macrophages by cannabinoid receptor 2 agonists. J Leukoc Biol. 2013;93(5):801–10.
Gelman BB, Chen T, Lisinicchia JG, Soukup VM, Carmical JR, Starkey JM, et al. The National NeuroAIDS Tissue Consortium Brain Gene Array: Two Types of HIV-Associated Neurocognitive Impairment. PLoS ONE. 2012;7(9):e46178.
Itoh K, Mehraein P, Weis S. Neuronal damage of the substantia nigra in HIV-1 infected brains. Acta Neuropathol. 2000;99(4):376–84.
Silvers JM, Aksenov MY, Aksenova MV, Beckley J, Olton P, Mactutus CF, et al. Dopaminergic marker proteins in the substantia nigra of human immunodeficiency virus type 1-infected brains. J Neurovirol. 2006;12(2):140–5.
Gelman BB, Spencer JA, Holzer CE, Soukup VM. Abnormal striatal dopaminergic synapses in National NeuroAIDS Tissue Consortium subjects with HIV encephalitis. J Neuroimmune Pharmacol. 2006;1(4):410–20.
Ferris MJ, Frederick-Duus D, Fadel J, Mactutus CF, Booze RM. In vivo microdialysis in awake, freely moving rats demonstrates HIV-1 tat-induced alterations in dopamine transmission. Synapse. 2009;63(3):181–5.
Meulendyke KA, Queen SE, Engle EL, Shirk EN, Liu J, Steiner JP, et al. Combination fluconazole/paroxetine treatment is neuroprotective despite ongoing neuroinflammation and viral replication in an SIV model of HIV neurological disease. J Neurovirol. 2014;20(6):591–602.
Sacktor N, Skolasky RL, Moxley R, Wang S, Mielke MM, Munro C, et al. Paroxetine and fluconazole therapy for HIV-associated neurocognitive impairment: results from a double-blind, placebo-controlled trial. J Neurovirol. 2018;24(1):16–27.
Hempelmann E. Hemozoin Biocrystallization in Plasmodium falciparum and the antimalarial activity of crystallization inhibitors. Parasitol Res. 2007;100(4):671–6.
Huang Z, Srinivasan S, Zhang J, Chen K, Li Y. Discovering thiamine transporters as targets of chloroquine using a novel functional genomics strategy. PLoS Genet. 2012;8(11):1003083.
Routy JP, Angel JB, Patel M, Kanagaratham C, Radzioch D, Kema I, et al. Assessment of chloroquine as a modulator of immune activation to improve CD4 recovery in immune nonresponding HIV-infected patients receiving antiretroviral therapy. HIV Med. 2015;16(1):48–56.
Paton NI, Goodall RL, Dunn DT, Franzen S, Collaco-moraes Y, Williams IG, et al. Effects of hydroxychloroquine on immune activation and disease progression among HIV-infected patients not receiving antiretroviral therapy: a randomized controlled trial. JAMA. 2012;308(4):353–61.
Gu C-J, Borjabad A, Hadas E, Kelschenbach J, Kim B-H, Chao W, et al. EcoHIV infection of mice establishes latent viral reservoirs in T cells and active viral reservoirs in macrophages that are sufficient for induction of neurocognitive impairment. PLOS Pathog. 2018;14(6):e1007061.
Weed MR, Gold LH, Polis I, Koob GF, Fox HS, Taffe MA. Impaired performance on a rhesus monkey neuropsychological testing battery following simian immunodeficiency virus infection. Aids Res Hum Retrovir. 2004;20:77–89.
Ambrosius B, Faissner S, Guse K, von Lehe M, Grunwald T, Gold R, et al. Teriflunomide and monomethylfumarate target HIV-induced neuroinflammation and neurotoxicity. J Neuroinflamm. 2017;14(1):51.
Gill AJ, Kovacsics CE, Cross SA, Vance PJ, Kolson LL, Jordan-Sciutto KL, et al. Heme oxygenase-1 deficiency accompanies neuropathogenesis of HIV-associated neurocognitive disorders. J Clin Invest. 2014;124(10):4459–72.
Acknowledgements
The authors would like to thank all of the funding agencies who supported this work.
Funding
This work is supported by an operating grant from the Canadian Institutes of Health Research (MOP-56976) awarded to Dr. Reina Bendayan. Dr. Reina Bendayan is a career scientist of Ontario HIV Treatment Network (OHTN), Ministry of Health Ontario. Amila Omeragic is the recipient of the Ontario Graduate Scholarship and the Leslie Dan Faculty of Pharmacy Dean’s Scholarship.
Author information
Authors and Affiliations
Contributions
AO and OK wrote the initial draft. TH contributed to the preparation of the revised manuscript. RB contributed to overall conceptual and editorial review of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Confirmed.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Omeragic, A., Kayode, O., Hoque, M.T. et al. Potential pharmacological approaches for the treatment of HIV-1 associated neurocognitive disorders. Fluids Barriers CNS 17, 42 (2020). https://doi.org/10.1186/s12987-020-00204-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12987-020-00204-5