
Abstract
Bacterial meningitis is a serious life threatening infection of the CNS. To cause meningitis, blood–borne bacteria need to interact with and penetrate brain endothelial cells (BECs) that comprise the blood–brain barrier. BECs help maintain brain homeostasis and they possess an array of efflux transporters, such as P-glycoprotein (P-gp), that function to efflux potentially harmful compounds from the CNS back into the circulation. Oftentimes, efflux also serves to limit the brain uptake of therapeutic drugs, representing a major hurdle for CNS drug delivery. During meningitis, BEC barrier integrity is compromised; however, little is known about efflux transport perturbations during infection. Thus, understanding the impact of bacterial infection on P-gp function would be important for potential routes of therapeutic intervention. To this end, the meningeal bacterial pathogen, Streptococcus agalactiae, was found to inhibit P-gp activity in human induced pluripotent stem cell-derived BECs, and live bacteria were required for the observed inhibition. This observation was correlated to decreased P-gp expression both in vitro and during infection in vivo using a mouse model of bacterial meningitis. Given the impact of bacterial interactions on P-gp function, it will be important to incorporate these findings into analyses of drug delivery paradigms for bacterial infections of the CNS.
Similar content being viewed by others


Introduction
The blood–brain barrier (BBB) and other brain barriers such as the meningeal blood-cerebrospinal fluid barrier are comprised of highly specialized brain endothelial cells (BECs) that promote proper brain function by separating the circulation from the central nervous system (CNS) [1,2,3]. BECs possess unique phenotypes that include the presence of specialized efflux transporters, complex tight junctions, and low rates of endocytosis [1,2,3]. Together, these properties maintain brain homeostasis and help to prevent the entry of pathogens and toxins into the CNS. P-glycoprotein (P-gp) is a major efflux transporter expressed in BECs that is able to efflux a wide variety of lipophilic molecules back into the bloodstream and because of this, can regulate the CNS accumulation of drugs [4,5,6,7,8,9]. Much work has been conducted to understand and modulate P-gp function at the BBB in order to enhance drugs access to the brain [4,5,6,7,8,9]. Moreover, the effects of disease conditions on P-gp function are increasingly being explored [10,11,12,13].
Bacterial meningitis is a serious infection of the CNS that occurs when blood-borne bacteria are able to breach BECs and cause inflammation [14,15,16,17]. Certain pathogens possess virulence factors that promote BEC interaction, and previous studies have characterized the molecular interactions that result in penetration of brain endothelium [14,15,16,17]. Streptococcus agalactiae, also known as Group B Streptococcus (GBS), is a Gram-positive bacterium that is the leading cause of neonatal meningitis [18]. GBS disruption of the BECs and other brain barriers during meningitis has been documented, and it has been shown that tight junctions are disrupted and endocytosis pathways altered [19, 20]. However, little is known about the effect of bacterial infection on P-gp function. Here, we show that bacterial infection can alter P-gp function in BECs, suggesting another mechanism by which bacterial pathogens contribute to BEC dysfunction.
Results
Group B Streptococcus infection inhibits BEC P-gp function
We first sought to determine if P-gp function was altered during infection. To do so, we utilized induced pluripotent stem cell (iPSC)-derived BEC-like cells that have been shown to possess P-gp activity [21,22,23]. iPSC-derived BECs were differentiated and express expected endothelial markers as previously described (Additional file 1: Figure S1A-F) [21,22,23]. Using a substrate accumulation assay and consistent with prior observations, BECs treated with the P-gp inhibitor Cyclosporine A (CsA) accumulated more of the P-gp substrate Rhodamine 123 (R123) than non-CsA-treated cells, indicating that P-gp is active in these BECs. Following GBS infection, we observed a significant increase of R123 accumulation in BECs when compared to uninfected BECs, to levels matching those with CsA inhibition (Fig. 1a). Addition of CsA to the infected condition made no impact on accumulation, and the combined data suggest P-gp function is diminished during GBS infection. To determine if the observation was substrate specific, similar experiments were performed with a different P-gp substrate, FLUO-3-AM, and a similar increase in substrate accumulation in GBS-infected BECs was observed (Fig. 1b). In addition, inhibition of P-gp with the second generation, more specific inhibitor PSC-833 [6] yielded similar results to CsA inhibition (Fig. 1c). To determine if this impact on P-gp function is specific to meningeal pathogens, P-gp function was assayed following incubation with a genetically similar non-pathogenic bacterium, Lactococcus lactis. In contrast to GBS effects, L. lactis did not inhibit P-gp function in an R123 accumulation assay (Fig. 1d). We and others have previously shown that inhibition of BCRP or MRP family proteins with Ko143 or MK571 respectively, in iPSC-BECs resulted in functional inhibition of those transporters [21, 22, 24,25,26,27,28,29]. Inhibition of BCRP or MRPs using Ko143 or MK571, did not result in an increase in R123 accumulation (Fig. 1e). This suggests that R123 efflux, in our model, is primarily mediated by P-gp and that accumulation can be increased by CsA. Taken together, these observations suggest that GBS infection results in reduced P-gp function in BECs as measured by substrate accumulation.

GBS effects on iPSC-derived BEC P-gp function. a BECs were either uninfected (control) or infected with GBS (MOI = 10) for 5 h. After infection, P-gp activity was measured by observing R123 accumulation with or without CsA inhibition. b P-gp activity measured using FLUO-3-AM as a P-gp substrate. Experimental groups and infection times are as described in (a). c Just as in (a), BECs were either left uninfected (control) or infected with GBS (MOI = 10) for 5 h. P-gp activity was measured by observing R123 accumulation with or without PSC-833 (PSC) as the inhibitor d. P-gp activity assay monitoring R123 accumulation as in (a) for BECs incubated with non-pathogenic L. lactis (MOI = 10) for 5 h. e BECs tested for R123 accumulation after treatment with the P-gp inhibitor CsA, BCRP inhibitor Ko143, and MRP family inhibitor MK571. Experiments were performed at least in triplicate on three independent differentiations (a–c, e, n = 9), or in triplicate on two independent differentiations (d, n = 6). All data are presented, and expressed as % of uninfected control BEC accumulation for all experiments, and bars represent mean ± SD. **p < 0.01, ***p < 0.001, NS p > 0.05, versus uninfected control; ANOVA followed by Dunnett’s multiple comparisons test
Group B Streptococcus infection inhibits function in P-gp overexpressing cells
To ensure that the observed decrease in P-gp was not a function of the iPSC origin of BECs, we also examined P-gp function in a human P-gp-overexpressing Madin-Darby canine kidney (MDCK) cell line [30]. As a result of the overexpression of human P-gp, there is a greater relative increase of R123 accumulation upon CsA inhibition (Fig. 2a). Despite this increase in P-gp activity, GBS-mediated P-gp inhibition could still be observed with complete inhibition of P-gp activity at high multiplicity of infection (MOI) (Fig. 2a). These data suggest that P-gp inhibition is related to the balance of P-gp expression and level of bacterial interaction, and that GBS-mediated P-gp inhibition may not be specific to BECs.

Bacterial interactions regulating P-gp inhibition. a Effects of GBS infection on R123 accumulation in an MDCK cell line that overexpresses human MDR1. Multiplicity of infection (MOI) was varied and after 5 h of infection, P-gp function was compared to uninfected cells and CsA treated cells. b Effects of live GBS, GBS-conditioned medium (CM) and heat-treated non-viable (Heat) or paraformaldehyde-fixed (PFA) GBS on R123 accumulation in iPSC-derived BECs after 5 h of treatment. c iPSC-derived BECs were infected with GBS mutants or wild-type (WT) (MOI = 10) for 5 h, and R123 accumulation compared to uninfected controls. Experiments were performed in triplicate on three independent biological replicates (a) or differentiations (b, c). All raw data are presented (n = 9), and are expressed as % of uninfected control BEC accumulation. All experiments and bars represent mean ± SD. **p < 0.01, ***p < 0.001, NS p > 0.05, versus uninfected control unless otherwise specified; ANOVA followed by Dunnett’s multiple comparisons test
Live GBS is required for inhibition of P-gp activity
We performed accumulation experiments to examine if a secreted factor was responsible for inhibiting P-gp activity. Exposure of BECs to GBS-conditioned medium had no influence on P-gp function, indicating a requirement for GBS-BEC interactions (Fig. 2b). Next, we investigated if bacterial components, but not living bacteria, could generate P-gp inhibition. Interaction of heat-treated, nonviable GBS or paraformaldehyde-fixed GBS with BECs also did not affect P-gp function (Fig. 2b). These results suggest that interaction with live GBS is required for P-gp inhibition as secreted GBS components or nonviable forms of GBS were insufficient to decrease P-gp function. Given this requirement, and the fact that previous work has identified various GBS virulence factors that contribute to overall bacterial-BEC interactions, we next tested a number of bacterial mutants identified to contribute to bacterial interaction with BECs. We examined GBS mutants lacking surface expressed adhesins SfbA and Srr2, and an invasion associated gene (iagA) that functions to properly anchor bacterial lipoteichoic acid, to see if any of the virulence factors contributed to P-gp inhibition during infection [31,32,33,34]. BECs infected by each of the mutants exhibit similar P-gp substrate accumulation to BECs infected by the wild-type (WT) GBS, and all show significantly increased accumulation over uninfected controls (Fig. 2c). Taken together, these data suggest that live GBS is required for P-gp inhibition, and SfbA, Srr2 or iagA alone does not mediate that P-gp inhibition.
P-gp expression and abundance decreases during GBS infection
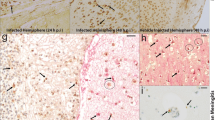
To examine whether the decrease in P-gp activity correlated with a decrease in P-gp abundance, western blot and flow cytometry were used to quantify P-gp expression. BECs were infected with GBS, and western blot analysis was conducted on BEC lysates. We observed that overall P-gp abundance decreased during GBS infection (Fig. 3a, b). These results were confirmed by flow cytometry for P-gp where a decrease in P-gp expression was measured (Fig. 3c, d). RT-qPCR conducted on lysates further demonstrated that P-gp-encoding ABCB1 expression was decreased upon GBS infection (Fig. 3e). To determine if this decrease was observed in vivo, we employed our murine model of GBS meningitis [19, 33, 35]. We observed that during GBS infection mice exhibited less P-gp immunolabeling that co-localized with endothelial cells (Fig. 3f, g, Additional file 2: Figure S2). Taken together, these data suggest that the disruption in P-gp activity in BECs after GBS infection may be due to a decrease in P-gp abundance.

Effects of GBS infection on P-gp expression in BECs. a BECs were infected with GBS (MOI = 10) for 5 h, and protein lysates were collected. Western blot analysis was performed on lysates using COXIV as a loading control, representative blot shown. b Densitometry was conducted to determine fold change in P-gp expression using FIJI ImageJ software after normalization to respective COXIV bands on all experiments performed in triplicate on three independent differentiations (n = 9). c Representative flow cytometry histogram of BECs stained for total P-gp after 5 h of GBS infection (MOI = 10) compared with uninfected BECs (Control). d P-gp abundance is plotted as the median fluorescence intensity generated by flow cytometry normalized to uninfected control on all experiments performed at least in duplicate on three independent differentiations (n = 7). e qPCR performed for ABCB1 (P-gp) and normalized to 18S rRNA for BECs with or without GBS infection at an MOI of 10 for 5 h. Data presented are from three experiments performed in triplicate. f Quantitation of relative P-gp pixel intensity overlayed with vascular lectin staining in mouse brains. Control mice (n = 5) with 12 FOV had 481 lectin positive vessels analyzed, GBS mice (n = 6) with 12 FOV had 544 lectin positive vessels analyzed. g Representative images of mouse brains stained for lectin (green), P-gp (red), and DAPI (blue) after GBS infection. Control mouse with no GBS in the brain (top), and mouse with GBS in the brain (bottom). Scale bar represents 50 μm. Data represent mean ± SD. ***p < 0.001; Student’s t test
Discussion
Previous work has shown that GBS can interact with BECs resulting in tight junction disruption, an innate immune response, and alteration of other cellular processes [14, 19, 20, 36]. Here we observe for the first time that GBS is also able to disrupt BEC P-gp efflux transport. Using iPSC-derived BECs, an MDR1 (P-gp) overexpressing MDCK line, and a mouse model of GBS infection we have established that P-gp function appears to be diminished during GBS infection.
The impact of GBS on P-gp was independent of substrate given the increased accumulation of two different substrates, R123 and FLUO-3-AM (Fig. 1a, b). Previously, we and others have demonstrated that iPSC-BECs possess functional BCRP and MRP that can be inhibited by Ko143 or MK571 respectively [21, 22, 24,25,26,27,28,29]. Inhibition of BCRP or MRP family proteins did not result in a significant increase in R123 accumulation suggesting that the CsA- induced increase in R123 accumulation is mediated by P-gp in the iPSC-BEC model (Fig. 1e). The more specific second-generation P-gp inhibitor PSC-833 (Valspodar) [6], showed a similar impact on R123 accumulation (Fig. 1c). In addition, the effects appear to be dependent on what bacterium is interacting with the BECs. For instance, while GBS affected P-gp function, another Gram-positive bacterium, L. lactis, that is non-pathogenic, did not impact P-gp function. More work is required to identify the specific bacterial factor in GBS responsible for the loss of P-gp function during infection, as several known GBS invasion mutants did not affect the observed P-gp inhibition (Fig. 2c). However, our data suggest that direct live bacterial challenge is required since neither conditioned medium nor nonviable bacteria resulted in the same functional inhibition of P-gp. Meanwhile, direct interaction of live GBS on a non-BEC cell line overexpressing P-gp was also sufficient for loss of function indicating that the effects may not be BEC-specific. Moreover in BECs, the decrease in P-gp function accompanied a decrease in P-gp expression.
P-gp function in BECs impacting neurological disorders such as Alzheimer’s disease has been examined, where decreased function is associated with more Aβ deposition in the brain [10, 12]. Interestingly, while modeling Huntington’s disease in iPSC-derived BECs, it was observed that P-gp function was decreased but expression was increased [11]. P-gp function is also altered in various inflammatory states. Isolation and treatment of primary BECs from guinea pigs with pro-inflammatory cytokines led to a decrease in P-gp function and expression similar to our observations [37]. In vivo, inflammatory molecules such as lipopolysaccharide (LPS) and cytokines have also been shown to decrease P-gp function while increasing P-gp expression in a murine model [38]. However in rats, LPS treatment resulted in both lower expression and function of P-gp in the brain, suggesting model and/or species dependent differences [39]. This disjointed observation between expression levels and function is further confirmed in other P-gp expressing tissues that have been challenged with LPS or bacteria with varying expression levels after treatment [40,41,42,43,44,45]. Further work will be required to determine mechanisms by which GBS infection diminishes P-gp function. We observed decreased expression of P-gp in BECs on both transcript and protein levels as a result of GBS infection (Fig. 3a–e). However, in the MDCK-MDR1 cells, human P-gp is overexpressed and is not under the control of endogenous P-gp gene regulatory systems [30], and yet we still observed disruption of P-gp function (Fig. 2a). These data then suggest that in addition to the decrease in expression, a bacterial factor may also directly interact with and contribute to the inhibition of P-gp.
In barrier forming intestinal epithelial cells, Listeria monocytogenes and Salmonella enterica serovar Typhimurium have been shown to disrupt P-gp function [46, 47]. In the case of Listeria monocytogenes, P-gp acted as protective factor to invasive infection, and Listerial proteins inhibited normal P-gp function by acting as a competitive substrate [47]. Additionally, S. typhimurium infection resulted in R123 accumulation into epithelial monolayers [46]. GBS is carried in up to 30% of healthy individuals in the gastrointestinal tract and could potentially be interacting with similar P-gp expressing epithelial cells in a colonization state [14, 48]. Invasive GBS late-onset disease in newborns manifesting in sepsis and meningitis up to 3 months postnatally coincides with increasing P-gp expression in the brain that reaches maximum levels between 3 and 6 months of life [49, 50]. It is possible that functional P-gp may possess BEC barrier protecting functions, such as in the case with L. monocytogenes in the gut [47], and may contribute to the decrease in invasive GBS disease after 3 months of life. Here we demonstrate that GBS can disrupt P-gp function in BECs, however further investigation is required to understand how inhibition might impact BEC barrier function and bacterial invasion.
Our study suggests that a generalized response to bacteria, such as through Toll-like receptor agonists, may not be sufficient to decrease P-gp function since non-pathogenic live bacteria, and nonviable GBS do not decrease P-gp function (Fig. 2b). Future work will be required to determine the precise mechanism of P-gp dysfunction during infection, signaling pathways involved with P-gp downregulation, and impacts other transporters in BECs. Finally, these findings suggest that P-gp inhibition should be taken into consideration when exploring therapeutic strategies for bacterial infection of the CNS.
Materials and methods
Bacterial strains and cell lines used
Induced pluripotent stem cell (iPSC) line IMR-90-C4 (WiCell) was maintained per previous reports and grown on Matrigel (WiCell) coated plates (Corning) in mTeSR1 medium (WiCell) changed daily. IMR-90 iPSCs were passaged twice a week as needed [21,22,23, 26, 36, 51]. MDCK-MDR1 [52] (ATCC) cells were maintained on tissue culture treated 25 cm2 flasks in DMEM (Life Technologies) + 10% Fetal Bovine Serum (WiCell). Group B Streptococcus (GBS, Streptococcus agalactiae) hypervirulent clinical isolate COH1 (serotype III, multilocus sequence type 17) strain was used [53]. Previously described COH1 mutants Δiag [33], Δsrr2 [54, 55], and ΔsfbA [34] were employed for mutant analysis. All GBS strains were grown in static Todd-Hewitt broth (THB) at 37 °C. Lactococcus lactis was grown in M17 medium at 30 °C in static culture as previously described [56]. L. lactis was prepared exactly like GBS as mentioned above.
Brain endothelial cell differentiation
iPSC-derived brain microvascular endothelial cells (BECs) were differentiated as previously described [21,22,23, 26, 36, 51]. To ensure the quality of the cultures, mycoplasma testing was conducted periodically through services from WiCell, and PCR based kit (PanReac AppliChem). Briefly, single cell suspension of iPSCs were seeded at a density of 10,000/cm2 onto Matrigel (WiCell) cell culture plates or flasks (Corning) and expanded for 3 days in mTeSR1 changing media daily. Initiation of differentiation was conducted by exchanging to unconditioned media (UM; DMEM-F12 base medium [Life Technologies], 20% Knockout serum replacement [Life Technologies], 1% minimal essential medium-nonessential amino acids [Life Technologies], 0.5% Glutamax [Life Technologies], and 0.07% beta-mercaptoethanol [Sigma]), for 6 days changing media daily. After the 6 days, media was then changed to EC medium (human endothelial cell serum-free media [Life Technologies], 1% platelet-poor plasma derived serum [Fisher], 500 ng/ml basic fibroblast growth factor, and 10 μM all trans-retinoic acid [Sigma]) for 2 days. Finally, differentiated BECs were purified onto collagen IV (Sigma) and Fibronectin (Sigma) coated plates and transwells inserts (Corning), and the following day, media was changed to EC medium without basic fibroblast growth factor or retinoic acid. BECs were analyzed for TEER using an EVOM2 instrument (World Precision) to ensure high electrical resistance unique to BECs.
Bacterial infection
iPSC-derived BECs that are purified onto collagen-fibronectin 24 well plates at 500 k cells/well were grown until Day 10 of the differentiation [23]. A multiplicity of infection (MOI) of 10 was used for all experiments unless specifically noted otherwise as previously described [36]. Overnight cultures of GBS were grown in THB media at 37 °C + 5% CO2. The following day bacteria are subcultured and grown to an optical density at 600 nm (OD600) of 0.400–0.600. Bacteria are then spun down and washed in phosphate-buffered saline (PBS) prior to infecting BECs. BECs were infected for 5 h at 37 °C at 5% CO2 followed by sample collection or assays. Heat killed and formalin-fixed GBS were prepared by growing WT GBS to an OD600 of 0.400–0.600 in THB followed by centrifugation and resuspension of the pellet in PBS and treated by heating to 95 °C for 15 min, or treatment of GBS with 4% paraformaldehyde for 15 min. Bacteria were then washed and used to treat BECs at an estimated MOI of 10 for 5 h like live infections described above. Killed GBS preparations were confirmed by plating undiluted fractions onto THB plates where no growth was observed overnight at 37 °C + 5% CO2. GBS conditioned media was generated by growing WT GBS in endothelial cell assay media for 5 h, then sterile filtered through a 0.2 μM filter as previously described [19]. Conditioned media preparations were also confirmed sterile by plating undiluted fractions onto THB plates where no growth was observed overnight at 37 °C + 5% CO2.
P-gp activity assay
P-gp activity is assessed by the accumulation of the fluorescent substrates Rhodamine 123 (R123) (Sigma) and FLUO-3-AM (Thermo) [23, 57]. The specific P-gp inhibitor cyclosporine A (CsA) is used as a control to monitor P-gp activity [23]. For other inhibitor experiments PSC-833 (Sigma), MK571 (Sigma), or Ko143 (Sigma) were used to inhibit P-gp, MRPs, and BCRP respectively. Uninfected BECs are used as a control for comparison. After infection, cells are washed with Hank’s Balanced Salt Solution (HBSS) (Thermo) and pre-incubated with or without inhibitor (10 μM CsA, 10 μM PSC-833, 10 μM MK571, or 1 μM Ko143) for 1 h at 37 °C + 5% CO2. Following pre-incubation, cells were incubated with 10 μM R123 or FLUO-3-AM with or without inhibitors mentioned for 2 h at 37 °C + 5% CO2. After incubation, cells are washed twice in PBS and 200 μl of RIPA buffer (Thermo) is added and placed on a shaker for 10 min at room temperature protected from light. Fluorescence was measured on a plate reader (Tecan). Next, a BCA protein assay (Thermo) was conducted on each well and fluorescence values are normalized to BCA to account for relative cell number as previously described [23].
Flow cytometry
To assay for the expression of P-gp, BECs were either infected with GBS or left uninfected as a control. After the infection, cells are washed 3× in PBS and 100 μl of Accutase to each well and incubate at 37 °C + 5% CO2 to remove cells from the plate. Cells are then fixed by adding 900 μl of 1% paraformaldehyde in PBS for 15 min at room temperature. Cells are then washed twice in wash buffer (5% bovine serum albumin (BSA), 0.1% Triton-X in PBS) to block and permeabilize for the staining of total P-gp. Samples were stained in the wash buffer with anti-P-gp clone F4 (Thermo) (1 μg/million cells) overnight at 4 °C. Following primary stain, samples were washed and stained with secondary anti-mouse 488 (Thermo) at 1:5000 in wash buffer for 1 h at room temperature. Cells were washed and data was collected on a MACSQuant Analyzer 10 (Miltenyi Biotec) and analyzed on FlowJo v10.
Western blotting
BECs were infected as described above for 5 h at an MOI of 10. After infection, cells were washed three times in PBS and lysates were taken using RIPA buffer (Thermo) plus HALT protease inhibitor cocktail (Thermo). Proteins were quantified using a standard BCA assay kit (Thermo), and equal amounts of protein were loaded onto Nu-PAGE gels (Thermo) and transferred onto nitrocellulose membranes. Membranes were blocked in 5% milk in tris-buffered saline with 0.1% Tween 20 (TBST) and anti-COX-IV antibody 1:1000 (Cell Signaling Technologies) was used as a protein loading control, and anti-P-gp (F4) (Thermo Fisher) was used to determine P-gp abundance. To visualize protein abundance horseradish peroxidase (HRP)-conjugated secondary antibodies (Jackson Laboratory) and a BioRad ChemiDoc XRS + instrument were used to image blots.
RNA isolation and quantitative PCR
Monolayers of BECs were either left uninfected, or infected with GBS for 5 h at an MOI of 10. After infection, cell lysates and RNA was purified using a NucleoSpin RNA kit (Machery Nagel). cDNA was generated using VILO first-strand synthesis kit (Thermo Fisher), or LunaScript RT (New England BioLabs). SYBR green qPCR was conducted for human ABCB1 (P-gp, MDR1) forward primer 5′-GAAGAGATTGTGAGGGCAGC-3′, and reverse primer 5′-CCACCAGAGAGCTGAGTTCC-3′. 18S rRNA was used to normalize results and primers used, forward primer 5′-GTAACCCGTTGAACCCCATT-3′ and reverse primer 5′-CCATCCAATCGGTAGTAGCG-3′ were previously described [58]. qPCR data was collected on an Applied Biosystems StepOnePlus and data are presented as fold change using the cycle threshold (ΔΔCt) calculation.
Murine model and staining
Animal experiments were approved by the committee on the use and care of animals at the University of Colorado School of Medicine (protocol #00316) and performed using accepted veterinary standards. The University of Colorado School of Medicine is AAALAC accredited and the facilities meet and adhere to the standards in the “Guide for the Care and Use of Laboratory Animals”. We utilized a mouse GBS infection model as previously described [19, 33, 35]. Briefly, 8 week old male CD-1 mice (Charles River) were injected intravenously with 108–109 of GBS. At the experimental endpoint mice were euthanized and brain tissue was collected. One half of the brain was frozen in OCT compound (Sakura) and sectioned using a CM1950 cryostat (Leica). Sections were fixed with ice cold methanol (Sigma) for 20 min, blocked with Mouse on Mouse blocking reagent (Vector Labs), and incubated with mouse anti P-glycoprotein antibody clone C219 (ThermoFisher) overnight at 4 °C followed by goat anti-mouse conjugated to Cy3 (Jackson Immunoresearch) and tomato lectin conjugated to DyLight488 (Vector Labs). Coverslips were mounted with Fluoroshield + DAPI (Abcam) and images were taken using a BZ-X710 microscope (Keyence). Pixel intensities of P-gp staining were estimated by using the FIJI image analysis program. Briefly, lectin positive vessels were traced manually and the selection was transposed onto the P-gp image and mean pixel intensity was measured.
Immunofluorescence
iPSC-BECs were fixed and stained for markers exactly as described previously [23], with the exceptions that anti-CD-31 (Abcam, cat# ab32457), and anti-ZO1 (Proteintech, cat# 21773-1-AP) were utilized in place of referenced reagents. Briefly, BECs were fixed for 15 min in cold methanol and stained overnight at 4 °C. The following day, secondary antibodies anti-mouse 488 (Invitrogen, cat# A11001), and anti-rabbit 555 (Invitrogen, cat# A31572) were used at a dilution of 1:200. Samples were then visualized on a Nikon Eclipse Ti using Nikon NIS image acquisition software.
Statistics
GraphPad Prism version 5.0 (GraphPad Software Inc.) was used for all statistical analysis. For pairwise comparison, 2-tailed Student’s t test was used where appropriate. For multiple comparisons, analysis of variance (ANOVA) followed by Dunnett’s multiple comparisons test was used where appropriate. Data are represented as mean, ± standard deviation (SD) since all raw values are presented. Statistical significance was accepted at a P value of less than 0.05.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request. Specifically regarding the representative western blots, flow cytometry histogram, and brain images from mice (Fig. 3). All other data sets generated or analyzed during this study are included in this published article.
Abbreviations
- BECs:
-
brain endothelial cells
- CNS:
-
central nervous system
- P-gp:
-
P-glycoprotein
- iPSC:
-
induced pluripotent stem cell
- CsA:
-
cyclosporine A
- GBS:
-
group B Streptococcus
- MOI:
-
multiplicity of infection
- R123:
-
rhodamine 123
- WT:
-
wild-type
- MDCK:
-
Madin–Darby Canine Kidney
- qPCR:
-
quantitative polymerase chain reaction
- OCT:
-
optimal cutting temperature
- CFU:
-
colony forming unit
- LPS:
-
lipopolysaccharide
- FOV:
-
field of view
- BCRP:
-
breast cancer resistance protein
- MRP:
-
multi-drug resistance protein
References
Rua R, McGavern DB. Advances in meningeal immunity. Trends Mol Med. 2018;24:542–59.
Abbott NJ. Blood–brain barrier structure and function and the challenges for CNS drug delivery. J Inherit Metab Dis. 2013;36(3):437–49.
Abbott NJ, Patabendige AAK, Dolman DEM, Yusof SR, Begley DJ. Structure and function of the blood–brain barrier. Neurobiol Dis. 2010;37:13–25.
Doan KMM. Passive permeability and P-glycoprotein-mediated efflux differentiate central nervous system (CNS) and non-CNS marketed drugs. J Pharmacol Exp Ther. 2002;303(3):1029–37.
Davis TP, Sanchez-Covarubias L, Tome ME. P-glycoprotein trafficking as a therapeutic target to optimize CNS drug delivery. Adv Pharmacol. 2014;71:25–44.
Amin ML. P-glycoprotein inhibition for optimal drug delivery. Drug Target Insights. 2013;2013:27–34.
Lin JH, Yamazaki M. Role of P-glycoprotein in pharmacokinetics: clinical implications. Clin Pharmacokinet. 2003;42:59–98.
Bauer B, Hartz AMS, Fricker G, Miller DS. Modulation of p-glycoprotein transport function at the blood–brain barrier. Exp Biol Med. 2005;230:118–27.
Miller DS, Bauer B, Hartz AMS. Modulation of P-glycoprotein at the blood–brain barrier: opportunities to improve central nervous system pharmacotherapy. Pharmacol Rev. 2008;60(2):196–209.
Van Assema DME, Lubberink M, Bauer M, Van Der Flier WM, Schuit RC, Windhorst AD, et al. Blood–brain barrier P-glycoprotein function in Alzheimer’s disease. Brain. 2012;135(1):181–9.
Lim RG, Quan C, Reyes-Ortiz AM, Lutz SE, Kedaigle AJ, Gipson TA, et al. Huntington’s disease iPSC-derived brain microvascular endothelial cells reveal WNT-mediated angiogenic and blood–brain barrier deficits. Cell Rep. 2017;19(7):1365–77.
Cirrito JR, Deane R, Fagan AM, Spinner ML, Parsadanian M, Finn MB, et al. P-glycoprotein deficiency at the blood–brain barrier increases amyloid-β deposition in an Alzheimer disease mouse model. J Clin Invest. 2005;115(11):3285–90.
Kao YH, Chern Y, Yang HT, Chen HM, Lin CJ. Regulation of P-glycoprotein expression in brain capillaries in Huntington’s disease and its impact on brain availability of antipsychotic agents risperidone and paliperidone. J Cereb Blood Flow Metab. 2016;36(8):1412–23.
Doran KS, Fulde M, Gratz N, Kim BJ, Nau R, Prasadarao N, et al. Host–pathogen interactions in bacterial meningitis. Acta Neuropathol. 2016;131:185–209.
Kim KS. Microbial translocation of the blood–brain barrier. Int J Parasitol. 2006;36:607–14.
Coureuil M, Lécuyer H, Bourdoulous S, Nassif X. A journey into the brain: insight into how bacterial pathogens cross blood–brain barriers. Nat Rev Microbiol. 2017;15:149–59.
van Sorge NM, Doran KS. Defense at the border: the blood–brain barrier versus bacterial foreigners. Future Microbiol. 2012;7(3):383–94.
Maisey HC, Doran KS, Nizet V. Recent advances in understanding the molecular basis of group B Streptococcus virulence. Exp Rev Mol Med. 2008;10:e27.
Kim BJ, Hancock BM, Bermudez A, Del Cid N, Reyes E, Van Sorge NM, et al. Bacterial induction of Snail1 contributes to blood–brain barrier disruption. J Clin Invest. 2015;125(6):2473–83.
Cutting AS, Del Rosario Y, Mu R, Rodriguez A, Till A, Subramani S, et al. The role of autophagy during Group B Streptococcus infection of blood–brain barrier endothelium. J Biol Chem. 2014;289(52):35711–23.
Lippmann ES, Azarin SM, Kay JE, Nessler RA, Wilson HK, Al-Ahmad A, et al. Derivation of blood–brain barrier endothelial cells from human pluripotent stem cells. Nat Biotechnol. 2012;30(8):783–91. https://doi.org/10.1038/nbt.2247.
Lippmann ES, Al-Ahmad A, Azarin SM, Palecek SP, Shusta EV. A retinoic acid-enhanced, multicellular human blood–brain barrier model derived from stem cell sources. Sci Rep. 2014;4:4160.
Stebbins MJ, Wilson HK, Canfield SG, Qian T, Palecek SP, Shusta EV. Differentiation and characterization of human pluripotent stem cell-derived brain microvascular endothelial cells. Methods. 2016;101:93–102.
Clark PA, Al-Ahmad AJ, Qian T, Zhang RR, Wilson HK, Weichert JP, et al. Analysis of cancer-targeting alkylphosphocholine analogue permeability characteristics using a human induced pluripotent stem cell blood–brain barrier model. Mol Pharm. 2016;13(9):3341–9.
Vatine GD, Al-Ahmad A, Barriga BK, Svendsen S, Salim A, Garcia L, et al. Modeling psychomotor retardation using iPSCs from MCT8-deficient patients indicates a prominent role for the blood–brain barrier. Cell Stem Cell. 2016;20(6):831–43.
Wilson HK, Canfield SG, Hjortness MK, Palecek SP, Shusta EV. Exploring the effects of cell seeding density on the differentiation of human pluripotent stem cells to brain microvascular endothelial cells. Fluids Barriers CNS. 2015;12(1):13.
Qian T, Maguire SE, Canfield SG, Bao X, Olson WR, Shusta EV, et al. Directed differentiation of human pluripotent stem cells to blood–brain barrier endothelial cells. Sci Adv. 2017;3(11):e1701679.
Neal EH, Marinelli NA, Shi Y, McClatchey PM, Balotin KM, Gullett DR, et al. A simplified, fully defined differentiation scheme for producing blood–brain barrier endothelial cells from human iPSCs. Stem Cell Rep. 2019;12(6):1380–8. https://doi.org/10.1016/j.stemcr.2019.05.008.
Hollmann EK, Bailey AK, Potharazu AV, Neely MD, Bowman AB, Lippmann ES. Accelerated differentiation of human induced pluripotent stem cells to blood–brain barrier endothelial cells. Fluids Barriers CNS. 2017;14:9.
Pastan I, Gottesman MM, Ueda K, Lovelace E, Rutherford AV, Willingham MC. A retrovirus carrying an MDR1 cDNA confers multidrug resistance and polarized expression of P-glycoprotein in MDCK cells. Proc Natl Acad Sci USA. 1988;85(12):4486–90.
Six A, Bellais S, Bouaboud A, Fouet A, Gabriel C, Tazi A, et al. Srr2, a multifaceted adhesin expressed by ST-17 hypervirulent Group B Streptococcus involved in binding to both fibrinogen and plasminogen. Mol Microbiol. 2015;97(6):1209–22.
Seo HS, Minasov G, Seepersaud R, Doran KS, Dubrovska I, Shuvalova L, et al. Characterization of fibrinogen binding by glycoproteins Srr1 and Srr2 of Streptococcus agalactiae. J Biol Chem. 2013;288(50):35982–96.
Doran KS, Engelson EJ, Khosravi A, Maisey HC, Fedtke I, Equils O, et al. Blood–brain barrier invasion by group B Streptococcus depends upon proper cell-surface anchoring of lipoteichoic acid. J Clin Invest. 2005;115(9):2499–507.
Mu R, Kim BJ, Paco C, Del Rosario YD, Courtney HS, Doran KS. Identification of a group B streptococcal fibronectin binding protein, SfbA, that contributes to invasion of brain endothelium and development of meningitis. Infect Immun. 2014;82(6):2276–86.
Banerjee A, Kim BJ, Carmona EM, Cutting AS, Gurney M, Carlos C, et al. Bacterial Pili exploit integrin machinery to promote immune activation and efficient blood–brain barrier penetration. Nat Commun. 2011;2:462. https://doi.org/10.1038/ncomms1474.
Kim BJ, Bee OB, Mcdonagh MA, Stebbins MJ, Palecek SP, Doran KS, et al. Modeling Group B Streptococcus and induced pluripotent stem cell-derived brain endothelial cells. 2017;2(6):1–12.
Iqbal M, Ho HL, Petropoulos S, Moisiadis VG, Gibb W, Matthews SG. Pro-inflammatory cytokine regulation of P-glycoprotein in the developing blood–brain barrier. PLoS ONE. 2012;7(8):e43022.
Salkeni MA, Lynch JL, Otamis-Price T, Banks WA. Lipopolysaccharide impairs blood–brain barrier P-glycoprotein function in mice through prostaglandin- and nitric oxide-independent pathways. J Neuroimmune Pharmacol. 2009;4(2):276–82.
Wang JH, Scollard DA, Teng S, Reilly RM, Piquette-Miller M. Detection of P-glycoprotein activity in endotoxemic rats by 99mTc-sestamibi imaging. J NuclMed. 2005;46(9):1537–45.
Babic Z, Svoboda-Beusan I, Kucisec-Tepes N, Dekaris D, Troskot R. Increased activity of Pgp multidrug transporter in patients with Helicobacter pylori infection. World J Gastroenterol. 2005;11(18):2720.
Lye P, Bloise E, Javam M, Gibb W, Lye SJ, Matthews SG. Impact of bacterial and viral challenge on multidrug resistance in first- and third-trimester human placenta. Am J Pathol. 2015;185(6):1666–75.
Hirsch-Ernst KI, Ziemann C, Foth H, Kozian D, Schmitz-Salue C, Kahl GF. Induction of mdr1b mRNA and P-glycoprotein expression by tumor necrosis factor alpha in primary rat hepatocyte cultures. J Cell Physiol. 1998;176(3):506–15.
Goralski KB, Hartmann G, Piquette-Miller M, Renton KW. Downregulation of mdr1a expression in the brain and liver during CNS inflammation alters the in vivo disposition of digoxin. Br J Pharmacol. 2003;139(1):35–48.
Mishra J, Zhang Q, Rosson JL, Moran J, Dopp JM, Neudeck BL. Lipopolysaccharide increases cell surface P-glycoprotein that exhibits diminished activity in intestinal epithelial cells. Drug Metab Dispos. 2008;36(10):2145–79.
Stein U, Walther W, Shoemaker RH. Modulation of mdr1 expression by cytokines in human colon carcinoma cells: an approach for reversal of multidrug resistance. Br J Cancer. 1996;74(9):1384–91.
Siccardi D, Mumy KL, Wall DM, Bien JD, McCormick BA. Salmonella enterica serovar Typhimurium modulates P-glycoprotein in the intestinal epithelium. Am J Physiol Liver Physiol. 2008;294(6):G1392–400.
Neudeck BL, Loeb JM, Faith NG, Czuprynski CJ. Intestinal P glycoprotein acts as a natural defense mechanism against Listeria monocytogenes. Infect Immun. 2004;72(7):3849–54.
Berggren S, Gall C, Wollnitz N, Ekelund M, Karibom U, Hoogstraate J, et al. Gene and protein expression of P-glycoprotein, MRP1, MRP2, and CYP3A4 in the small and large human intestine. Mol Pharm. 2007;4(2):252–7.
Edmond KM, Kortsalioudaki C, Scott S, Schrag SJ, Zaidi AK, Cousens S, et al. Group B streptococcal disease in infants aged younger than 3 months: systematic review and meta-analysis. Lancet. 2012;379(9815):547–56.
Lam J, Baello S, Iqbal M, Kelly LE, Shannon PT, Chitayat D, et al. The ontogeny of P-glycoprotein in the developing human blood–brain barrier: implication for opioid toxicity in neonates. Pediatr Res. 2015;78(4):417.
Canfield SG, Stebbins MJ, Morales BS, Asai SW, Vatine GD, Svendsen CN, et al. An isogenic blood–brain barrier model comprising brain endothelial cells, astrocytes, and neurons derived from human induced pluripotent stem cells. J Neurochem. 2017;140:874–88. https://doi.org/10.1111/jnc.13923.
Evers R, Kool M, Smith AJ, van Deemter L, de Haas M, Borst P. Inhibitory effect of the reversal agents V-104, GF120918 and Pluronic L61 on MDR1 Pgp-, MRP1- and MRP2-mediated transport. Br J Cancer. 2000;83(3):366–74.
Rubens CE, Wessels MR, Heggen LM, Kasper DL. Transposon mutagenesis of type III group B Streptococcus: correlation of capsule expression with virulence. Proc Natl Acad Sci USA. 1987;84(20):7208–12.
Sheen TR, Jimenez A, Wang NY, Banerjee A, van Sorge NM, Doran KS. Serine-rich repeat proteins and pili promote Streptococcus agalactiae colonization of the vaginal tract. J Bacteriol. 2011;193(24):6834–42.
Wang NY, Patras KA, Seo HS, Cavaco CK, Rösler B, Neely MN, et al. Group B streptococcal serine-rich repeat proteins promote interaction with fibrinogen and vaginal colonization. J Infect Dis. 2014;210:982–91.
Maisey HC, Hensler M, Nizet V, Doran KS. Group B streptococcal pilus proteins contribute to adherence to and invasion of brain microvascular endothelial cells. J Bacteriol. 2007;189:1464–7.
Orlický J, Sulová Z, Dovinová I, Fiala R, Zahradníková A, Breier A. Functional fluo-3/AM assay on P-glycoprotein transport activity in L1210/VCR cells by confocal microscopy. Gen Physiol Biophys. 2004;23(3):357–66.
Rho HW, Lee BC, Choi ES, Choi IJ, Lee YS, Goh SH. Identification of valid reference genes for gene expression studies of human stomach cancer by reverse transcription-qPCR. BMC Cancer. 2010;10(1):240.
Acknowledgements
We would like to thank Dr. William Elmquist (University of Minnesota) for supplying the MDCK-MDR1 cell line, and Charlotte Goldeman (University of Copenhagen) for the ABCB1 primer design and validation. We also thank Dr. Sean Palecek (University of Wisconsin) for his constructive comments. Finally, we would like to thank Lena Wolter (University of Würzburg), our medical laboratory technician for assistance with iPSC-BEC culture.
Funding
Work was supported by Defense Threat Reduction Agency Grant HDTRA1-15-0047 and National Institutes of Health Grants R01NS083688 to EVS and R01NS051247 to KSD, BJK was partially supported by postdoctoral fellowships from the Wisconsin Stem Cell and Regenerative Medicine Center, and the Alexander von Humboldt Foundation. MAM was partially supported by a Wisconsin Stem Cell Roundtable Fellowship at the University of Wisconsin. BDG was partially supported by National Institutes of Health Biotechnology Training Program grant T32 GM008349 and the National Science Foundation Graduate Research Fellowship Program under grant number 1747503. ASU is partially supported by the German Research Foundation (DFG) research training group “3D-Tissue Models for Studying Microbial Infections by Human Pathogens” (RTG 2157).
Author information
Authors and Affiliations
Contributions
BJK, MAM, LD, and BDG performed experiments and collected data. ASU provided materials, input on data, and support for the medical laboratory technician. BJK, KSD, and EVS provided materials, input on data analysis and interpretation, contributed to experimental design, and wrote the paper. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files

Additional file 1: Figure S1.
Characterization of iPSC-BECs. (A-F) Representative immunofluorescence images of differentiated iPSC-BECs. (A) VE-cadherin, (B) CD-31, (C) Claudin-5, (D) Occludin, (E) ZO-1, (F) Glut-1. Images were taken using a 20× objective and scale bar represents 50 μm.

Additional file 2: Figure S2.
Additional staining of different mice for lectin (green) and P-gp (red), and DAPI (blue). Two representative images from two control mice (top). Two representative images from two GBS mice (bottom). Scale bar represents 50 μm.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

{kind=link}
{kind=link}
Cite this article
Kim, B.J., McDonagh, M.A., Deng, L. et al. Streptococcus agalactiae disrupts P-glycoprotein function in brain endothelial cells. Fluids Barriers CNS 16, 26 (2019). https://doi.org/10.1186/s12987-019-0146-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12987-019-0146-5