
Abstract
Feline calicivirus (FCV) is a highly contagious virus in cats, which typically causes respiratory tract and oral infections. Despite vaccination against FCV being a regular practice in China, new FCV cases still occur. Antigenic diversity of FCV hinders the effective control by vaccination. This is first report which aims to investigate the molecular epidemiology and molecular characteristics of FCV in Kunshan, China. The nasopharyngeal swabs were collected from cats showing variable clinical signs from different animal clinics in Kunshan from 2022 to 2023. Preliminary detection and sequencing of the FCV capsid gene were performed to study genetic diversity and evolutionary characteristics. FCV-RNA was identified in 52 (26%) of the samples using RT-PCR. A significant association was found between FCV-positive detection rate, age, gender, vaccination status and living environment, while a non-significant association was found with breed of cats. Nucleotide analysis revealed two genotypes, GI and GII. GII predominated in Kunshan, with diverse strains and amino acid variations potentially affecting vaccination efficacy and FCV detection. Notably, analysis pinpointed certain strains’ association with FCV-virulent systemic disease pathotypes. This investigation sheds light on FCV dynamics, which may aid in developing better prevention strategies and future vaccine designs against circulating FCV genotypes.
Similar content being viewed by others


Introduction
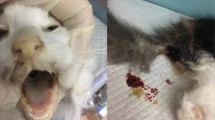
Feline calicivirus (FCV), a member of the Caliciviridae family and Vesivirus genus, is a highly contagious pathogen affecting both domestic cats and wild felines worldwide [1]. FCV was first reported in 1957 and has since been isolated in many countries in Asia, America, and Europe [2,3,4]. Typical clinical syndromes of FCV infection include upper respiratory tract disease (URTD), oral ulcers, conjunctivitis, rhinitis, fever, diarrhea, and lethargy [5,6,7]. Without vaccine protection kittens are more susceptible to severe pneumonia following FCV infection [1, 8]. Nevertheless, new virulent mutants of FCV (VS-FCVs) have become increasingly prevalent worldwide in recent years [4, 9]. In contrast to classical FCV infection, VS-FCVs is associated with severe virulent systemic diseases (VSD), including persistent fever, limb edema, bronchitis, and pneumonia [4, 10]. Even adult or vaccinated cats can be infected by these strains and die [1, 4, 11]. Even months or years after recovery from FCV infection, these asymptomatic carrier cats can shed the virus, leading to more epidemics of FCV [12, 13].
The FCV virus has a small, non-enveloped, single-stranded RNA genome approximately 7700 bp in length. Its genome of positive polarity allows the virus to evolve quickly, and its structure, which lacks a lipid envelope, helps it persist in the environment for a relatively long time [1]. FCV contain three functional open reading frames (ORFs): ORF1, ORF2, and ORF3. ORF1 encodes a polyprotein that is post-translationally cleaved into non-structural proteins such as proteases and polymerase [1]. ORF2 encodes a polyprotein that is subsequently processed to produce the major capsid protein (VP1), and can be divided into six regions, A to F, based on their amino acid sequence variability [1, 14]. ORF3 encodes the smaller capsid protein (VP2), which is vital for replication, viral particle assembly, and delivery of the FCV genome into its host cells [15]. In ORF2, genomic regions A, B, D, and F are relatively conserved, whereas regions C and E are more variable [14].
The VP1 protein is the main component of structural proteins in FCV. More importantly, the variable regions of VP1 have one of the highest rates of molecular evolution ever recorded [12]. Conventionally, phylogenetic analysis was performed on VP1 gene [10]. Considering the genetic diversity of the VP1 gene, FCVs worldwide can be classified into two groups: genogroup I and genogroup II [5, 16]. According to previous studies, most FCV strains found in China belong to genogroup II [11]. Therefore, to gain a better understanding of the epidemiology and pathogenesis of FCV it is vital to understand the genetic diversity and characterization of VP1.
In addition, the study of capsid proteins can largely contribute to the development of effective FCV vaccines. The best defense against this virus has always been vaccination. However, with an increasing number of new FCV variant strains frequently emerging in recent years, traditional and existing vaccines are no longer able to provide sufficient protection against them [10]). The aim of this investigation was to explore the epidemiological status and molecular characteristics of FCV in Kunshan, Jiangsu Province, China, as well as to further improve the theoretical basis for the development of new FCV vaccines and better strategies for the prevention and control of FCV. The situation of FCV in Kunshan has not been reported before; thus, this is the first report on the genetic and phylogenetic analysis of FCV strains in Kunshan, China.
Materials and methods
Sample collection
Ethical approval from the Institutional Animal Care and Use Committee (IACUC) was not needed for this study as long as the animals investigated in this study were not identifiable in the retrospective records. A total of 200 nasopharyngeal swab samples were collected from cats using sterile cotton swabs from nine animal clinics in Kunshan from June 2022 to June 2023 (Fig. 1). Nasopharyngeal swab from a single cat were put into one 5 ml sterile tube containing Dulbecco’s Modified Eagle’s Medium (DMEM, Thermo Fisher Scientific, San Jose, CA, USA). Clinic staff recorded data on the breed, age, sex, clinical symptoms, residence, and vaccination status of all the sampled cats. The samples were then immediately transported on wet ice to the Laboratory. All specimens were stored at -80 °C for subsequent RT-PCR and sequence analysis.

Map showing sampling sites in Kunshan, China
RNA extraction and RT-PCR screening for FCV
RNA was extracted from the swabs, and complementary DNA (cDNA) strands were synthesized from RNA templates using commercially available kits for viral RNA extraction and reverse transcription (TaKaRa MiniBEST Viral RNA/DNA Extraction Kit, Takara, Dalian, China). Preliminary screening of FCV in the samples was performed using FCV-F1 (5′-GTTGACCCTTACTCATACAC-3′) and FCV-R1 (5′-CCCTGGGGTTAGGCGC-3′) as previously described [17]. RT-PCR was performed to amplify and detect a small segment of the VP1 gene using the above-mentioned primers, 2x rapid Taq master mix (Cat# P222, Vazyme, Nanjing, China). and LifeEco Bioer Thermal Cycler (LifeEco TC 96, Bioer, Hangzhou, China). The following thermocycling conditions were used for amplification: 95 °C for 5 min, followed by 40 cycles at 95 °C for 30 s, 52 °C for 30 s, and 72 °C for 30 s, and a 5 min final extension at 72 °C. PCR amplicons were analyzed on 2% agarose gels and approximately 132 bp of PCR amplicons were observed using the GenoSens 1880 gel imaging analysis system (GenoSens 1880, Clinx, Shanghai, China). FCV positive specimens were also tested for coinfection of feline herpesvirus.
VP1 gene sequencing for mutation detection
To enhance the specificity of the PCR, we slightly modified previously reported primer combinations that amplified the partial length of ORF2 (1980 bp) [12, 17]. The forward and reverse primers were 5′-CCTHCACTGTGATGTGTTCGA-3′ and 5′-GAATTCCCATGTAGGAGGC-3′, respectively. The VP1 gene of the positive samples was amplified using the following protocol: 95 °C for 3 min, 45 cycles of 95 °C for 30 s, 58 °C for 30 s, 72 °C for 2 min, and 72 °C for 10 min. All PCR runs contained a negative template control (nuclease-free water) and a corresponding synthetic positive control sample. PCR was performed using a two-step RT-PCR kit (Takara, Dalian, China). Positive PCR products were sequenced using Sanger sequencing methods.
Sequence analysis
Nucleotide sequences were analysed, edited, and aligned for phylogenetic analysis using Bio Edit Software version 7.2 9 (Ibis Biosciences, Carlsbad, CA, USA). Sequence electropherograms were carefully analysed, and nucleotide ambiguities were excluded. To make the sequencing data more reliable, we aligned the forward and reverse sequences together to generate a consensus sequence. Multiple sequences were assembled and aligned using ClustalW version 2.0. To understand the molecular epidemiology of the identified FCV in this study, reference sequences were retrieved from the National Centre for Biotechnology Information (NCBI) nucleotide database (http://www.ncbi.nlm.nih.gov) to infer the overall detected virus phylogeny as of July 2023. All sequences were trimmed and aligned according to the VP1 gene. Molecular Evolutionary Genetic Analysis (MEGA version 11.0) was used to construct and analyse the phylogenetic tree [18]. Phylogenetic trees were constructed using the Neighbour Joining method with the Kimura 2-parameter model. These sequences were deposited in GenBank (accession numbers OR472393–OR472444).
Statistical analysis
Association analyses of FCV infection with gender, living environment, breed, vaccination status and age were performed using the Chi-square test in SPSS V. 19.0. A multi-cat environment was defined as the number of cats of ≥ 2. A value of p < 0.05 and p > 0.05 were considered statistically significant and non-significant, respectively.
Results
Detection of FCV, signalment and clinical findings
Of the 200 nasopharyngeal swab specimens, 52 (52/200, 26%) were positive for FCV, while 148 (148/200, 74%) were negative for FCV. Among these 52, 17 (32.7%) were vaccinated cats and 35 (67.3%) were non-vaccinated cats. Among the 52 positive specimens, 45 (86.5%) were from cats aged below 1 year, and 7 (13.5%) were from cats older than 1 year. A total of 24 (46.1%) and 28 (53.9%) specimens were positive for FCV in male (Tom) and female (Queen) cats, respectively (Tables 1 and 2). Detection rates of 63.5% (33/52) and 36.5% (19/52) were observed in cats living in groups and singly, respectively. Various clinical signs were observed in cats, including respiratory distress, sneezing, coughing, lacrimation, nasal discharge, mouth ulcers, anorexia, and fever. The clinical signs such as nasal discharge, sneezing and mouth ulcers were generally more severe in non-vaccinated cats than in vaccinated cats (personal observation). Chi-square test analysis revealed a significant association (p < 0.05) between age, gender, living environment and vaccination status with FCV-positive detection rate, while a non-significant association was observed with breed of cats (p > 0.05) (Table 2). Eight FCV positive specimens were coinfected with feline herpesvirus. Mucopurulent ocular discharge was an additional clinical sign in coinfected cats and clinical parameters were more severe in mixed infections than in mono-infections.
Nucleotide homology analysis of FCV VP1 gene
All sequences (n = 52) were blasted against each other and previously reported sequences from the NCBI GenBank database. These sequences were compared for nucleotide homology within themselves using pairwise alignment using EMBOSS needle software. Pairwise comparative analysis revealed a homology of 69.2–100%, and 79.5–100% for nucleotide and deduced amino acid sequences, respectively, between the 52 FCV strains identified in this study. Several substitutions were observed in all sequences compared to the reference FCV strains (vaccine strain F9/F4/255). Similarly, sequence identity was 72.3–89.0%, and 81.5–93.7% between 52 FCV strains detected in this study and representative reference strains. The 52 new FCV strains detected in this study revealed 69.4–79.8%, 71.9–79.2%, and 73.5–78.4% nucleotide sequence homology, respectively, and 82.6– 90.3%, 80.7–91.4%, and 82.7–87.6% amino acid sequence identity, respectively, when compared with FCV vaccine strains F9 (M86379), F4 (D31836), and 255 (KM111171). FCV strains identified in this study revealed 72.3–83.2% nucleotide sequence identity and 83.7.4–91.5% amino acid sequence homology when compared with VSD (KM111557) representative strains respectively. Similarly, a nucleotide identity of 74.6–85.7% and amino acid sequence homology 84.1.4–93.2% were observed when compared with the ORD (AY560113) representative strain. The specific FCV strains from worldwide that shared the highest nucleotide identity with the 52 Kunshan FCV strains are shown in Table 3.
Phylogenetic analysis of VP1 gene
To reveal the evolutionary characteristics of FCV, we constructed a phylogenetic tree among 52 new FCV strains identified in this study and 43 reference FCV strains downloaded from the GenBank database. Phylogenetic analysis clustered the FCV strains into two genotypes, GI and GII, which are denoted by red and blue circles, respectively. A total of 37 sequences were clustered into the GII genotype clade, whereas only 15 sequences were clustered within the GI genotype clade. FCV-GII strains identified in this study demonstrated the closest genetic relationship with strains reported from different provinces in China (Shanghai, Fujian, Sichuan, Hubei, Shandong, and Nanjing). Interestingly, the FCV-G1 strains identified in this study were more closely related to the FCV strains previously reported in Beijing and Hubei (Fig. 2).

Phylogenetic trees were constructed based on ORF2 in the study. The phylogenetic tree was constructed using the neighbor-joining method for 1000 bootstrap values. The sequences identified in this study were labeled with red (GI) and blue (GII) circles
Deduced amino acid analysis of VP1 gene
Amino acid positions within the hypervariable region E of the VP1 protein were analyzed and compared with those of previously reported reference strains of FCV. ORF2 encodes the VP1 protein, which comprises 668–671 amino acid residues. The amino acid sites between positions 426 and 523 were designated region E of the VP1 protein. FCV GI and GII identified in this study varied at several amino acid sites within region E of the VP1 protein (Table 4). Notably, 52 FCV strains in the present study exhibited multiple site mutations in region E of the VP1 protein when compared with the vaccine strain F9 (M86379)(439 N → S/T, 441T → R/N/S, 446T → I, 448 A → S, 449T → K/N/S, 450G → E/Q, 453T → S/A, 455D → G, 465G → S, 492I → V, 521 K → S/E/T, 522 K → A/D/E/G, and 523 A → V/T). Furthermore, 52 new FCV strains identified in this study were compared with the VSD and ORD reference strains. Different amino acids were detected at seven virulence factor-related loci within the E region of the VP1 protein of 52 isolates (amino acid sites:438, 440, 448, 452,455,465, 492). A comparison of amino acids at seven virulence factor-related sites between FCV genotypes in this study and reference strains (M86379, D31836, KM111171, KM111557, and AY560113) is presented in Table 5.
Discussion
Respiratory viruses frequently cause severe diseases in cats and have a significant impact on morbidity and mortality worldwide. FCV is a common contagious virus that causes feline infectious diseases with poor outcomes, especially in nonvaccinated cats. Over the past few decades, FCV have become a serious burden to the health of cats [19]. FCV tends to mutate more often than other viruses, mainly because of the lack of exonuclease proofreading activity displayed by their RNA polymerases, which may render vaccines less effective against FCV infections [17, 20]. Therefore, it is imperative to understand the epidemiological patterns and genetic characteristics of FCV in order to implement better therapeutic and preventive measures. Although, FCV has been reported from several regions of China [10,11,12, 17, 20,21,22,23,24], there are relatively a few published reports on evolutionary characteristics of FCV. From an evolutionary perspective, this study aimed to investigate circulating strains of FCV in Kunshan.
In this study, 200 nasopharyngeal swabs were collected from cats in Kunshan and subjected to FCV detection. Overall, the FCV detection rate was 26%, which was higher than that reported in previous studies conducted in China [21, 24, 25]. In the FCV positive cats, 32.7% were vaccinated, similar to previously reported [11, 17, 26]. However, vaccinated cats generally exhibit reduced clinical signs. This suggests that FCV might be undergoing frequent adaptive mutations, resulting in poor protective efficacy of routine vaccines. There was a significant higher positive rate (p < 0.05) among cats living in groups and having less than one year of age, which is consistent with previous studies [3, 11, 17, 27, 28]. However, no significant association (p > 0.05) was found between the FCV detection rate and breed of the cats. Notably, eight FCV positive specimens were found coinfected with feline herpesvirus. Other pathogens associated with feline respiratory diseases complex such as Chlamydia felis and Mycoplasma felis were not tested in this study.
The VP1 protein showed the highest variability and tended to undergo mutations more frequently than the other capsid proteins of FCV. VP1 protein consisted of six regions (A–F). Region E of the VP1 protein is considered hypervariable and contains major targets of virus-neutralizing antibodies (B-cell epitopes) ( [11, 29]. Because of these specific features, VP1 is often targeted for novel vaccine development and clinical diagnosis. Genetic and phylogenetic analyses of the VP1 gene revealed abundant genetic diversity among the FCV strains identified in this study and reference strains. 52 FCV strains in this study clustered into two genotypes, namely genotype I (GI) and genotype II (GII), with 15 (15/52, 28.84%) isolates clustered into the GI clade and 37 (37/52, 71.15%) isolates belonging to the GII clade, indicating that genotype II was the predominant genotype currently circulating in Kunshan City, which is consistent with the latest studies from China [17, 22]. However, this finding differs to some previous studies which reports predominance of genotype I in China [10,11,12, 25]. We did not notice any obvious link between genotypes and clinical signs, and variable clinical signs were noticed by clinicians in FCV-positive cats [17, 20, 22, 24].
Mutation is an important aspect of the evolutionary process and can lead to genetic variation in FCVs in a field in which the evolutionary process depends. However, it remains unclear whether genetic variations in FCVs alter the immune response to commercial vaccine antigens. Nucleotide and deduced amino acid analysis revealed several mutations in the VP1 capsid gene and genetic divergence of FCVs strains in this study with reference strains, which was in line with previous reports [11, 17, 20, 22]. FCV strains in this study shared 70.8–88.0% nucleotide identity with the reference strains. Notably, all the detected FCV strains in this study showed a distant relationship with vaccine strains (M86379, D31836, and KM111171), suggesting that these vaccines may not provide effective cross-protection against the FCV strains circulating in Kunshan, China. In this study, several amino acid substitutions were observed at seven virulence factor-related positions among the new FCV isolates, VSD, and ORD strains. These mutations may influence antigenicity of the virus by inducing stronger binding or neutralizing antibody responses. In addition, these mutations can assist viruses in adapting to evolutionary pressure. Vaccines appear to exert evolutionary pressure on FCV, resulting in the emergence of new FCV variants. These new FCV genotypes are thought to disrupt vaccine-induced defenses among immunized cats, leading to clinical cases in vaccinated cat populations. The protective efficacy of current vaccines (based on Genotype I of FCV) against genotype II strains in China is unknown; therefore, experimental studies are needed for confirmation. In addition, it is imperative to update vaccine strains on a regular basis according to the circulating genotypes of FCV for better preventive measures.
Our study has some limitations. First, we could not collect specimens from other cities in Jiangsu Province; therefore, a clear prevalence and comparison of FCV genotypes in Jiangsu Province cannot be drawn. Second, due to limited funding, we could not culture FCV isolates or test recombination events among the vaccine and field strains of FCV. Third, we could not perform a serum virus neutralization assay to assess cross-reactivity between different field and vaccine strains.
Conclusion
In summary, the present study describes the molecular epidemiology and evolutionary characteristics of FCV in Kunshan, China. FCV strains from the present study exhibited high genetic diversity and were grouped into two genotypes, with genotype II being the predominant genotype currently circulating in Kunshan city. Continuous laboratory-based surveillance programs in other parts of China are warranted to provide new insights into the evolutionary characteristics of FCV and develop and implement better vaccination strategies in China.
References
Hofmann-Lehmann R, Hosie MJ, Hartmann K, Egberink H, Truyen U, Tasker S, Belák S, Boucraut-Baralon C, Frymus T, Lloret A, et al. Calicivirus infection in cats. Viruses. 2022;14:937.
Fastier LB. A new feline virus isolated in tissue culture. Am J Vet Res. 1957;18:382–9.
Bannasch MJ, Foley JE. Epidemiologic evaluation of multiple respiratory pathogens in cats in animal shelters. J Feline Med Surg. 2005;7:109–19.
Battilani M, Vaccari F, Carelle MS, Morandi F, Benazzi C, Kipar A, Dondi F, Scagliarini A. Virulent feline calicivirus disease in a shelter in Italy: a case description. Res Vet Sci. 2013;95:283–90.
Komina A, Krasnikov N, Kucheruk O, Zhukova E, Yuzhakov A, Gulyukin A. Distribution and genetic diversity of feline calicivirus in Moscow metropolitan area. J Vet Sci. 2022;23:e92.
Hurley KF, Sykes JE. Update on feline calicivirus: new trends. Vet Clin North Am Small Anim Pract. 2003;33:759–72.
Radford AD, Coyne KP, Dawson S, Porter CJ, Gaskell RM. Feline calicivirus. Vet Res. 2007;38:319–35.
Slaviero M, Ehlers LP, Argenta FF, Savi C, Lopes BC, Pavarini SP, Driemeier D, Sonne L. Causes and lesions of fatal pneumonia in domestic cats. J Comp Pathol. 2021;189:59–71.
Reynolds BS, Poulet H, Pingret JL, Jas D, Brunet S, Lemeter C, Etievant M, Boucraut-Baralon C. A nosocomial outbreak of feline calicivirus associated virulent systemic disease in France. J Feline Med Surg. 2009;11:633–44.
Mao J, Ye S, Li Q, Bai Y, Wu J, Xu L, Wang Z, Wang J, Zhou P, Li S. Molecular characterization and phylogenetic analysis of feline calicivirus isolated in Guangdong Province, China from 2018 to 2022. Viruses. 2022;14.
Zhou L, Fu N, Ding L, Li Y, Huang J, Sha X, Zhou Q, Song X, Zhang B. Molecular characterization and cross-reactivity of feline calicivirus circulating in Southwestern China. Viruses. 2021;13.
Liang J, Zang M, Zhou Z. Genetic and phylogenetic analysis of capsid gene of feline calicivirus in Nanjing, China. Infect Genet Evol. 2022;103:105323.
Wilhelm S, Truyen U. Real-time reverse transcription polymerase chain reaction assay to detect a broad range of feline calicivirus isolates. J Virol Methods. 2006;133:105–8.
Seal BS, Ridpath JF, Mengeling WL. Analysis of feline calicivirus capsid protein genes: identification of variable antigenic determinant regions of the protein. J Gen Virol. 1993;74(Pt 11):2519–24.
Conley MJ, McElwee M, Azmi L, Gabrielsen M, Byron O, Goodfellow IG, Bhella D. Calicivirus VP2 forms a portal-like assembly following receptor engagement. Nature. 2019;565:377–81.
Sato Y, Ohe K, Murakami M, Fukuyama M, Furuhata K, Kishikawa S, Suzuki Y, Kiuchi A, Hara M, Ishikawa Y, Taneno A. Phylogenetic analysis of field isolates of feline calcivirus (FCV) in Japan by sequencing part of its capsid gene. Vet Res Commun. 2002;26:205–19.
Cao L, Li Q, Shi K, Wei L, Ouyang H, Ye Z, Du W, Ye J, Hui X, Li J, et al. Isolation and phylogenetic analysis of feline calicivirus strains from various region of China. Anim Dis. 2022;2:16.
Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35:1547–9.
Thomas S, Lappin DF, Spears J, Bennett D, Nile C, Riggio MP. Prevalence of feline calicivirus in cats with odontoclastic resorptive lesions and chronic gingivostomatitis. Res Vet Sci. 2017;111:124–6.
Guo H, Miao Q, Zhu J, Yang Z, Liu G. Isolation and molecular characterization of a virulent systemic feline calicivirus isolated in China. Infect Genet Evol. 2018;65:425–9.
Liu C, Liu Y, Qian P, Cao Y, Wang J, Sun C, Huang B, Cui N, Huo N, Wu H, et al. Molecular and serological investigation of cat viral infectious diseases in China from 2016 to 2019. Transbound Emerg Dis. 2020;67:2329–35.
Sun Y, Deng M, Peng Z, Hu R, Chen H, Wu B. Genetic and phylogenetic analysis of feline calicivirus isolates in China. Vet J. 2017;220:24–7.
Tian J, Liu D, Liu Y, Wu H, Jiang Y, Zu S, Liu C, Sun X, Liu J, Qu L. Molecular characterization of a feline calicivirus isolated from tiger and its pathogenesis in cats. Vet Microbiol. 2016;192:110–7.
Zhao Y, Chen X, Ying Y, Wang K, Dong H, Gao C, Yang S, Hu G. Isolation and phylogenetic analysis of three feline calicivirus strains from domestic cats in Jilin Province, China. Arch Virol. 2017;162:2579–89.
Guo J, Ding Y, Sun F, Zhou H, He P, Chen J, Guo J, Zeng H, Long J, Wei Z, et al. Co-circulation and evolution of genogroups I and II of respiratory and enteric feline calicivirus isolates in cats. Transbound Emerg Dis. 2022;69:2924–37.
Coyne KP, Jones BR, Kipar A, Chantrey J, Porter CJ, Barber PJ, Dawson S, Gaskell RM, Radford AD. Lethal outbreak of disease associated with feline calicivirus infection in cats. Vet Rec. 2006;158:544–50.
Coutts AJ, Dawson S, Willoughby K, Gaskell RM. Isolation of feline respiratory viruses from clinically healthy cats at UK cat shows. Vet Rec. 1994;135:555–6.
Radford AD, Turner PC, Bennett M, McArdle F, Dawson S, Glenn MA, Williams RA, Gaskell RM. Quasispecies evolution of a hypervariable region of the feline calicivirus capsid gene in cell culture and in persistently infected cats. J Gen Virol. 1998;79(Pt 1):1–10.
Seal BS, Neill JD. Capsid protein gene sequence of feline calicivirus isolates 255 and LLK: further evidence for capsid protein configuration among feline caliciviruses. Virus Genes. 1995;9:183–7.
Acknowledgements
We would like to thank all the staff, technicians, and veterinary doctors for collecting specimens and clinical data from infected cats at veterinary clinics in Kunshan.
Funding
This research was funded by the 2022 college students innovation and entrepreneurship training programs as a national-level innovation training project (project number: 202216406005Z), and the research work was conducted at Duke Kunshan University in Jiangsu province, China. The research study was also partially sponsored by the Kunshan Municipal Government research funding (Grant Number: 22KKSGR075).
Author information
Authors and Affiliations
Contributions
Conceptualization, S.U.; methodology, S.U.; validation, S.U.; formal analysis, A.Y., H.Y., S.U., X.L.; investigation, S.K., Y.C., Z.F.; resources, Z.Q., W.Y.; data curation, S.U.; writing—original draft preparation, A.Y., H.Y., S.U., X.L., Y.C.; writing—review and editing, H.Y., A.Y., S.K., S.U.; visualization, A.Y., H.Y., S.U., X.L.; supervision, S.U.; project administration, S.U.; funding acquisition, S.K., S.U., Y.C., Z.F. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Ethics statement
The authors confirm that the ethical policies of the journal, as noted on the journal’s author guidelines page, have been adhered to. No ethical approval was required.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Kim, S., Cheng, Y., Fang, Z. et al. Molecular epidemiology and phylogenetic analysis of feline calicivirus in Kunshan, China. Virol J 21, 50 (2024). https://doi.org/10.1186/s12985-024-02319-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12985-024-02319-9