
Abstract
Non-alcoholic fatty liver disease (NAFLD) is a chronic liver disease that is becoming increasingly prevalent, and it ranges from simple steatosis to cirrhosis. However, there is still a lack of pharmacotherapeutic strategies approved by the Food and Drug Administration, which results in a higher risk of death related to carcinoma and cardiovascular complications. Of note, it is well established that the pathogenesis of NAFLD is tightly associated with whole metabolic dysfunction. Thus, targeting interconnected metabolic conditions could present promising benefits to NAFLD, according to a number of clinical studies. Here, we summarize the metabolic characteristics of the development of NAFLD, including glucose metabolism, lipid metabolism and intestinal metabolism, and provide insight into pharmacological targets. In addition, we present updates on the progresses in the development of pharmacotherapeutic strategies based on metabolic intervention globally, which could lead to new opportunities for NAFLD drug development.
Similar content being viewed by others

Introduction
With well over 25% of the world’s population suffering from non-alcoholic fatty liver disease (NAFLD), it is currently the most prevalent chronic liver disease worldwide [1]. Moreover, it is proposed that the NAFLD population in China will increase by 29.1% to 314.58 million during 2016–2030 [2].
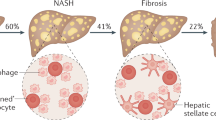
The development of NAFLD is progressive with a sophisticated clinicopathological classification system. Individuals with NAFLD mostly present hallmarks of steatosis. In 60% of NAFLD patients, non-alcoholic steatohepatitis develops and is associated with inflammatory infiltration and significant fibrosis [3]. Over time, 22% of NASH-related fibrosis patients progress to cirrhosis, and 2% progress to hepatocellular carcinoma [3][4]. The risk of cardiovascular conditions and malignant carcinoma associated with mortality is increased in individuals with NAFLD (Fig. 1a) [5, 6].

Multiple metabolic dysfunctions contribute to the progression of non-alcoholic fatty liver disease (NAFLD). a NAFLD is defined as intrahepatic triglyceride content exceeding 5.5% within hepatocytes and has a sophisticated clinicopathological classification system [8]. Gradually, excessive lipid levels could overwhelm the capacity to deal with inflammation and hepatocyte ballooning due to lipotoxicity, which are characteristic of non-alcoholic steatohepatitis (NASH). Progressively, hepatic stellate cells are actively responsible for inflammation and hepatocyte death. This results in fibrosis through the generation of fibrogenic myofibroblasts [221], and 22% of patients develop cirrhosis [4]. Finally, patients with severe cirrhosis patients progress to hepatocellular carcinoma (HCC). b Available evidence indicates that multiple metabolic dysfunctions, such as obesity, type 2 diabetes mellitus (T2DM) and dysfunction of the gut microbiota, are the main risk factors for the progression of NAFLD [3]
However, patients with NAFLD are typically asymptomatic until the disease progresses to cirrhosis [7]. Initially, symptoms of right upper quadrant pain and fatigue are most commonly noticed. Then, excessive triglyceride accumulation in the liver is detected by imaging examination [8], and increased levels of liver-related enzymes, alanine aminotransferase (ALT) and aspartate aminotransferase (AST), in serum typically reflect hepatocellular damage [9]. The clinical strategy is limited to ameliorating progression through diet modification and exercise; this strategy improves only simple steatosis due to the unsustainability of long-term intervention [10, 11]. In addition, while liver transplants are a reliable treatment for NASH, they are highly expensive, difficult to obtain and carry traumatizing risks. Thus, potential drugs that can replace this treatment in clinical practice are urgently needed [12]. Unfortunately, to date, there is still a lack of clinically approved drugs targeting NAFLD.
The metabolic disorders associated with NAFLD are characterized by dysregulation of lipid metabolism, glucose homeostasis [13] and intestinal-hepatic crosstalk [14], supporting the movement to rename NAFLD as metabolic-associated fatty liver disease (MAFLD) [15]. Moreover, it is helpful to decelerate the progression of NAFLD by improving whole-body metabolic homeostasis to improve associated conditions, such as diabetes and hypertension [16].
In this article, we mainly focus on the metabolic characteristics involved in the development of NAFLD, including glucose metabolism, lipid metabolism and intestinal metabolism, and propose some promising targets for further investigation. Moreover, we assess pharmaceutical targets for NAFLD from the perspective of metabolic intervention and development status at present globally, which might provide new drug development prospects.
Definition of NAFLD
The stages of NAFLD include non-alcoholic fatty liver (NAFL), NASH, liver fibrosis and liver cirrhosis. NAFLD is first characterized by intrahepatic triglyceride levels exceeding 5.5%, as detected by magnetic resonance spectroscopy or liver biopsy, and the exclusion of secondary causes, such as alcohol abuse, viral infection, other metabolic liver diseases including Wilson’s disease, and drugs, including tamoxifen and amiodarone [8, 17]. Broadly, NAFLD is divided into two pathological forms: NAFL, which shows macrovascular steatosis and mild lobular inflammation, and progressive NASH, which shows ballooning with or without perisinusoidal fibrosis [18]. It is difficult to identify NAFLD in the early stage because the majority of individuals are asymptomatic until they progress to cirrhosis. The most common symptom is right upper quadrant pain, which is then confirmed by ultrasonic evidence or MRI [8]. Consistent with these findings, the serum levels of liver enzymes and albumin are changed with the progression of NAFLD, and these levels reflect whole body dysfunction [19]. Due to the systemic nature of NAFLD, its incidence has been correlated with that of cardiovascular disease, cancer and other conditions, such as chronic kidney disease and obstructive sleep apnea [20]. Patients with severe liver fibrosis are more likely to develop subclinical carotid atherosclerosis, and cardiovascular diseases account for the majority of NAFLD-related mortality [21].
The metabolic risk of NAFLD
Principally, NAFLD is a systemic disease that can be controlled by whole-body homeostasis, so other diseases, such as polycythemia, hyperuricemia, hypothyroidism, hypopituitarism and polycystic ovary syndrome, could be independent risk factors for its occurrence and development [22,23,24,25]. Importantly, the consumption of diets rich in fat and sugar with insufficient exercise may contribute to NAFLD; this may explain the increased prevalence of NAFLD with metabolic impairments [26]. NAFLD is frequently associated with obesity and type 2 diabetes mellitus (T2DM) in China. The proportions of individuals with NAFLD in the obesity and T2DM groups were 60-90% and 28-70%, respectively. Moreover, 51.3% of NAFLD patients had obesity and 22.5% had T2DM [27], which reflects systemic metabolic disorders. Of note, it is increasingly appreciated that the microbiota plays a functional role in regulating metabolic homeostasis, such as that in NAFLD [28], as evidenced by different gut bacteria between obese and lean humans [29] (Fig. 1b).
Dysregulated metabolism in NAFLD
Lipid metabolism in NAFLD
In the development of NAFLD, the imbalance between lipid input and output leads to the accumulation of lipids in the liver. Triglycerides (TGs) are the main form of lipids that are stored in the liver and are synthesized by the esterification of free fatty acids (FFAs) [30]. Excessive FFAs impair the liver through lipotoxicity [31,32,33], mitochondrial dysfunction [34], stimulation of signaling pathways related to metabolism and inflammation [35] and even direct activation of receptors that promote inflammation [36]. Apart from FFAs, intermediates of DNL, such as diacylglycerol, also disrupt metabolic homeostasis [37, 38] through increased reactive oxygen species (ROS) derived from weakening mitochondrial activity [39, 40]. To avoid damage caused by excessive FFAs, the liver will initiate a series of self-protection mechanisms. FFAs can be esterified and transported into serum via very low-density lipoprotein (VLDL). Additionally, FFAs can be oxidized and converted to other substrates. However, in the NASH stage, overwhelmed mitochondria produce ROS, which further aggravates NAFLD [39, 41, 42].
Considering the role of FFAs in NAFLD, it is vital to understand the three main sources of FFAs. The first is an increase in the spontaneous lipolysis of adipose tissue (59%). The canonical pathway for lipolysis promotes cyclic adenosine monophosphate (cAMP) generation, and then protein kinase A (PKA) is activated to phosphorylate lipases phospho-hormone sensitive lipase (p-HSL) and phospho-perilipin 1 (p-PLIN1). This pathway can be suppressed by insulin [43]. Following their release into circulation, FFAs are taken up by the liver [44]. A number of studies have demonstrated that the lipolysis of adipose tissue in NAFLD, regardless of the existence of diabetes, is increased [45,46,47]. In obese individuals, due to factors such as adipocyte hypertrophy and insulin resistance, increased lipolysis produces more FFAs, and these FFAs are then transported to the liver (Fig. 2b) [48].

Lipid metabolism in non-alcoholic fatty liver disease (NAFLD). a. Under physiological conditions, lipase breaks down triacylglycerol into monoacylglycerol and FFAs, which are then absorbed by intestinal epithelial enterocytes. Then, FFAs and monoacylglycerol are used to resynthesize triacylglycerol by two key enzymatic steps: the first by mannoside acetylglucosaminyltransferase (MGAT) and the second by diglyceride acyltransferase (DGAT). Triacylglycerols are incorporated into chylomicrons (CMs) and secreted into the lymphatic vessels. After catalyzed by lipase, the remnants of CMs absorbed by liver [68, 69]. b. Insulin promotes lipid storage by inhibiting lipolysis via adipose triglyceride lipase (ATGL), phosphodiesterase 3B (PDE3B) and protein kinase A (PKA)-controlled hormone-sensitive lipase (HSL) and perilipins (PLINs). However, in regard to insulin resistance conditions (such as obesity or type 2 diabetes mellitus [T2DM]), lower insulin sensitivity stimulates lipolysis, which then leads to more NEFA flux to the liver. c. Several key enzymes (such as acetyl-CoA carboxylase [ACC], fatty acid synthase [FAS], stearoyl-CoA desaturase [SCD1] and DGAT2) are involved in de novo lipogenesis in the liver [222]
The second source of FFAs is de novo lipogenesis (DNL) (26%). DNL starts with acetyl-CoA subunits, which are mainly derived from glucose [49], and further condensation occurs with the glycerol backbone of these products [50]. There are two major proteins, sterol response element binding protein (SREBP1c) and carbohydrate response element binding protein (ChREBP), that are involved in the transcriptional regulation of DNL [51, 52]. Then, several genes, including fatty acid synthase (FAS), acetyl-CoA carboxylase (ACC) and stearoyl-CoA desaturase 1 (SCD1), are upregulated. Malonyl-CoA is produced from an acetyl-CoA precursor under the controlled catalytic activity of ACC at the beginning of this process [53]. Acyl carrier protein (ACP), which belongs to the FAS domain, transports malonyl-CoA to the prosthetic phosphopantetheine group of the acyl carrier protein [54,55,56]. Through the prosthetic phosphopantetheine arm of ACP, the elongating FA chain can be shuttled to the different catalytic centers in the active site cleft of FAS by its rotation [57,58,59]. The malonyl moiety bound to ACP is the additive monomer for elongating the substrate acyl chain, resulting in an elongated 16- or 18-carbon FFA chain [60, 61]. In the initial step of triacylglycerol (TG) synthesis, FFAs are incorporated into glycerol-3-phosphate via primary acylation, resulting in lysophosphatidic acid (LPA) via glycerol-phosphate acyl transferase (GPAT) [50]. In the following step, after desaturated, acylglycerol-phosphate acyl transferase catalyzes LPA to produce phosphatidic acid (PA), which is then dephosphorylated by phosphatidic acid phosphorylase (PAP) to produce diacylglycerol (DG) [62]. Through the catalytic activity of diacylglycerol acyltransferase (DGAT), DG is acylated to TG [63]. DNL not only increases the synthesis of FFAs but also inhibits β-oxidation by its intermediate product malonyl coenzyme (Fig. 2c) [64].
The third source is excessive dietary fatty acids (15%). Hepatocytes take up chylomicron (CM) particle remnants, which contain FFAs [65], and increased absorption of CM remnants leads to the excessive accumulation of lipids in the liver [66, 67]. Mechanically, triacylglycerol is broken down into FFAs and monoacylglycerol by pancreatic lipase. Enterocytes resynthesize triacylglycerol through two sequential acylation steps: first by monoacylglycerol acyltransferase 2 (MGAT2) and then by DGAT. Then, chylomicrons are secreted into lymphatic vessels and incorporated with triacylglycerol. After catalysis by lipases, the FFAs are stored in adipose tissue or utilized by muscle tissue as an energy source. The remnants of CM are transported into the liver. There, they form triglycerides and are packaged into VLDL particles, which are released into the bloodstream (Fig. 2a) [68, 69].
Glucose and fructose metabolism in NAFLD
Compared with normoglycemic NAFLD patients, hyperglycemic NAFLD patients more rapidly progress from NAFL to NASH [70, 71], indicating that glucose metabolism is tightly associated with NAFLD. Recently, it was found that the levels of key enzymes in glycolysis were significantly higher in NAFLD in parallel with enhanced glycolytic capacity in NAFLD patients. Moreover, overexpression of hexokinase 2 (HK2) and pyruvate kinase isozyme type M2 (PKM2), which are involved in glycolysis, could promote the accumulation of triglycerides in hepatocytes [72, 73]. The Warburg effect produces lactic acids in the presence of oxygen. Tumors often adapt this process, and it also occurs in NAFLD (Fig. 3a) [72]. High levels of lactic acid stimulate the uptake of FFAs by hepatocytes and promote the expression of lipogenic genes [74]. In contrast to the Warburg effect, the effect of the TCA cycle on NAFLD remains controversial. However, there is no doubt that oxidative stress and DNA damage in the NASH stage impair mitochondrial function and worsen the TCA cycle [75].

Glucose and fructose metabolism in non-alcoholic fatty liver disease (NAFLD). a Increases in glucose transport results in enhanced glycolysis in the liver. There, pyruvate is converted to oxaloacetate, which provides more substrates for de novo lipogenesis (DNL), or lactate, which stimulates the DNL pathway via decreased activity of histone deacetylase (HDAC) [223]. b In addition, fructose is phosphorylated to fructose-1-phosphate (F-1-P) by ketohexokinase (KHK) upon entering hepatocytes, which have high-rate activity and bypass more limited steps [224]. Moreover, substrates, such as adenosine diphosphate (ADP) derived from adenosine triphosphate (ATP) during hydrolysis activity, are converted into uric acid, which impairs the liver by stimulating DNL [225, 226]. c Insulin regulates the liver directly by upregulating sterol regulatory element-binding protein 1c (SREBP1c) and carbohydrate-responsive element-binding protein (ChREBP); it also decreases the production of very-low-density lipoprotein (VLDL) via the downregulation of microsomal triglyceride transfer protein (MTTP) and apolipoprotein B (ApoB) [78, 79]
Insulin resistance is a prominent feature of NAFLD that can regulate NAFLD directly, as evidenced by the observation that the short-term consumption of high-fat diets leads to hepatic insulin resistance without peripheral insulin resistance [76]. Insulin resistance impairs the inhibition of gluconeogenesis [48]. This leads to increased production of glucose [77], which is the main source of DNL. Insulin also promotes DNL by stimulating liver X receptor (LXR), which further upregulates Chrebp1 and Srebp1 [78]. Additionally, insulin inhibits microsomal triglyceride transport protein (MTTP) and promotes apolipoprotein B (ApoB) degradation to regulate VLDL production. In regard to insulin resistance, the increased production of MTTP results from decreased phosphorylation of forkhead box transcription factor 1 (FoxO1) [79] and the degradation of ApoB resulting from the decreased insulin sensitivity and increased uptake of FFAs by the liver (Fig. 3a) [80].
The effect of fructose on NAFLD has also attracted considerable attention recently. Fructose is regarded as the “sweet killer” to metabolic homeostasis [81], and abundant evidence demonstrates that long-term fructose intake aggravates hepatic steatosis [82]. In contrast to glucose, fructose bypasses some regulatory steps in glycolysis. It is catalyzed by phosphofructokinase in the liver and provides more substrates for the DNL pathway [83]. Moreover, the silencing of the feedback cycle in fructose metabolism leads to a continuous decrease in ATP and phosphate [84,85,86,87]. This ultimately results in redundant uric acid and deficiency of ATP [88]. Furthermore, ATP deficiency leads to a series of adverse reactions that include inhibitory effects on protein synthesis and oxidative stress [84, 89]. It has also been shown that fructose stimulates the DNL pathway but inhibits β-oxidation by stimulating ChREBP and SREBP1c. This results in a decrease in FFAs consumption [89, 90], thereby worsening NAFLD (Fig. 3b) [84]. In parallel, fructose not only disturbs gut microbiota homeostasis to stimulate hepatic steatosis by regulating the production of short-chain fatty acids (SCFAs) but also destroys tight junctions, which promotes endotoxin exposure to the liver [91,92,93].
Gut microbiota in NAFLD
The gut microbiota plays a vital role in barrier protection, immunity and metabolic homeostasis in the host. The main factor that affects the gut microbiota is overnutrition [94]. Gut microbiota dysfunction increases susceptibility to various diseases, including metabolic diseases such as NAFLD [95]. NAFLD is reported to be characterized by chronic low-grade inflammation. Inflammatory mediators, such as endotoxin, are derived from gut microbiota [96], and a high-fat diet increases the proportion of endotoxin [97, 98]. Recent studies on the gut microbiota in NAFLD have found that a high-fat diet increased specific bacteria, such as Enterobacter cloacae B29, Escherichia coli py102 and Klebsiella pneumoniae A7, which impair the progression of NAFLD [99]. Moreover, in regard to the advanced stage, the abundances of Proteus and Escherichia coli were increased, while the abundances of Firmicutes and fecal bacteria were significantly decreased [100]. Additionally, Ruminococcaceae and Veronibacteriaceae were found to be risk factors for liver fibrosis [101]. It has also been found that dysfunction of the gut microbiota dominated by Enterobacteriaceae, Escherichia coli and Shigella is associated with NAFLD progression [102].
A number of studies have demonstrated that metabolic dysfunction is associated with decreased concentrations of bacteria that produce SCFAs, propionate and butyrate [103]. On the one hand, butyrate could act as a substrate to stimulate β-oxidation to maintain the anaerobic environment for the microbiota [104] and suppress the expression of nitric oxide synthase via nuclear receptor peroxisome proliferator-activated receptor gamma (PPARγ). This results in a decrease in NO, which inhibits Enterobacteriaceae [105, 106]. On the other hand, butyrate can moderate inflammatory conditions by activating immune cells, such as regulatory T cells (Tregs) [107]. In addition, SCFAs are beneficial for maintaining intestinal permeability and insulin secretion and sensitivity via increased secretion of glucagon-like peptide-1 (GLP-1) and peptide YY (PYY) (Fig. 4c) [108,109,110]. Unfortunately, dysfunction of the gut microbiota aggravates NAFLD due to a decrease in SCFAs [111]. Specifically, F. prausnitzii (Faecalibacterium), A. muciniphila (Akkermansia) and Dysosmobacter welbionis are involved in this decrease in SCFAs [112]. Moreover, disorder of the gut microbiota inhibits intestinal epithelial cells from secreting a lipoprotein lipase inhibitor, fasting-induced adipose factor (FIAF), which increases FFAs levels in the liver [28].

Gut dysbiosis and bile acid metabolism in non-alcoholic fatty liver disease (NAFLD). a. Hepatocytes produce primary bile acids via the classic and alternative pathways. The classic pathway starts with cholesterol 7α-hydroxylase (CYP7A1) and the action of sterol 12α-hydroxylase (CYP8B1), which produces cholic acid (CA) or chenodeoxycholic acid (CDCA) through sterol 27 hydroxylase (CYP27A1) [227, 228]. The alternative pathway is initiated by CYP27A1 and produces CDCA through the action of oxysterol 7α-hydroxylase (CYP7B1) [229]. After a meal, the release of cholecystokinin from the pancreas causes bile stored in the gallbladder to be released into the duodenum. Then, ~ 95% of the bile acids involved in the hepatic intestinal circulation are reabsorbed by enterocytes via the apical sodium-dependent bile salt transporter (ASBT) [230] and excreted into the portal vein via organic solute transporter-α and -β (OSTα and OSTβ) [231, 232]. Finally, ~ 5% of bile acids are transported into the systemic circulation from hepatocytes via multidrug resistance-associated protein 3 (MRP3), MRP4, OSTα and OSTβ. b. Two kinds of farnesoid X receptor (FXR)-dependent pathways have been proposed for the feedback regulation of bile acid synthesis. Activation of hepatic FXR in the liver increases the expression of the small heterodimer partner (SHP), which inhibits CYP7A1 and CYP8B1 expression [233, 234]. In addition, FXR plays a key role in regulating metabolism in the liver by suppressing de novo lipogenesis (DNL), promoting β-oxidation and producing very-low-density lipoprotein (VLDL) [235,236,237]. In addition, activation of FXR in the intestine stimulates the production of FGF15/19, which inhibits CYP7A1 and activates the DNL pathway [238]. Another vital receptor for bile acids is Takeda G protein-coupled receptor 5 (TGR5), which promotes the production of glucagon-like peptide-1 (GLP-1) through increased cyclic adenosine monophosphate (cAMP) [239, 240]. c. In healthy conditions, the production of butyrate aids in the consumption of oxygen to maintain anaerobic conditions through β-oxidation and decreases the production of nitrate, which is available for specific pathogens via conjunction with peroxisome proliferator activated receptor gamma (PPARγ). Short-chain fatty acids (SCFAs), another beneficial product derived from nondigestible carbohydrates [241], help to maintain metabolic homeostasis through the secretion of GLP-1 and Yin-Yang 1 (YY1) [110, 242]. However, under pathogenic conditions, decreased butyrate and SCFA levels disturb metabolic homeostasis
Bile acids metabolism in NAFLD
Systemic homeostasis is influenced by the gut microbiota, partially by regulating bile acids (BAs) metabolism and signal transduction via BAs receptors [113]. Studies have shown that BAs metabolic disorder could aggravate chronic liver diseases [114], and BAs metabolic disorder progresses to NAFLD independent of obesity and diabetes [115]. These findings show the importance of the regulation of BAs in NAFLD. Approximately 95% of BAs are involved in enterohepatic circulation, while the remaining 5% are excreted in the feces [116]. To maintain the BAs pool, the number of newly synthesized BAs should be equal to that of BAs excreted in the feces. Therefore, inhibiting the reabsorption of BAs will increase the excretion of BAs in the feces. Thus, more cholesterol will be converted to BAs, which lowers the risk of obesity [117].
There are two synthesis pathways of BAs. The first is the canonical pathway, also named the neutral pathway (75%), which is regulated by CYP8B1 after cholesterol is hydroxylated by CYP7A. Another pathway is the alternative pathway, also named the acidic pathway (25%). This pathway is controlled by CYP7B1, which is triggered by CYP27A1; as a result, mainly CDCA is produced [118]. It has been reported that activation of the alternative pathway produces more BAs, which benefits the consumption of cholesterol [119]. Additionally, significant increases in CYP8B1 in db/db mice and the overexpression of CYP8B1 have been shown to upregulate lipogenesis-related genes, and this process is dependent on SREBP1. However, the loss of CYP8B1 could ameliorate NAFLD [120, 121].
Moreover, BAs could directly regulate hepatic metabolism as a signal molecule through the activation of farnesoid X receptor (FXR). Hepatic FXR inactivates the lipogenesis pathway by inhibiting SREBP1c. It also induces β-oxidation by activating peroxisome proliferator-activated receptor-α (PPARα) and clears VLDL in plasma, ultimately ameliorating NAFLD [122,123,124]. Moreover, hepatic FXR stimulates FFAs oxidation and ketogenesis, which is dependent on fibroblast growth factor 21 (FGF21) [125, 126]. However, the activation of intestinal FXR stimulates intestinal epithelial cells to secrete FGF15/19 into the liver, which potently reduces hepatic steatosis and improves insulin resistance [127,128,129,130]. However, the contribution of FXR to NAFLD is still under debate due to its wide distribution in various tissues. Recently, it was found that when FXR was globally knocked out, the insulin sensitivity of ob/ob and HFD mice was improved. This may be because the long-term activation of FXR reduces energy consumption and aggravates HFD-induced glucose intolerance (Fig. 4a) [131,132,133]. However, in liver-specific FXR knockout mice, the above effect was not observed, indicating that intestinal FXR contributes significantly [134]. In parallel, increases in level of T-β MCA, an intestinal FXR antagonist, ameliorates NAFLD through increased BAs synthesis [135,136,137], and GLP-1 secretion decreases via the activation of intestinal FXR [138]. As a result, the coordination of intestinal FXR in maintaining metabolic homeostasis still needs to be further confirmed (Fig. 4b).
Another bile acid receptor, Takeda G protein-coupled receptor 5 (TGR5), is mainly expressed in the gallbladder, adipose tissue, intestine, and liver and is activated primarily by secondary BAs [139]. Once TGR5 is activated in muscles or brown adipose tissue, it stimulates energy consumption, and in the intestine, it increases the secretion of GLP-1 (Fig. 4b) [114, 140, 141]. Moreover, recent studies found that TGR5 prefers to influence NAFLD-related hypothyroidism regardless of the level of thyroid hormone [142], and researchers found that thyroid hormone β receptor (TRβ) regulates the synthesis of BAs by interfering with SHP [143, 144] or CYP7A1 directly in the liver [145]. Additionally, it has been reported that activation of TRβ reduces systemic lipid content and increases lipid oxidation to improve hepatic lipid homeostasis [146].
Treatments for NAFLD
Diet and lifestyle intervention
Several recent studies have demonstrated that steatohepatitis improves in 58% of cases in which the patient lost > 5% of their body weight and in 90% of cases in which the patient lost > 10% of their body weight [10]. Patients are encouraged to adapt a diet pattern of low-fat, low-carbohydrate or Mediterranean type, with a daily energy intake of 500–1000 kcal. It has also been demonstrated that isocaloric diets with high protein content could reduce hepatic steatosis and inflammation in T2DM patients [147].
Exercise
Exercise has been demonstrated to reduce hepatic steatosis independently of diet changes [148]; additionally, exercise has also been found to improve liver stiffness [149]. Over the course of five years of follow-up, moderate-vigorous exercise was shown to prevent fatty liver in 233,676 subjects who participated in this study [150]. Specifically, a dose‒response relationship was demonstrated between exercise volume and reduction in hepatic steatosis, with individuals exercising over 250 min a week experiencing higher responses [151]. In terms of the type of exercise, sufficient exercise could ameliorate NAFLD regardless of whether aerobic exercise is performed [152].
Bariatric and metabolic surgery
To date, there is debate regarding the adaptation of foregut bariatric surgery to NAFLD treatment [8], and surgery is only provided for NAFLD patients with other severe obesity-related comorbidities [153]. After surgery, 75% of patients with steatohepatitis showed improvements in ballooning and lobular inflammation [154]. However, the risk of potential complications of secondary steatohepatitis and liver fibrosis is increased [155].
Updated metabolism-targeted drugs for NAFLD
As the most prevalent chronic liver disease, there is an urgent need for available drugs approved by the FDA for the treatment of NAFLD. In the following, we summarize the emerging pharmacotherapeutic targets and related clinical experimental information regarding metabolic interventions globally (Table 1).
Regulating lipid metabolism
ACC inhibitors
Firsocostat, an ACC inhibitor, effectively reduces lipid accumulation and improves fibrosis by inhibiting the DNL pathway after 12 weeks of intervention, but it increased the risk of hypertriglyceridemia [156]. In addition, PF-05221304, developed by Pfizer, is another potent and reversible dual ACC1/2 inhibitor. In a 16-week phase II clinical trial, at least 10 mg of this drug per day dose-dependently reduced lipid accumulation in the liver. The highest percentage of reduction was 65%, but the adverse effect was a dose-dependent increase in triglycerides in serum in 8% of subjects [157].
FASN inhibitors
TVB 2640 is an inhibitor of FASN. Patients were randomly divided into groups that received placebo or 25 mg or 50 mg of the drug orally every day for 12 weeks in a phase II clinical trial. Lipid accumulation increased by an average of 4.5% compared to baseline in the control group. However, lipid accumulation was decreased by 9.6% in the TVB 2640-25 mg group and decreased by 28.1% in the 50 mg group. Additionally, the ALT levels decreased in a dose-dependent and time-dependent manner. Moreover, serum LDL levels were decreased in the groups receiving the drug, and no drug-related toxicity was observed in organs. However, this study is limited by the small sample size, and further evaluation of liver histology is needed [158]. Currently, another IIb clinical trial is recruiting volunteers for further evaluation.
SCD1 inhibitors
Aramchol, an inhibitor of hepatic stearoyl-CoA desaturase (SCD1), can reduce steatosis, steatohepatitis and liver fibrosis in rodents. Moreover, in a phase II clinical trial, aramchol improved NAFLD, with a 12.5% reduction in hepatic lipid accumulation after 3 months of treatment [159]. Additionally, in a phase IIb clinical trial with more participants, a double-blind trial of 600 mg/per day for 52 weeks, individuals with NAFLD receiving drug intervention showed a 16.7% reduction in hepatic lipid accumulation compared to only a 5% reduction in the placebo group. Moreover, a 29.1% decrease in serum ALT less and a marked improvement in fibrosis less than 1 grade were observed. However, these differences did not reach statistical significance. This drug is considered safe to use because the probability of adverse events is less than 5%. However, the decrease in hepatic lipids was not robust enough, and the differences were not statistically significant [160]. The drug is currently undergoing phase III clinical trials, but outcomes have yet to be reported.
DGAT inhibitors
At the end of triglyceride synthesis, DGAT catalyzes the conversion of DAG to triglycerides. This enzyme is classified into two isoforms: DGAT1 and DGAT2. The isoforms have different expression patterns and substrate specificities [161]. Liver-specific DGAT2-deficient mice exhibited reduced hepatic lipid accumulation compared to normal mice [162], and PF-06865571 (a DGAT2 inhibitor) was also shown to reduce the accumulation of lipids in the liver in a phase I clinical trial. Unfortunately, PF-06865571 increases the risk of diarrhea [163]. Currently, another phase II clinical trial has recruited volunteers [164].
MGAT2 inhibitors
It has been reported that MGAT2 is overexpressed in the small intestine and liver [165, 166]. Considering the redundancy of the MGAT2 enzyme system, selective inhibition of MGAT2 will only partially impede triacylglycerol synthesis in the intestine. Therefore, this will delay the absorption of fat rather than prevent it completely. As a result, the inhibitor diminishes the risk of diarrhea and other side effects associated with lipid synthesis targets. Moreover, the use of this inhibitor benefits NASH indirectly through weight loss. It has been proposed that MGAT2 contributes to the accumulation of endogenous cannabinoid 2-arachidonoylglycerol, which exhibits anti-inflammatory and antifibrotic effects [167]. Recently, a new selective MGAT2 inhibitor, BMS-963,272, showed benefits in improving liver inflammation and fibrosis without diarrhea in NASH mice. Moreover, BMS-963,272 decreased body weight and increased GLP-1 and PYY levels without adverse effects in a phase I trial [168].
Statins
Hyperlipidemia is characterized by increases in triglyceride-rich and cholesterol-rich lipoproteins in the serum. Hyperlipidemia plays a critical role in promoting NAFLD by increasing the transport of lipids to the liver. It has been reported that in prospective clinical trials, statins reduced the risk of hepatic steatosis and fibrosis [169]. Moreover, in a randomized clinical trial, a significant improvement in NAS evaluation after drug treatment was observed in patients with NAFLD [170]. Another small pilot prospective clinical trial demonstrated that the hypolipidemic drug atorvastatin decreases the level of ALT and improves hepatic steatosis [171]. Rosuvastatin also reduces ALT and AST levels and ameliorates liver fibrosis [172]. However, large clinical trials for statins are currently underway to confirm these benefits.
Hypoglycemic drugs and targeting intermediary metabolism of glucose
PPAR agonists
There are three types of PPARs, PPAR-α, PPAR-δ and PPAR-γ, that regulate lipid and glucose metabolism; agonists of PPARs have been shown to ameliorate NAFLD [173]. PPAR-γ greatly regulates adipocyte differentiation and lipid and glucose metabolism and inhibits inflammation [174]. Thiazolidinediones are potent activators of PPAR-γ that are used for the treatment of diabetes, and a further benefit is their ability to reduce plasma FFAs and hepatic lipid accumulation by improving insulin sensitivity [175]. Additionally, thiazolidinediones have been shown to improve fibrosis by directly inhibiting the activation of hepatic stellate cells [176]. Pioglitazone is a mild PPAR-γ activator that ameliorates steatosis and reduces liver enzymes without affecting fibrosis [177]. However, its use is controversial due to the risk of weight gain and edema [178, 179]. This treatment is currently undergoing a phase III clinical trial for treating NAFLD. Elafibranor is a dual agonist of PPARα/δ. It was shown to reduce hepatic lipid accumulation and improve inflammation and fibrosis [180]. When obese patients were treated with elafibranor, liver enzymes decreased and insulin sensitivity improved [181]. However, the latest phase III trial was terminated in advance because the predefined primary surrogate efficacy endpoint was unmet. The dual agonist of PPARα/γ, saroglitazar, significantly reduced hepatic lipid accumulation in mice and is currently used for the treatment of diabetic dyslipidemia in India [182]. However, clinical trials for its use for NAFLD are currently recruiting participants. The pan-PPAR agonist lanifibranor decreased hepatic lipid accumulation, liver enzyme levels, and biomarkers of inflammation in plasma and improved fibrosis in an IIb clinical trial. However, the adverse effects of gastrointestinal reactions and weight gain were greater than those in the control group [182]. A phase III trial is currently recruiting volunteers.
Sodium-dependent glucose transporters-2 (SGLT-2) inhibitors
SGLT-2 is a glucose transporter that is dependent on sodium and is responsible for most glucose reabsorption after filtration in the kidney [183]. Because it is not expressed in the liver [183], SGLT-2 indirectly decreases hepatic lipid accumulation through weight loss or metabolic improvement. Additionally, the SGLT-2 inhibitor dapagliflozin reduces hepatic lipid accumulation without significant effects on insulin sensitivity [184, 185]. In patients with type 2 diabetes, empagliflozin reduces liver enzyme levels in plasma and reduces the hepatic accumulation of lipids. It is considered an early treatment for type 2 diabetes patients with NAFLD [186], and it simultaneously reduces the risk of lower extremity amputation and diabetic ketoacidosis [187].
GLP-1 modulators
GLP-1 is an endogenous gut hormone that stimulates insulin production and release directly. It also inhibits glucagon secretion indirectly and reduces appetite. GLP-1 receptors are widely distributed but not significantly expressed in the liver [188]. In addition, the improvement in NAFLD by GLP-1 correlates with weight loss and other metabolic improvements, and the benefit of GLP-1 agonists for NAFLD may be an indirect effect that acts by improving systemic metabolism, such as improved insulin sensitivity and appetite suppression. However, exenatide increases hepatocyte uptake of glucose under oral glucose stimulation, suggesting that it directly affects the liver [189]. Until now, it has been debated whether GLP-1 improves NAFLD by regulating the liver directly. T2DM is currently treated with GLP-1R agonists, such as exenatide and liraglutide [190]. Liraglutide not only improves insulin sensitivity [191] but also ameliorates NAFLD with 39% efficacy [192]. Another GLP-1 receptor agonist, exenatide, stimulates β-oxidation and conversely downregulates genes related to lipogenesis, ultimately improving NAFLD [193, 194]. The phases II clinical trial for this drug has ended [195].
Dimethyl peptidase 4 (DPP4) inhibitors
DPP4 is widely expressed on a variety of cell surfaces and selectively cleaves a variety of substrates, including GLP-1, to inactivate and thereby regulate diabetes [196]. A decrease in DPP4 activity increases GLP-1 activity. In patients with NAFLD, DPP4 is elevated and positively correlated with hepatocyte apoptosis and fibrosis [197]. Mice with NASH have been shown to benefit from DPP4 inhibitors, as inflammation and fibrosis of the liver was improved [198]. However, in a phase II trial, the DPP inhibitor sitagliptin failed to reduce hepatic lipid accumulation and NAS assessment [199], which means that it is not a reliable strategy for treating NAFLD.
Ketohexokinase (KHK) inhibitors
As the rate-limiting enzyme in fructose metabolism, KHK catalyzes the conversion of fructose to fructose 1-phosphate. Excessive fructose is always accompanied by increased hexokinase levels, impaired fatty acid oxidation, enhanced DNL, aggravated hepatic steatosis and impaired insulin signal transduction [200]. When hexokinase is specifically knocked out in the liver, it will moderate the hepatic damage caused by excessive fructose [201]. In an early clinical trial, the hexokinase inhibitor PF-06835919 decreased hepatic lipid accumulation, but no improvement in insulin resistance was observed [202]. To date, a longer-term phase II RCT of PF-06835919 has been carried out in the NAFLD population.
Drugs targeting the gut-liver crosstalk
Microbiota transplantation
Fecal transplantation has emerged as a treatment option for NAFLD, as the gut microbiota differ between NAFLD patients and healthy people. In a phase II RCT, 21 patients with NAFLD received allogeneic or autologous fecal transplantation through endoscopy, but there was no change in hepatic lipid accumulation after six months [203]. Therefore, the feasibility of fecal transplantation needs further investigation. Of note, more studies acknowledge that the appropriate supplementation of butyrate could improve NAFLD. In a randomized controlled trial, a single dose injection of A. soehngenii to the duodenum in Mets patients showed robust GLP-1 production and peripheral glycemic homeostasis [204].
FXR agonists
It was shown that OCA, a classic FXR agonist, reduced inflammation, hepatic lipid accumulation, and liver enzyme activity in NAFLD patients. In an ongoing global phase III RCT, liver fibrosis was significantly improved after 18 months of treatment with 25 mg OCA per day, but there was a mild to moderate incidence of adverse effects, such as pruritus [205]. Cilofexor is another FXR agonist. In a completed phase II RCT, 24 weeks of oral administration of 30 mg of cilofexor per day in NASH patients significantly improved steatosis and reduced the content of primary BAs without significant changes in liver fibrosis. In patients taking 100 mg, however, moderate to severe pruritus was experienced [206]. EDP-305 is another FXR agonist. A phase II RCT showed that the ALT level and hepatic lipid accumulation of NAFLD patients were both decreased after 12 weeks of treatment with EDP-305, but the incidence of side effects, including pruritus and nausea, was also higher [207].
TRβ agonists
Resmetirom is an oral TRβ agonist that specifically targets the liver to ameliorate NAFLD by improving lipid metabolism and lipotoxicity. In a 36-week phase II RCT, patients receiving 80 mg resmetirom per day had significantly reduced hepatic lipid accumulation, but transient mild diarrhea and nausea were also more common [208]. At present, a phase III RCT for its use as a treatment for NAFLD is recruiting worldwide.
FGF19 analogs
Aldafermin is an analog of FGF19 that inhibits BAs synthesis and regulates metabolic homeostasis. In a 24-week phase II RCT conducted in patients with NASH, the results showed that hepatic lipid accumulation decreased by 7.7%, and liver fibrosis trended toward improvement after treatment with aldafermin in NAFLD patients [209]. Another phase IIb RCT revealed that aldafermin was well tolerated, but there was no significant dose-dependent response in fibrosis [210]. Presently, another clinical trial is underway to further support this hypothesis.
FGF21 analogs
Fibroblast growth factor 21 (FGF21) is the most prominent hepatokine. It regulates overall metabolic homeostasis by targeting multiple tissues, and its production is highly dependent on nutritional stress, including starvation, a high-fat diet and a nutritional restriction diet [211, 212]. It has been reported that FGF21 exerts beneficial effects in treating obesity due to the potential for increased energy consumption and insulin sensitivity [213], which therefore indirectly benefits hepatic metabolism. Surprisingly, FGF21 has also been reported to directly improve NAFLD, even though the specific mechanism is still unclear [214]. Thus, it is regarded as a promising target for NAFLD. There is a PEGylated analog of FGF21 known as pegbelfermin (PGBF). In a phase II trial, hepatic lipid accumulation in NAFLD patients decreased significantly after subcutaneous injection with PGBF for 16 weeks. While the histology of the liver was still under evaluation, 16% of patients presented adverse effects, such as nausea [215]. Another phase IIb RCT to evaluate the effect of PGBF on fibrosis in NAFLD has ended, but the results have not been reported [216]. Additionally, for 11 weeks, subcutaneous injection of B1344 (another analog of FGF21) significantly reduced hepatic steatosis, inflammation and fibrosis in cynomolgus monkeys suffering from nonalcoholic fatty liver disease (NAFLD), and an evaluation of FGF21 analog administration in nonhuman primate species undergoing liver biopsies for the treatment of NAFLD is first reported in this study [217].
Conclusion
The threat of NAFLD to human health is gradually increasing. However, to date, there is a lack of specific drugs for treating NAFLD; thus, researchers need to continue to explore potential targets of NAFLD. The results of many studies show that NAFLD patients suffer from diverse metabolic disorders, including lipid, glucose and BAs disorders, which further aggravate NAFLD. The inseparable relationship between metabolism and NAFLD shows the necessity for metabolic therapy. Here, we described the characteristics of lipid metabolism, glucose metabolism, the gut microbiota and BAs metabolism in NAFLD. Various metabolites, including intermediates during the process, can affect the corresponding signaling pathways as signaling molecules. Moreover, different metabolic pathways can act independently or interact with each other to affect NAFLD. The systemic metabolic complexity of NAFLD implies the risk of systemic adverse effects and reveals the challenge of its treatment. Over the past few years, drugs have been tested in clinical trials worldwide. We summarized the therapeutic targets of NAFLD and the corresponding drugs. Due to the complexity of NAFLD, targeted drugs have the defect of a single function. Additionally, a single target has the adverse effect of activating a variety of signaling pathways. As a result, no specific drug is currently available for the treatment of NAFLD. However, from a positive point of view, the metabolic complexity of NAFLD also provides researchers with a combination of drugs and tissue-targeted specific strategies. Currently, clinical trials of multitarget combination therapy and more in-depth investigations in specific tissues of known targets have been ongoing globally. Such studies include GLP-1 receptor agonists combined with DPP4 inhibitors.
It should be noted that NAFLD not only has metabolic dysregulation but also relates to the immunity closely, which could provide aims at the immunotherapy such as the anti-inflammatory and anti-fibrosis agents. Moreover, the beneficial immune factors also could ameliorate NAFLD. Furthermore, the genetic and epigenetic factors have been proved to promote the progression of NAFLD, providing the new therapeutic strategies including RNAi or mRNA vaccines to ameliorate NAFLD. Additionally, we can’t ignore that NAFLD is a whole metabolic homeostatic disease which is link with other diseases, so it is in need for us to detail the underlying mechanisms and find more specific crosstalk factors, which could greatly provide the new targets or therapeutic strategies. Additionally, we should consider using targeted drugs for other closely related diseases in combination with targeted drugs for NAFLD Meanwhile, despite numerous drugs have showed potential in NAFLD in preclinical research, they still fail to achieve the great outcomes in clinical trials, suggesting us revise the experimental models and test strategies to recapitulate the NAFLD pathology in human as realistic as possible, which could tremendously accelerate the drug development of NAFLD. These studies could bring new hope for overcoming NAFLD.
Data availability
Not applicable.
References
Younossi ZM. Non-alcoholic fatty liver disease - A global public health perspective. J Hepatol. 2019;70(3):531–44.
Estes C, Anstee QM, Arias-Loste MT, Bantel H, Bellentani S, Caballeria J, et al. Modeling NAFLD disease burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the period 2016–2030. J Hepatol. 2018;69(4):896–904.
Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016;64(1):73–84.
Sanyal AJ, Harrison SA, Ratziu V, Abdelmalek MF, Diehl AM, Caldwell S, et al. The natural history of Advanced Fibrosis due to nonalcoholic steatohepatitis: data from the Simtuzumab trials. Hepatology. 2019;70(6):1913–27.
Targher G, Byrne CD, Tilg H. NAFLD and increased risk of cardiovascular disease: clinical associations, pathophysiological mechanisms and pharmacological implications. Gut. 2020;69(9):1691–705.
Simon TG, Roelstraete B, Khalili H, Hagstrom H, Ludvigsson JF. Mortality in biopsy-confirmed nonalcoholic fatty liver disease: results from a nationwide cohort. Gut. 2021;70(7):1375–82.
Spengler EK, Loomba R. Recommendations for Diagnosis, Referral for Liver Biopsy, and Treatment of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Mayo Clinic Proceedings. 2015;90(9):1233-46.
Chalasani N, Younossi Z, Lavine JE, Charlton M, Cusi K, Rinella M, et al. The diagnosis and management of nonalcoholic fatty liver disease: practice guidance from the American Association for the study of Liver Diseases. Hepatology. 2018;67(1):328–57.
Torres DM, Williams CD, HarrisonO SA. Features, diagnosis, and treatment of nonalcoholic fatty liver disease. Clinical Gastroenterology and Hepatology. 2012;10(8):837–58.
Vilar-Gomez E, Martinez-Perez Y, Calzadilla-Bertot L, Torres-Gonzalez A, Gra-Oramas B, Gonzalez-Fabian L, et al. Weight loss through lifestyle modification significantly reduces features of nonalcoholic steatohepatitis. Gastroenterology. 2015;149(2):367-+.
Kistler KD, Brunt EM, Clark JM, Diehl AM, Sallis JF, Schwimmer JB, et al. Physical activity recommendations, exercise intensity, and histological severity of nonalcoholic fatty liver disease. Am J Gastroenterol. 2011;106(3):460–8; quiz 9.
Wong RJ, Aguilar M, Cheung R, Perumpail RB, Harrison SA, Younossi ZM, et al. Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the United States. Gastroenterology. 2015;148(3):547–55.
Samuel VT, Shulman GI. Nonalcoholic fatty liver disease as a Nexus of metabolic and hepatic Diseases. Cell Metabolism. 2018;27(1):22–41.
Jia W, Xie G, Jia W. Bile acid-microbiota crosstalk in gastrointestinal inflammation and carcinogenesis. Nature Reviews Gastroenterology & Hepatology. 2018;15(2):111–28.
Eslam M, Sanyal AJ, George J, Int Consensus P. MAFLD: A Consensus-Driven proposed nomenclature for metabolic Associated fatty liver disease. Gastroenterology. 2020;158(7):1999-+.
Chitturi S, Wong VW-S, Chan W-K, Wong GL-H, Wong SK-H, Sollano J, et al. The Asia-Pacific Working Party on non-alcoholic fatty liver Disease guidelines 2017Part 2: management and special groups. Journal of Gastroenterology and Hepatology. 2018;33(1):86–98.
Szczepaniak LS, Nurenberg P, Leonard D, Browning JD, Reingold JS, Grundy S, et al. Magnetic resonance spectroscopy to measure hepatic triglyceride content: prevalence of hepatic steatosis in the general population. American Journal of Physiology-Endocrinology and Metabolism. 2005;288(2):E462-E8.
Singh S, Allen AM, Wang Z, Prokop LJ, Murad MH, Loomba R. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: a systematic review and Meta-analysis of Paired-Biopsy Studies. Clinical Gastroenterology and Hepatology. 2015;13(4):643-+.
Loomba R, Adams LA. The 20% rule of NASH Progression: the natural history of Advanced Fibrosis and Cirrhosis caused by NASH. Hepatology. 2019;70(6):1885–8.
Zhou Y-Y, Zhou X-D, Wu S-J, Fan D-H, Van Poucke S, Chen Y-P, et al. Nonalcoholic fatty liver Disease contributes to subclinical atherosclerosis: a systematic review and Meta-analysis. Hepatology Communications. 2018;2(4):376–92.
Angulo P, Kleiner DE, Dam-Larsen S, Adams LA, Bjornsson ES, Charatcharoenwitthaya P, et al. Liver fibrosis, but No other histologic features, is Associated with Long-term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology. 2015;149(2):389-+.
Xu L, Ma H, Miao M, Li Y. Impact of subclinical hypothyroidism on the development of non-alcoholic fatty liver disease: a prospective case-control study. J Hepatol. 2012;57(5):1153–4.
Xu C, Yu C, Xu L, Miao M, Li Y. High serum uric acid increases the risk for nonalcoholic fatty liver disease: a prospective observational study. Plos One. 2010;5(7).
Ma H, Xu C, Xu L, Yu C, Mao M, Li Y. Independent association of HbA1c and nonalcoholic fatty liver disease in an elderly chinese population. Bmc Gastroenterology. 2013;13.
Xu C, Wan X, Xu L, Miao M, Li Y, Yu C. Xanthine Oxidase promotes hyperuricemia and nonalcoholic fatty liver disease in patients and mice. Gastroenterology. 2015;148(4):S1053-S.
Wehmeyer MH, Zyriax B-C, Jagemann B, Roth E, Windler E, Zur Wiesch JS, et al. Nonalcoholic fatty liver disease is associated with excessive calorie intake rather than a distinctive dietary pattern. Medicine. 2016;95(23).
Kwok R, Choi KC, Wong GL-H, Zhang Y, Chan HL-Y, Luk AO-Y, et al. Screening diabetic patients for non-alcoholic fatty liver disease with controlled attenuation parameter and liver stiffness measurements: a prospective cohort study. Gut. 2016;65(8):1359–68.
Leung C, Rivera L, Furness JB, Angus PW. The role of the gut microbiota in NAFLD. Nature Reviews Gastroenterology & Hepatology. 2016;13(7):412–25.
Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, et al. Human gut microbiome viewed across age and geography. Nature. 2012;486(7402):222-+.
Bian X, Liu R, Meng Y, Xing D, Xu D, Lu Z. Cancer focus lipid metabolism and cancer. Journal of Experimental Medicine. 2021;218(1).
Hirsova P, Ibrabim SH, Gores GJ, Malhi H. Lipotoxic lethal and sublethal stress signaling in hepatocytes: relevance to NASH pathogenesis. J Lipid Res. 2016;57(10):1758–70.
Ralston JC, Lyons CL, Kennedy EB, Kirwan AM, Roche HM. Fatty acids and NLRP3 inflammasome-mediated inflammation in metabolic tissues. Annu Rev Nutr. 2017;37:77–102.
Lomonaco R, Ortiz-Lopez C, Orsak B, Webb A, Hardies J, Darland C, et al. Effect of adipose tissue insulin resistance on metabolic parameters and liver histology in obese patients with nonalcoholic fatty liver disease. Hepatology. 2012;55(5):1389–97.
Fu S, Watkins SM, Hotamisligil GS. The role of endoplasmic reticulum in hepatic lipid homeostasis and stress signaling. Cell Metabolism. 2012;15(5):623–34.
Perry RJ, Samuel VT, Petersen KF, Shulman GI. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature. 2014;510(7503):84–91.
Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. Journal of Clinical Investigation. 2006;116(11):3015–25.
Turpin SM, Nicholls HT, Willmes DM, Mourier A, Brodesser S, Wunderlich CM, et al. Obesity-Induced CerS6-Dependent C-16:0 Ceramide Production promotes weight gain and glucose intolerance. Cell Metabolism. 2014;20(4):678–86.
Xia JY, Holland WL, Kusminski CM, Sun K, Sharma AX, Pearson MJ, et al. Targeted induction of Ceramide Degradation leads to Improved systemic metabolism and reduced hepatic steatosis. Cell Metabolism. 2015;22(2):266–78.
GarciaRuiz C, Colell A, Mari M, Morales A, FernandezCheca JC. Direct effect of ceramide on the mitochondrial electron transport chain leads to generation of reactive oxygen species - role of mitochondrial glutathione. Journal of Biological Chemistry. 1997;272(17):11369–77.
Martinez L, Torres S, Baulies A, Alarcon-Vila C, Elena M, Fabrias G, et al. Myristic acid potentiates palmitic acid-induced lipotoxicity and steatohepatitis associated with lipodystrophy by sustaning de novo ceramide synthesis. Oncotarget. 2015;6(39):41479–96.
Perez-Carreras M, Del Hoyo P, Martin MA, Rubio JC, Martin A, Castellano G, et al. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology. 2003;38(4):999–1007.
Sanyal AJ, Campbell-Sargent C, Mirshahi F, Rizzo WB, Contos MJ, Sterling RK, et al. Nonalcoholic steatohepatitis: Association of insulin resistance and mitochondrial abnormalities. Gastroenterology. 2001;120(5):1183–92.
DiPilato LM, Ahmad F, Harms M, Seale P, Manganiello V, Birnbaum MJ. The role of PDE3B phosphorylation in the inhibition of lipolysis by insulin. Molecular and Cellular Biology. 2015;35(16):2752–60.
Ceddia RP, Collins S. A compendium of G-protein-coupled receptors and cyclic nucleotide regulation of adipose tissue metabolism and energy expenditure. Clin Sci (Lond). 2020;134(5):473–512.
Kotronen A, Juurinen L, Tiikkainen M, Vehkavaara S, Yki-Jarvinen H. Increased liver fat, impaired insulin clearance, and hepatic and adipose tissue insulin resistance in type 2 diabetes. Gastroenterology. 2008;135(1):122–30.
Fabbrini E, Magkos F, Mohammed BS, Pietka T, Abumrad NA, Patterson BW, et al. Intrahepatic fat, not visceral fat, is linked with metabolic complications of obesity. Proc Natl Acad Sci U S A. 2009;106(36):15430–5.
Gastaldelli A, Cusi K, Pettiti M, Hardies J, Miyazaki Y, Berria R, et al. Relationship between hepatic/visceral fat and hepatic insulin resistance in nondiabetic and type 2 diabetic subjects. Gastroenterology. 2007;133(2):496–506.
Samuel VT, Shulman GI. The pathogenesis of insulin resistance: integrating signaling pathways and substrate flux. Journal of Clinical Investigation. 2016;126(1):12–22.
Lambert JE, Ramos-Roman MA, Browning JD, Parks EJ. Increased De Novo Lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology. 2014;146(3):726–35.
Coleman RA, Lee DP. Enzymes of triacylglycerol synthesis and their regulation. Progress in Lipid Research. 2004;43(2):134–76.
Kawano Y, Cohen DE. Mechanisms of hepatic triglyceride accumulation in non-alcoholic fatty liver disease. Journal of Gastroenterology. 2013;48(4):434–41.
Oosterveer MH, Schoonjans K. Hepatic glucose sensing and integrative pathways in the liver. Cellular and Molecular Life Sciences. 2014;71(8):1453–67.
Bianchi A, Evans JL, Iverson AJ, Nordlund AC, Watts TD, Witters LA. Identification of an isozymic form of acetyl-CoA carboxylase. J Biol Chem. 1990;265(3):1502–9.
Majerus PW, Alberts AW, Vagelos PR. The acyl carrier protein of fatty acid synthesis: purification, Physical Properties, and substrate binding site. Proc Natl Acad Sci U S A. 1964;51:1231–8.
Brindley DN, Matsumura S, Bloch K. Mycobacterium phlei fatty acid Synthetase—A bacterial Multienzyme Complex. Nature. 1969;224(5220):666–9.
Smith S. The animal fatty acid synthase: one gene, one polypeptide, seven enzymes. FASEB J. 1994;8(15):1248–59.
Wakil SJ. Fatty acid synthase, a proficient multifunctional enzyme. Biochemistry. 1989;28(11):4523–30.
Smith S, Tsai SC. The type I fatty acid and polyketide synthases: a tale of two megasynthases. Nat Prod Rep. 2007;24(5):1041–72.
Maier T, Leibundgut M, Ba n N. The crystal structure of a mammalian fatty acid synthase. Science. 2008;321(5894):1315–22.
Foster DW, Bloom B. The synthesis of fatty acids by rat liver slices in tritiated water. J Biol Chem. 1963;238:888–92.
Carey EM, Dils R, Hansen HJ. Short communications. Chain-length specificity for termination of atty acid biosynthesis by fatty acid synthetase complexes from lactating rabbit mamary gland and rat liver. Biochem J. 1970;117(3):633–5.
Aguado B, Campbell RD. Characterization of a human lysophosphatidic acid acyltransferase that is encoded by a gene located in the class III region of the human major histocompatibility complex. J Biol Chem. 1998;273(7):4096–105.
Shi Y, Cheng D. Beyond triglyceride synthesis: the dynamic functional roles of MGAT and DGAT enzymes in energy metabolism. American Journal of Physiology-Endocrinology and Metabolism. 2009;297(1):E10-E8.
McGarry JD, Mannaerts GP, Foster DW. A possible role for malonyl-CoA in the regulation of hepatic fatty acid oxidation and ketogenesis. The Journal of clinical investigation. 1977;60(1):265–70.
Frayn KN, Arner P, Yki-Jarvinen H. Fatty acid metabolism in adipose tissue, muscle and liver in health and disease. In: Wagenmakers AJM, editor. Essays in Biochemistry, Vol 42: The Biochemical Basis of the Health Effects of Exercise. Essays in Biochemistry. 422006. p. 89–103.
Laurencikiene J, Skurk T, Kulyte A, Heden P, Astrom G, Sjolin E, et al. Regulation of Lipolysis in Small and large Fat cells of the same subject. Journal of Clinical Endocrinology & Metabolism. 2011;96(12):E2045-E9.
Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. Journal of Clinical Investigation. 2005;115(5):1343–51.
Shi Y, Cheng D. Beyond triglyceride synthesis: the dynamic functional roles of MGAT and DGAT enzymes in energy metabolism. Am J Physiol Endocrinol Metab. 2009;297(1):E10-8.
Yen CE, Nelson DW, Yen MI. Intestinal triacylglycerol synthesis in fat absorption and systemic energy metabolism. J Lipid Res. 2015;56(3):489–501.
Bazick J, Donithan M, Neuschwander-Tetri BA, Kleiner D, Brunt EM, Wilson L, et al. Clinical model for NASH and Advanced Fibrosis in adult patients with diabetes and NAFLD: guidelines for Referral in NAFLD. Diabetes Care. 2015;38(7):1347–55.
Portillo-Sanchez P, Bril F, Maximos M, Lomonaco R, Biernacki D, Orsak B, et al. High prevalence of nonalcoholic fatty liver disease in patients with type 2 diabetes Mellitus and normal plasma aminotransferase levels. Journal of Clinical Endocrinology & Metabolism. 2015;100(6):2231–8.
Liu J, Jiang S, Zhao Y, Sun Q, Zhang J, Shen D, et al. Geranylgeranyl diphosphate synthase (GGPPS) regulates non-alcoholic fatty liver disease (NAFLD)-fibrosis progression by determining hepatic glucose/fatty acid preference under high-fat diet conditions. Journal of Pathology. 2018;246(3):277–88.
Kim H-S, Xiao C, Wang R-H, Lahusen T, Xu X, Vassilopoulos A, et al. Hepatic-specific disruption of SIRT6 in mice results in fatty liver formation due to enhanced glycolysis and triglyceride synthesis. Cell Metabolism. 2010;12(3):224–36.
Wang T, Chen K, Yao W, Zheng R, He Q, Xia J, et al. Acetylation of lactate dehydrogenase B drives NAFLD progression by impairing lactate clearance. J Hepatol. 2021;74(5):1038–52.
Satapati S, Sunny NE, Kucejova B, Fu X, He TT, Mendez-Lucas A, et al. Elevated TCA cycle function in the pathology of diet-induced hepatic insulin resistance and fatty liver. Journal of Lipid Research. 2012;53(6):1080–92.
Samuel VT, Liu ZX, Qu XQ, Elder BD, Bilz S, Befroy D, et al. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J Biol Chem. 2004;279(31):32345–53.
Sunny NE, Parks EJ, Browning JD, Burgess SC. Excessive hepatic mitochondrial TCA cycle and gluconeogenesis in humans with nonalcoholic fatty liver disease. Cell Metabolism. 2011;14(6):804–10.
Beaven SW, Matveyenko A, Wroblewski K, Chao L, Wilpitz D, Hsu TW, et al. Reciprocal regulation of hepatic and adipose lipogenesis by liver X receptors in obesity and insulin resistance. Cell Metab. 2013;18(1):106–17.
Kamagate A, Qu S, Perdomo G, Su D, Kim DH, Slusher S, et al. FoxO1 mediates insulin-dependent regulation of hepatic VLDL production in mice. J Clin Invest. 2008;118(6):2347–64.
Avramoglu RK, Basciano H, Adeli K. Lipid and lipoprotein dysregulation in insulin resistant states. Clin Chim Acta. 2006;368(1–2):1–19.
Febbraio MA, Karin M. “Sweet death”: Fructose as a metabolic toxin that targets the gut-liver axis. Cell Metab. 2021;33(12):2316–28.
Tappy L, Le KA. Metabolic effects of fructose and the worldwide increase in obesity. Physiol Rev. 2010;90(1):23–46.
Tappy L, Le K-A. Metabolic Effects of Fructose and the Worldwide increase in obesity. Physiological Reviews. 2010;90(1):23–46.
Maenpaa PH, Raivio KO, Kekomaki MP. Liver adenine nucleotides: fructose-induced depletion and its effect on protein synthesis. Science (New York, NY). 1968;161(3847):1253–4.
Smith CM, Rovamo LM, Raivio KO. Fructose-induced adenine nucleotide catabolism in isolated rat hepatocytes. Canadian journal of biochemistry. 1977;55(12):1237–40.
van den Berghe G, Bronfman M, Vanneste R, Hers HG. The mechanism of adenosine triphosphate depletion in the liver after a load of fructose. A kinetic study of liver adenylate deaminase. The Biochemical journal. 1977;162(3):601–9.
Kurtz TW, Kabra PM, Booth BE, Al-Bander HA, Portale AA, Serena BG, et al. Liquid-chromatographic measurements of inosine, hypoxanthine, and xanthine in studies of fructose-induced degradation of adenine nucleotides in humans and rats. Clinical chemistry. 1986;32(5):782–6.
Bawden SJ, Stephenson MC, Ciampi E, Hunter K, Marciani L, Macdonald IA, et al. Investigating the effects of an oral fructose challenge on hepatic ATP reserves in healthy volunteers: a P-31 MRS study. Clinical Nutrition. 2016;35(3):645–9.
Lanaspa MA, Sanchez-Lozada LG, Choi Y-J, Cicerchi C, Kanbay M, Roncal-Jimenez CA, et al. Uric acid induces hepatic steatosis by generation of mitochondrial oxidative stress POTENTIAL ROLE IN FRUCTOSE-DEPENDENT AND -INDEPENDENT FATTY LIVER. Journal of Biological Chemistry. 2012;287(48):40732–44.
Softic S, Cohen DE, Kahn CR. Role of Dietary Fructose and hepatic De Novo Lipogenesis in fatty liver disease. Digestive Diseases and Sciences. 2016;61(5):1282–93.
Jegatheesan P, Beutheu S, Freese K, Waligora-Dupriet A-J, Nubret E, Butel M-J, et al. Preventive effects of citrulline on Western diet-induced non-alcoholic fatty liver disease in rats. British Journal of Nutrition. 2016;116(2):191–203.
Jegatheesan P, Beutheu S, Ventura G, Sarfati G, Nubret E, Kapel N, et al. Effect of specific amino acids on hepatic lipid metabolism in fructose-induced non-alcoholic fatty liver disease. Clinical Nutrition. 2016;35(1):175–82.
Ritze Y, Bardos G, Claus A, Ehrmann V, Bergheim I, Schwiertz A, et al. Lactobacillus rhamnosus GG protects against non-alcoholic fatty liver disease in mice. Plos One. 2014;9(1).
Sharpton SR, Schnabl B, Knight R, Loomba R. Current Concepts, Opportunities, and Challenges of Gut Microbiome-Based Personalized Medicine in nonalcoholic fatty liver disease. Cell Metabolism. 2021;33(1):21–32.
Sommer F, Baeckhed F. The gut microbiota - masters of host development and physiology. Nature Reviews Microbiology. 2013;11(4):227–38.
Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444(7121):860–7.
Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56(7):1761–72.
Cani PD, Bibiloni R, Knauf C, Neyrinck AM, Neyrinck AM, Delzenne NM, et al. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. 2008;57(6):1470–81.
Fei N, Bruneau A, Zhang X, Wang R, Wang J, Rabot S, et al. Endotoxin Producers Overgrowing in Human Gut Microbiota as the causative agents for nonalcoholic fatty liver disease. Mbio. 2020;11(1).
Loomba R, Seguritan V, Li W, Long T, Klitgord N, Bhatt A, et al. Gut microbiome-based metagenomic signature for non-invasive detection of Advanced Fibrosis in Human nonalcoholic fatty liver disease. Cell Metabolism. 2017;25(5):1054-+.
Lee G, You HJ, Bajaj JS, Joo SK, Yu J, Park S, et al. Distinct signatures of gut microbiome and metabolites associated with significant fibrosis in non-obese NAFLD. Nature Communications. 2020;11(1).
Frost F, Kacprowski T, Ruehlemann M, Pietzner M, Bang C, Franke A, et al. Long-term instability of the intestinal microbiome is associated with metabolic liver disease, low microbiota diversity, diabetes mellitus and impaired exocrine pancreatic function. Gut. 2021;70(3):522–30.
Blaak EE, Canfora EE, Theis S, Frost G, Groen AK, Mithieux G, et al. Short chain fatty acids in human gut and metabolic health. Benef Microbes. 2020;11(5):411–55.
den Besten G, Bleeker A, Gerding A, van Eunen K, Havinga R, van Dijk TH, et al. Short-chain fatty acids protect Against High-Fat Diet-Induced obesity via a PPARgamma-Dependent switch from lipogenesis to Fat Oxidation. Diabetes. 2015;64(7):2398–408.
Litvak Y, Byndloss MX, Tsolis RM, Baumler AJ. Dysbiotic Proteobacteria expansion: a microbial signature of epithelial dysfunction. Curr Opin Microbiol. 2017;39:1–6.
Litvak Y, Mon KKZ, Nguyen H, Chanthavixay G, Liou M, Velazquez EM, et al. Commensal Enterobacteriaceae protect against Salmonella colonization through Oxygen Competition. Cell Host Microbe. 2019;25(1):128–39 e5.
de Vos WM, Tilg H, Van Hul M, Cani PD. Gut microbiome and health: mechanistic insights. Gut. 2022;71(5):1020–32.
Aziz AA, Kenney LS, Goulet B, Abdel-Aal el S. Dietary starch type affects body weight and glycemic control in freely fed but not energy-restricted obese rats. J Nutr. 2009;139(10):1881–9.
Keenan MJ, Zhou J, McCutcheon KL, Raggio AM, Bateman HG, Todd E, et al. Effects of resistant starch, a non-digestible fermentable fiber, on reducing body fat. Obesity (Silver Spring). 2006;14(9):1523–34.
Zhou J, Martin RJ, Tulley RT, Raggio AM, McCutcheon KL, Shen L, et al. Dietary resistant starch upregulates total GLP-1 and PYY in a sustained day-long manner through fermentation in rodents. Am J Physiol Endocrinol Metab. 2008;295(5):E1160-6.
Cho I, Yamanishi S, Cox L, Methe BA, Zavadil J, Li K, et al. Antibiotics in early life alter the murine colonic microbiome and adiposity. Nature. 2012;488(7413):621–6.
Le Roy T, de Hase EM, Van Hul M, Paquot A, Pelicaen R, Regnier M, et al. Dysosmobacter welbionis is a newly isolated human commensal bacterium preventing diet-induced obesity and metabolic disorders in mice. Gut. 2022;71(3):534–43.
Massafra V, Pellicciari R, Gioiello A, van Mil SWC. Progress and challenges of selective farnesoid X receptor modulation. Pharmacology & Therapeutics. 2018;191:162–77.
Chavez-Talavera O, Tailleux A, Lefebvre P, Staels B. Bile acid control of metabolism and inflammation in obesity, type 2 diabetes, Dyslipidemia, and nonalcoholic fatty liver disease. Gastroenterology. 2017;152(7):1679-+.
Jung Y, Koo BK, Jang SY, Kim D, Lee H, Lee DH, et al. Association between circulating bile acid alterations and nonalcoholic steatohepatitis independent of obesity and diabetes mellitus. Liver International. 2021;41(12):2892–902.
Chiang JYL, Ferrell JM. Bile acids as metabolic regulators and nutrient sensors. Annu Rev Nutr. 2019;39:175–200.
Rao A, Kosters A, Mells JE, Zhang W, Setchell KDR, Amanso AM, et al. Inhibition of ileal bile acid uptake protects against nonalcoholic fatty liver disease in high-fat diet-fed mice. Science Translational Medicine. 2016;8(357).
Takahashi S, Fukami T, Masuo Y, Brocker CN, Xie C, Krausz KW, et al. Cyp2c70 is responsible for the species difference in bile acid metabolism between mice and humans. Journal of Lipid Research. 2016;57(12):2130–7.
Jia W, Wei M, Rajani C, Zheng X. Targeting the alternative bile acid synthetic pathway for metabolic diseases. Protein & Cell. 2021;12(5):411–25.
Zhang Y, Jiang R, Zheng X, Lei S, Huang F, Xie G, et al. Ursodeoxycholic acid accelerates bile acid enterohepatic circulation. British Journal of Pharmacology. 2019;176(16):2848–63.
Bertaggia E, Jensen KK, Castro-Perez J, Xu Y, Di Paolo G, Chan RB, et al. Cyp8b1 ablation prevents Western diet-induced weight gain and hepatic steatosis because of impaired fat absorption. American Journal of Physiology-Endocrinology and Metabolism. 2017;313(2):E121-E33.
Torra IP, Claudel T, Duval C, Kosykh V, Fruchart JC, Staels B. Bile acids induce the expression of the human peroxisome proliferator-activated receptor alpha gene via activation of the farnesoid X receptor. Molecular Endocrinology. 2003;17(2):259–72.
Watanabe M, Houten SM, Wang L, Moschetta A, Mangelsdorf DJ, Heyman RA, et al. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. Journal of Clinical Investigation. 2004;113(10):1408–18.
Savkur RS, Bramlett KS, Michael LF, Burris TP. Regulation of pyruvate dehydrogenase kinase expression by the farnesoid X receptor. Biochemical and Biophysical Research Communications. 2005;329(1):391–6.
Li Y, Wong K, Walsh K, Gao B, Zang M. Retinoic acid receptor beta stimulates hepatic induction of fibroblast growth factor 21 to promote fatty acid oxidation and control whole-body energy homeostasis in mice. J Biol Chem. 2013;288(15):10490–504.
Badman MK, Pissios P, Kennedy AR, Koukos G, Flier JS, Maratos-Flier E. Hepatic fibroblast growth factor 21 is regulated by PPAR alpha and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metabolism. 2007;5(6):426–37.
Tomlinson E, Fu L, John L, Hultgren B, Huang XJ, Renz M, et al. Transgenic mice expressing human fibroblast growth factor-19 display increased metabolic rate and decreased adiposity. Endocrinology. 2002;143(5):1741–7.
Alvarez-Sola G, Uriarte I, Ujue Latasa M, Fernandez-Barrena MG, Urtasun R, Elizalde M, et al. Fibroblast growth factor 15/19 (FGF15/19) protects from diet-induced hepatic steatosis: development of an FGF19-based chimeric molecule to promote fatty liver regeneration. Gut. 2017;66(10):1818–28.
Fu L, John LM, Adams SH, Yu XX, Tomlinson E, Renz M, et al. Fibroblast growth factor 19 increases metabolic rate I and reverses dietary and leptlin-deficient diabetes. Endocrinology. 2004;145(6):2594–603.
Kir S, Beddow SA, Samuel VT, Miller P, Previs SF, Suino-Powell K, et al. FGF19 as a Postprandial, insulin-independent activator of hepatic protein and glycogen synthesis. Science. 2011;331(6024):1621–4.
Prawitt J, Abdelkarim M, Stroeve JHM, Popescu I, Duez H, Velagapudi VR, et al. Farnesoid X receptor Deficiency improves glucose homeostasis in mouse models of obesity. Diabetes. 2011;60(7):1861–71.
Zhang Y, Ge X, Heemstra LA, Chen W-D, Xu J, Smith JL, et al. Loss of FXR protects against Diet-Induced obesity and accelerates liver carcinogenesis in ob/ob mice. Molecular Endocrinology. 2012;26(2):272–80.
Watanabe M, Horai Y, Houten SM, Morimoto K, Sugizaki T, Arita E, et al. Lowering bile Acid Pool size with a synthetic farnesoid X receptor (FXR) agonist induces obesity and diabetes through reduced energy expenditure. J Biol Chem. 2011;286(30):26913–20.
Park YJ, Kim SC, Kim J, Anakk S, Lee JM, Tseng H-T, et al. Dissociation of diabetes and obesity in mice lacking orphan nuclear receptor small heterodimer partner. Journal of Lipid Research. 2011;52(12):2234–44.
Sayin SI, Wahlstrom A, Felin J, Jantti S, Marschall H-U, Bamberg K, et al. Gut microbiota regulates bile acid metabolism by reducing the levels of Tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metabolism. 2013;17(2):225–35.
Jiang C, Xie C, Li F, Zhang L, Nichols RG, Krausz KW, et al. Intestinal farnesoid X receptor signaling promotes nonalcoholic fatty liver disease. Journal of Clinical Investigation. 2015;125(1):386–402.
Xie C, Jiang C, Shi J, Gan X, Sun D, Sun L, et al. An intestinal farnesoid X Receptor-Ceramide Signaling Axis modulates hepatic gluconeogenesis in mice. Diabetes. 2017;66(3):613–26.
Trabelsi M-S, Daoudi M, Prawitt J, Ducastel S, Touche V, Sayin SI, et al. Farnesoid X receptor inhibits glucagon-like peptide-1 production by enteroendocrine L cells. Nature Communications. 2015;6.
Kawamata Y, Fujii R, Hosoya M, Harada M, Yoshida H, Miwa M, et al. A G protein-coupled receptor responsive to bile acids. J Biol Chem. 2003;278(11):9435–40.
Yuan L, Bambha K. Bile acid receptors and nonalcoholic fatty liver disease. World journal of hepatology. 2015;7(28):2811–8.
van Nierop FS, Scheltema MJ, Eggink HM, Pols TW, Sonne DP, Knop FK, et al. Clinical relevance of the bile acid receptor TGR5 in metabolism. Lancet Diabetes & Endocrinology. 2017;5(3):224–33.
Guo Z, Li M, Han B, Qi X. Association of non-alcoholic fatty liver disease with thyroid function: a systematic review and meta-analysis. Digestive and Liver Disease. 2018;50(11):1153–62.
Forrest D, Vennstrom B. Functions of thyroid hormone receptors in mice. Thyroid. 2000;10(1):41–52.
Lindemann JAL, Angajala A, Engler DA, Webb P, Ayers SD. Thyroid hormone induction of human cholesterol 7 alpha-hydroxylase (Cyp7a1) in vitro. Molecular and Cellular Endocrinology. 2014;388(1–2):32–40.
Ahn HY, Kim HH, Kim YA, Kim M, Ohn JH, Chung SS, et al. Thyroid hormone regulates the mRNA expression of small Heterodimer Partner through Liver receptor Homolog-1. Endocrinology and metabolism (Seoul, Korea). 2015;30(4):584–92.
Sinha RA, Bruinstroop E, Singh BK, Yen PM. Nonalcoholic fatty liver Disease and Hypercholesterolemia: roles of thyroid hormones, metabolites, and agonists. Thyroid. 2019;29(9):1173–91.
Markova M, Pivovarova O, Hornemann S, Sucher S, Frahnow T, Wegner K, et al. Isocaloric diets high in animal or plant protein reduce Liver Fat and inflammation in individuals with type 2 diabetes. Gastroenterology. 2017;152(3):571-+.
Orci LA, Gariani K, Oldani G, Delaune V, Morel P, Toso C. Exercise-based interventions for nonalcoholic fatty liver disease: a Meta-analysis and Meta-regression. Clinical Gastroenterology and Hepatology. 2016;14(10):1398–411.
Oh S, So R, Shida T, Matsuo T, Kim B, Akiyama K, et al. High-intensity Aerobic Exercise improves both hepatic Fat Content and Stiffness in Sedentary obese men with nonalcoholic fatty liver disease. Scientific reports. 2017;7:1–12.
Sung K-C, Ryu S, Lee J-Y, Kim J-Y, Wild SH, Byrne CD. Effect of exercise on the development of new fatty liver and the resolution of existing fatty liver. J Hepatol. 2016;65(4):791–7.
Oh S, Shida T, Yamagishi K, Tanaka K, So R, Tsujimoto T, et al. Moderate to vigorous physical activity volume is an important factor for managing nonalcoholic fatty liver disease: a retrospective study. Hepatology. 2015;61(4):1205–15.
Hashida R, Kawaguchi T, Bekki M, Omoto M, Matsuse H, Nago T, et al. Aerobic vs. resistance exercise in non-alcoholic fatty liver disease: a systematic review. J Hepatol. 2017;66(1):142–52.
Chavaez-Tapia NC, Tellez-Avila FI, Barrientos-Gutierrez T, Mendez-Sanchez N, Lizardi-Cervera J, Uribe M. Bariatric surgery for non-alcoholic steatohepatitis in obese patients. Cochrane Database of Systematic Reviews. 2010(1).
Lee Y, Doumouras AG, Yu J, Brar K, Banfield L, Gmora S, et al. Complete resolution of nonalcoholic fatty liver Disease after bariatric surgery: a systematic review and Meta-analysis. Clinical Gastroenterology and Hepatology. 2019;17(6):1040-+.
Aguilar-Olivos NE, Almeda-Valdes P, Aguilar-Salinas CA, Uribe M, Mendez-Sanchez N. The role of bariatric surgery in the management of nonalcoholic fatty liver disease and metabolic syndrome. Metabolism-Clinical and Experimental. 2016;65(8):1196–207.
Lawitz EJ, Poordad F, Coste A, Loo N, Djedjos CS, McColgan B, et al. Acetyl-CoA carboxylase (ACC) inhibitor GS-0976 leads to suppression of hepatic de novo lipogenesis and significant improvements in MRI-PDFF, MRE, and markers of fibrosis after 12 weeks of therapy in patients with NASH. J Hepatol. 2017;66(1):S34-S.
Calle RA, Amin NB, Carvajal-Gonzalez S, Ross TT, Bergman A, Aggarwal S, et al. ACC inhibitor alone or co-administered with a DGAT2 inhibitor in patients with non-alcoholic fatty liver disease: two parallel, placebo-controlled, randomized phase 2a trials. Nat Med. 2021;27(10):1836-+.
Loomba R, Mohseni R, Lucas KJ, Gutierrez JA, Perry RG, Trotter JF, et al. TVB-2640 (FASN inhibitor) for the treatment of nonalcoholic steatohepatitis: FASCINATE-1, a Randomized, Placebo-Controlled Phase 2a Trial. Gastroenterology. 2021;161(5):1475–86.
Safadi R, Konikoff FM, Mahamid M, Zelber-Sagi S, Halpern M, Gilat T, et al. The fatty acid-bile Acid Conjugate Aramchol reduces Liver Fat Content in patients with nonalcoholic fatty liver disease. Clinical Gastroenterology and Hepatology. 2014;12(12):2085-U365.
Ratziu V, de Guevara L, Safadi R, Poordad F, Fuster F, Flores-Figueroa J, et al. Aramchol in patients with nonalcoholic steatohepatitis: a randomized, double-blind, placebo-controlled phase 2b trial. Nat Med. 2021;27(10):1825-+.
Yen C-LE, Stone SJ, Koliwad S, Harris C, Farese RV, Jr. DGAT enzymes and triacylglycerol biosynthesis. Journal of Lipid Research. 2008;49(11):2283–301.
Gluchowski NL, Gabriel KR, Chitraju C, Bronson RT, Mejhert N, Boland S, et al. Hepatocyte deletion of triglyceride-synthesis enzyme Acyl CoA: Diacylglycerol Acyltransferase 2 reduces steatosis without increasing inflammation or fibrosis in mice. Hepatology. 2019;70(6):1972–85.
Saxena A, Chidsey K, Somayaji V, Ogden A, Duvvuri S. DIACYLGLYCEROL ACYLTRANSFERASE 2 (DGAT2) INHIBITOR PF-06865571 REDUCES LIVER FAT BY MRI-PDFF AFTER 2 WEEKS IN ADULTS WITH NAFLD. Hepatology. 2019;70:1260A-A.
Amin NB, Darekar A, Anstee QM, Wong VW, Tacke F, Vourvahis M, et al. Efficacy and safety of an orally administered DGAT2 inhibitor alone or coadministered with a liver-targeted ACC inhibitor in adults with non-alcoholic steatohepatitis (NASH): rationale and design of the phase II, dose-ranging, dose-finding, randomised, placebo-controlled MIRNA (metabolic interventions to Resolve NASH with fibrosis) study. BMJ Open. 2022;12(3):e056159.
Yen CLE, Farese RV. MGAT2, a monoacylglycerol acyltransferase expressed in the small intestine. J Biol Chem. 2003;278(20):18532–7.
Cao J, Lockwood J, Burn P, Shi Y. Cloning and functional characterization of a mouse intestinal acyl-CoA:monoacylglycerol acyltransferase, MGAT2. The Journal of biological chemistry. 2003;278(16):13860–6.
Julien B, Grenard P, Teixeira-Clerc F, Van Nhieu JT, Li LY, Karsak M, et al. Antifibrogenic role of the cannabinoid receptor CB2 in the liver. Gastroenterology. 2005;128(3):742–55.
Cheng D, Zinker BA, Luo Y, Shipkova P, De Oliveira CH, Krishna G, et al. MGAT2 inhibitor decreases liver fibrosis and inflammation in murine NASH models and reduces body weight in human adults with obesity. Cell Metab. 2022;34(11):1732–48 e5.
Lee JI, Lee HW, Lee KS, Lee HS, Park JY. Effects of Statin Use on the development and progression of nonalcoholic fatty liver disease: a Nationwide Nested Case-Control Study. Am J Gastroenterol. 2021;116(1):116–24.
Sfikas G, Psallas M, Koumaras C, Imprialos K, Perdikakis E, Doumas M, et al. Prevalence, diagnosis, and treatment with 3 different statins of non-alco- holic fatty liver Disease/Non-alcoholic steatohepatitis in military Person- nel. Do Genetics play a role? Current Vascular Pharmacology. 2021;19(5):572–81.
Han KH, Rha SW, Kang HJ, Bae JW, Choi BJ, Choi SY, et al. Evaluation of short-term safety and efficacy of HMG-CoA reductase inhibitors in hypercholesterolemic patients with elevated serum alanine transaminase concentrations: PITCH study (PITavastatin versus atorvastatin to evaluate the effect on patients with hypercholesterolemia and mild to moderate hepatic damage). J Clin Lipidol. 2012;6(4):340–51.
Nakahara T, Hyogo H, Kimura Y, Ishitobi T, Arihiro K, Aikata H, et al. Efficacy of rosuvastatin for the treatment of non-alcoholic steatohepatitis with dyslipidemia: an open-label, pilot study. Hepatology Research. 2012;42(11):1065–72.
Pineda C, Rios R, Raya AI, Rodriguez M, Aguilera-Tejero E, Lopez I. Hypocaloric Diet prevents the decrease in FGF21 elicited by high Phosphorus Intake. Nutrients. 2018;10(10).
Kliewer SA, Mangelsdorf DJ. A Dozen Years of Discovery: insights into the physiology and pharmacology of FGF21. Cell Metab. 2019;29(2):246–53.
Xu J, Lloyd DJ, Hale C, Stanislaus S, Chen M, Sivits G, et al. Fibroblast growth factor 21 reverses hepatic steatosis, increases energy expenditure, and improves insulin sensitivity in diet-induced obese mice. Diabetes. 2009;58(1):250–9.
Gong Q, Hu Z, Zhang F, Cui A, Chen X, Jiang H, et al. Fibroblast growth factor 21 improves hepatic insulin sensitivity by inhibiting mammalian target of rapamycin complex 1 in mice. Hepatology. 2016;64(2):425–38.
Sanyal A, Charles ED, Neuschwander-Tetri BA, Loomba R, Harrison SA, Abdelmalek MF, et al. Pegbelfermin (BMS-986036), a PEGylated fibroblast growth factor 21 analogue, in patients with non-alcoholic steatohepatitis: a randomised, double-blind, placebo-controlled, phase 2a trial. Lancet. 2019;392(10165):2705–17.
Abdelmalek MF, Charles ED, Sanyal AJ, Harrison SA, Neuschwander-Tetri BA, Goodman Z, et al. The FALCON program: two phase 2b randomized, double-blind, placebo-controlled studies to assess the efficacy and safety of pegbelfermin in the treatment of patients with nonalcoholic steatohepatitis and bridging fibrosis or compensated cirrhosis. Contemporary Clinical Trials. 2021;104.
Cui A, Li J, Ji S, Ma F, Wang G, Xue Y, et al. The Effects of B1344, a novel fibroblast growth factor 21 Analog, on nonalcoholic steatohepatitis in Nonhuman Primates. Diabetes. 2020;69(8):1611–23.
Boeckmans J, Natale A, Rombaut M, Buyl K, Rogiers V, De Kock J, et al. Anti-NASH Drug Development hitches a lift on PPAR agonism. Cells. 2020;9(1).
Gross B, Pawlak M, Lefebvre P, Staels B. PPARs in obesity-induced T2DM, dyslipidaemia and NAFLD. Nature Reviews Endocrinology. 2017;13(1):36–49.
Mayerson AB, Hundal RS, Dufour S, Lebon V, Befroy D, Cline GW, et al. The effects of rosiglitazone on insulin sensitivity, lipolysis, and hepatic and skeletal muscle triglyceride content in patients with type 2 diabetes. Diabetes. 2002;51(3):797–802.
Galli A, Crabb DW, Ceni E, Salzano R, Mello T, Svegliati-Baroni G, et al. Antidiabetic thiazolidinediones inhibit collagen synthesis and hepatic stellate cell activation in vivo and in vitro. Gastroenterology. 2002;122(7):1924–40.
Belfort R, Harrison SA, Brown K, Darland C, Finch J, Hardies J, et al. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. New England Journal of Medicine. 2006;355(22):2297–307.
Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma). The Journal of biological chemistry. 1995;270(22):12953–6.
Juurlink DN, Gomes T, Lipscombe LL, Austin PC, Hux JE, Mamdani MM. Adverse cardiovascular events during treatment with pioglitazone and rosiglitazone: population based cohort study. BMJ. 2009;339:b2942.
Staels B, Rubenstrunk A, Noel B, Rigou G, Delataille P, Millatt LJ, et al. Hepatoprotective Effects of the dual peroxisome proliferator-activated receptor Alpha/Delta agonist, GFT505, in Rodent Models of nonalcoholic fatty liver Disease/Nonalcoholic steatohepatitis. Hepatology. 2013;58(6):1941–52.
Cariou B, Hanf R, Lambert-Porcheron S, Zair Y, Sauvinet V, Noel B, et al. Dual peroxisome proliferator-activated receptor alpha/delta agonist GFT505 improves hepatic and peripheral insulin sensitivity in abdominally obese subjects. Diabetes Care. 2013;36(10):2923–30.
Francque SM, Bedossa P, Ratziu V, Anstee QM, Bugianesi E, Sanyal AJ, et al. A Randomized, Controlled Trial of the Pan-PPAR agonist Lanifibranor in NASH. N Engl J Med. 2021;385(17):1547–58.
Heerspink HJL, Perkins BA, Fitchett DH, Husain M, Cherney DZI. Sodium glucose cotransporter 2 inhibitors in the treatment of diabetes Mellitus: Cardiovascular and kidney Effects, potential mechanisms, and clinical applications. Circulation. 2016;134(10):752–72.
Aso Y, Kato K, Sakurai S, Kishi H, Shimizu M, Jojima T, et al. Impact of dapagliflozin, an SGLT2 inhibitor, on serum levels of soluble dipeptidyl peptidase-4 in patients with type 2 diabetes and non-alcoholic fatty liver disease. International Journal of Clinical Practice. 2019;73(5).
Latva-Rasku A, Honka M-J, Kullberg J, Mononen N, Lehtimaeki T, Saltevo J, et al. The SGLT2 inhibitor Dapagliflozin reduces Liver Fat but does not affect tissue insulin sensitivity: a Randomized, Double-Blind, placebo-controlled study with 8-Week treatment in type 2 diabetes patients. Diabetes Care. 2019;42(5):931–7.
Kahl S, Gancheva S, Strassburger K, Herder C, Machann J, Katsuyama H, et al. Empagliflozin effectively lowers Liver Fat Content in Well-Controlled type 2 diabetes: a Randomized, Double-Blind, phase 4, placebo-controlled trial. Diabetes Care. 2020;43(2):298–305.
Ueda P, Svanstrom H, Melbye M, Eliasson B, Svensson A-M, Franzen S, et al. Sodium glucose cotransporter 2 inhibitors and risk of serious adverse events: nationwide register based cohort study. Bmj-British Medical Journal. 2018;363.
Muller TD, Finan B, Bloom SR, D’Alessio D, Drucker DJ, Flatt PR, et al. Glucagon-like peptide 1 (GLP-1). Mol Metab. 2019;30:72–130.
Gastaldelli A, Gaggini M, Daniele G, Ciociaro D, Cersosimo E, Tripathy D, et al. Exenatide improves both hepatic and adipose tissue insulin resistance: a dynamic Positron Emission Tomography Study. Hepatology. 2016;64(6):2028–37.
Gimeno RE, Briere DA, Seeley RJ. Leveraging the gut to treat metabolic disease. Cell Metabolism. 2020;31(4):679–98.
Mells JE, Fu PP, Sharma S, Olson D, Cheng L, Handy JA, et al. Glp-1 analog, liraglutide, ameliorates hepatic steatosis and cardiac hypertrophy in C57BL/6J mice fed a Western diet. American Journal of Physiology-Gastrointestinal and Liver Physiology. 2012;302(2):G225-G35.
Armstrong MJ, Hull D, Guo K, Barton D, Hazlehurst JM, Gathercole LL, et al. Glucagon-like peptide 1 decreases lipotoxicity in non-alcoholic steatohepatitis. J Hepatol. 2016;64(2):399–408.
Ding XK, Saxena NK, Lin SB, Gupta N, Anania FA. Exendin-4, a glucagon-like protein-1 (GLP-1) receptor agonist, reverses hepatic steatosis in ob/ob mice. Hepatology. 2006;43(1):173–81.
Svegliati-Baroni G, Saccomanno S, Rychlicki C, Agostinelli L, De Minicis S, Candelaresi C, et al. Glucagon-like peptide-1 receptor activation stimulates hepatic lipid oxidation and restores hepatic signalling alteration induced bya high-fat diet in nonalcoholic steatohepatitis. Liver International. 2011;31(9):1285–97.
Alkhouri N, Herring R, Kabler H, Kayali Z, Hassanein T, Kohli A, et al. Safety and efficacy of combination therapy with semaglutide, cilofexor and firsocostat in patients with non-alcoholic steatohepatitis: a randomised, open-label phase II trial. J Hepatol. 2022.
Roehrborn D, Wronkowitz N, Eckel J. DPP4 in diabetes. Frontiers in Immunology. 2015;6:1–20.
Williams KH, De Ribeiro AJV, Prakoso E, Veillard A-S, Shackel NA, Brooks B, et al. Circulating dipeptidyl peptidase-4 activity correlates with measures of hepatocyte apoptosis and fibrosis in non-alcoholic fatty liver disease in type 2 diabetes mellitus and obesity: a dual cohort cross-sectional study. Journal of Diabetes. 2015;7(6):809–19.
Kawakubo M, Tanaka M, Ochi K, Watanabe A, Saka-Tanaka M, Kanamori Y, et al. Dipeptidyl peptidase-4 inhibition prevents nonalcoholic steatohepatitis-associated liver fibrosis and tumor development in mice independently of its anti-diabetic effects. Scientific reports. 2020;10(1).
Joy TR, McKenzie CA, Tirona RG, Summers K, Seney S, Chakrabarti S, et al. Sitagliptin in patients with non-alcoholic steatohepatitis: a randomized, placebo-controlled trial. World Journal of Gastroenterology. 2017;23(1):141–50.
Jang C, Wada S, Yang S, Gosis B, Zeng X, Zhang Z, et al. The small intestine shields the liver from fructose-induced steatosis. Nature Metabolism. 2020;2(7):586-+.
Andres-Hernando A, Orlicky DJ, Kuwabara M, Ishimoto T, Nakagawa T, Johnson RJ, et al. Deletion of Fructokinase in the liver or in the intestine reveals Differential Effects on Sugar-Induced metabolic dysfunction. Cell Metabolism. 2020;32(1):117-+.
Calle R, Bergman A, Somayaji V, Chidsey K, Kazierad D. Ketohexokinase inhibitor PF-06835919 administered for 6 weeks reduces whole liver fat as measured by magnetic resonance imaging-proton density fat fraction in subjects with non-alcoholic fatty liver disease. J Hepatol. 2019;70(1):E69-E70.
Craven L, Rahman A, Nair Parvathy S, Beaton M, Silverman J, Qumosani K, et al. Allogenic fecal microbiota transplantation in patients with nonalcoholic fatty liver Disease improves abnormal small intestinal permeability: a Randomized Control Trial. Am J Gastroenterol. 2020;115(7):1055–65.
Koopen A, Witjes J, Wortelboer K, Majait S, Prodan A, Levin E, et al. Duodenal anaerobutyricum soehngenii infusion stimulates GLP-1 production, ameliorates glycaemic control and beneficially shapes the duodenal transcriptome in metabolic syndrome subjects: a randomised double-blind placebo-controlled cross-over study. Gut. 2022;71(8):1577–87.
Younossi ZM, Ratziu V, Loomba R, Rinella M, Anstee QM, Goodman Z, et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet. 2019;394(10215):2184–96.
Patel K, Harrison SA, Elkhashab M, Trotter JF, Herring R, Rojter SE, et al. Cilofexor, a NonsteroidalFXRAgonist, in patients with Noncirrhotic NASH: a phase 2 Randomized Controlled Trial. Hepatology. 2020;72(1):58–71.
Ratziu V, Rinella ME, Neuschwander-Tetri BA, Lawitz E, Denham D, Kayali Z, et al. EDP-305 in patients with NASH: a phase II double-blind placebo-controlled dose-ranging study. J Hepatol. 2022;76(3):506–17.
Harrison SA, Bashir MR, Guy CD, Zhou R, Moylan CA, Frias JP, et al. Resmetirom (MGL-3196) for the treatment of non-alcoholic steatohepatitis: a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet. 2019;394(10213):2012–24.
Harrison SA, Neff G, Guy CD, Bashir MR, Paredes AH, Frias JP, et al. Efficacy and safety of Aldafermin, an Engineered FGF19 Analog, in a Randomized, Double-Blind, placebo-controlled trial of patients with nonalcoholic steatohepatitis. Gastroenterology. 2021;160(1):219-+.
Harrison SA, Abdelmalek MF, Neff G, Gunn N, Guy CD, Alkhouri N, et al. Aldafermin in patients with non-alcoholic steatohepatitis (ALPINE 2/3): a randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Gastroenterol Hepatol. 2022.
Kargiotis K, Katsiki N, Athyros VG, Giouleme O, Patsiaoura K, Katsiki E, et al. Effect of rosuvastatin on non-alcoholic steatohepatitis in patients with metabolic syndrome and hypercholesterolaemia: a preliminary report. Curr Vasc Pharmacol. 2014;12(3):505–11.
Jain MR, Giri SR, Bhoi B, Trivedi C, Ranvir R, Kadam S, et al. Saroglitazar shows therapeutic benefits in mouse model of nonalcoholic fatty liver Disease (NAFLD) and nonalcoholic steatohepatitis (NASH). Diabetes. 2015;64:A503-A.
Scorletti E, Afolabi PR, Miles EA, Smith DE, Almehmadi A, Alshathry A, et al. Synbiotics Alter Fecal Microbiomes, but Not Liver Fat or Fibrosis, in a Randomized Trial of patients with nonalcoholic fatty liver disease. Gastroenterology. 2020;158(6):1597–610 e7.
Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol. 2017;14(7):397–411.
Samuel VT, Shulman GI. Nonalcoholic fatty liver disease as a Nexus of metabolic and hepatic Diseases. Cell Metab. 2018;27(1):22–41.
Wang T, Chen K, Yao W, Zheng R, He Q, Xia J, et al. Acetylation of lactate dehydrogenase B drives NAFLD progression by impairing lactate clearance. J Hepatol. 2021;74(5):1038–52.
Diggle CP, Shires M, Leitch D, Brooke D, Carr IM, Markham AF, et al. Ketohexokinase: expression and localization of the principal fructose-metabolizing enzyme. J Histochem Cytochem. 2009;57(8):763–74.
Debray FG, Seyssel K, Fadeur M, Tappy L, Paquot N, Tran C. Effect of a high fructose diet on metabolic parameters in carriers for hereditary fructose intolerance. Clin Nutr. 2021;40(6):4246–54.
Lanaspa MA, Sanchez-Lozada LG, Choi YJ, Cicerchi C, Kanbay M, Roncal-Jimenez CA, et al. Uric acid induces hepatic steatosis by generation of mitochondrial oxidative stress: potential role in fructose-dependent and -independent fatty liver. The Journal of biological chemistry. 2012;287(48):40732–44.
Axelson M, Mork B, Sjovall J. Occurrence of 3 beta-hydroxy-5-cholestenoic acid, 3 beta,7 alpha-dihydroxy-5-cholestenoic acid, and 7 alpha-hydroxy-3-oxo-4-cholestenoic acid as normal constituents in human blood. J Lipid Res. 1988;29(5):629–41.
Durward QJ, Cohen MM, Naiman SC. Intramural hematoma of the gastric cardia. Am J Gastroenterol. 1979;71(3):301–5.
Martin KO, Reiss AB, Lathe R, Javitt NB. 7 alpha-hydroxylation of 27-hydroxycholesterol: biologic role in the regulation of cholesterol synthesis. J Lipid Res. 1997;38(5):1053–8.
Shneider BL, Dawson PA, Christie DM, Hardikar W, Wong MH, Suchy FJ. Cloning and molecular characterization of the ontogeny of a rat ileal sodium-dependent bile acid transporter. J Clin Invest. 1995;95(2):745–54.
Rao A, Haywood J, Craddock AL, Belinsky MG, Kruh GD, Dawson PA. The organic solute transporter alpha-beta, Ostalpha-Ostbeta, is essential for intestinal bile acid transport and homeostasis. Proc Natl Acad Sci U S A. 2008;105(10):3891–6.
Dawson PA, Haywood J, Craddock AL, Wilson M, Tietjen M, Kluckman K, et al. Targeted deletion of the ileal bile acid transporter eliminates enterohepatic cycling of bile acids in mice. The Journal of biological chemistry. 2003;278(36):33920–7.
Goodwin B, Jones SA, Price RR, Watson MA, McKee DD, Moore LB, et al. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol Cell. 2000;6(3):517–26.
Zhang M, Chiang JY. Transcriptional regulation of the human sterol 12alpha-hydroxylase gene (CYP8B1): roles of heaptocyte nuclear factor 4alpha in mediating bile acid repression. The Journal of biological chemistry. 2001;276(45):41690–9.
Watanabe M, Houten SM, Wang L, Moschetta A, Mangelsdorf DJ, Heyman RA, et al. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J Clin Invest. 2004;113(10):1408–18.
Claudel T, Inoue Y, Barbier O, Duran-Sandoval D, Kosykh V, Fruchart J, et al. Farnesoid X receptor agonists suppress hepatic apolipoprotein CIII expression. Gastroenterology. 2003;125(2):544–55.
Pineda Torra I, Claudel T, Duval C, Kosykh V, Fruchart JC, Staels B. Bile acids induce the expression of the human peroxisome proliferator-activated receptor alpha gene via activation of the farnesoid X receptor. Mol Endocrinol. 2003;17(2):259–72.
Song KH, Li T, Owsley E, Strom S, Chiang JY. Bile acids activate fibroblast growth factor 19 signaling in human hepatocytes to inhibit cholesterol 7alpha-hydroxylase gene expression. Hepatology. 2009;49(1):297–305.
Katsuma S, Hirasawa A, Tsujimoto G. Bile acids promote glucagon-like peptide-1 secretion through TGR5 in a murine enteroendocrine cell line STC-1. Biochem Biophys Res Commun. 2005;329(1):386–90.
Thomas C, Gioiello A, Noriega L, Strehle A, Oury J, Rizzo G, et al. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 2009;10(3):167–77.
Chambers ES, Preston T, Frost G, Morrison DJ. Role of gut microbiota-generated short-chain fatty acids in metabolic and Cardiovascular Health. Curr Nutr Rep. 2018;7(4):198–206.
Leung C, Rivera L, Furness JB, Angus PW. The role of the gut microbiota in NAFLD. Nat Rev Gastroenterol Hepatol. 2016;13(7):412–25.
Acknowledgements
We gratefully thank to the “Key Laboratory of Cell Differentiation and Apoptosis of Chinese Ministry of Education” and “Shanghai Frontiers Science Center of Cellular Homeostasis and Human Diseases” for supporting the research platforms.
Funding
This study was supported by the following grants to Junli Liu: National Key R&D Program of China (2021YFA0804800, 2018YFA0800600); Shanghai Municipal Commission of Science and Technology (NO. 20410713200; NO. 21S11909000); National Facility for Translational Medicine (Shanghai) (TMSK-2020-102); Shanghai Municipal Education Commission (20SG10); and Training Program of the Major Research Plan of the National Natural Science Foundation (NO. 91857111), China; Innovative research team of high-level local universities in Shanghai (SHSMU-ZDCX20212501). This work was also supported by the following grants to Liu Han: China National Postdoctoral Program for Innovative Talents (BX2021191); China Postdoctoral Science Foundation (NO. 2021M702168); Shanghai Sixth People’s Hospital (ynqn202104). This work was also supported by the following grants to Suzhen Chen: National Natural Science Foundation of China (NO. 82170863); Shanghai Rising-Star Program (NO. 21QA1407000); Lingang Laboratory (NO. LG-QS-202205-06).
Author information
Authors and Affiliations
Contributions
YS conceptualized the review with help from JL and LH; YS wrote the original draft and interpretated the data with the assistance from LH, SC and JL; JL, SC, and LH provided fundings; YS revised the review with the help from JL and LH. All authors contributed to the article and approved the submitted version.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Informed consent and patient details
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Shao, Y., Chen, S., Han, L. et al. Pharmacotherapies of NAFLD: updated opportunities based on metabolic intervention. Nutr Metab (Lond) 20, 30 (2023). https://doi.org/10.1186/s12986-023-00748-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12986-023-00748-x