
Abstract
Background
An outbreak of aseptic meningitis occurred from June to August 2016, in Inner Mongolia Autonomous Region, China.
Methods
To determine its epidemiological characteristics, etiologic agent, and possible origin, specimens were collected for virus isolation and identification, followed by molecular epidemiological analysis.
Results
A total of 363 patients were clinically diagnosed from June 1st to August 31st 2016, and most cases (63.1%, n = 229) were identified between June 22nd and July 17th, with children aged 6 to 12 years constituting the highest percentage (68.9%, n = 250). All viral isolates from this study belonged to genotype C of echovirus 30 (E30), which dominated transmission in China. To date, two E30 transmission lineages have been identified in China, of which Lineage 2 was predominant. We observed fluctuant progress of E30 genetic diversity, with Lineage 2 contributing to increased genetic diversity after 2002, whereas Lineage 1 was significant for the genetic diversity of E30 before 2002.
Conclusions
We identified the epidemiological and etiological causes of an aseptic meningitis outbreak in Inner Mongolia in 2016, and found that Lineage 2 played an important role in recent outbreaks. Moreover, we found that Gansu province could play an important role in E30 spread and might be a possible origin site. Furthermore, Fujian, Shandong, Taiwan, and Zhejiang provinces also demonstrated significant involvement in E30 evolution and persistence over time in China.
Similar content being viewed by others


Background
Aseptic meningitis (AM) is a clinical entity with inflammation of the brain parenchyma, it is an acute disease with many infectious and non-infectious causes, among which viral infection is the most common. A variety of viruses, such as Enteroviruses, mumps virus, adenovirus, and herpes simplex virus, can cause AM. Among these viruses, enteroviruses have become among the most common pathogens of viral meningitis in China [1]. The clinical manifestations of AM are mainly symptoms of meningeal inflammation, including fever, headache, and vomiting, as well as possible severe coma or convulsions. In some cases, enteroviruses cause large outbreaks with many severe and fatal cases [2, 3].
Echovirus 30 (E30) is a member of genus Enterovirus, family Picornaviridae, and belongs to human enterovirus species B (EV-B) together with 62 other serotypes [4]. Globally, E30 is among the most frequently identified enteroviruses and a major cause of aseptic meningitis (AM) [5,6,7,8,9]. In China, E30 outbreaks have been reported in many provinces, including Zhejiang, Jiangsu, Shandong, Henan, Fujian, and Guangdong [7, 9,10,11,12,13]. Further, E30 has a high isolation rate from patients with acute flaccid paralysis (AFP) and those with hand, foot, and mouth disease (HFMD), which is a new emerging concern that should promote extensive study on the inherent mechanisms involved [5,6,7, 12,13,14]. The AFP symptoms in immunosuppressed transplant recipients and outbreaks of acute myalgia and rhabdomyolysis in Brazil revealed severe non-neuropathic findings, expanding the variety of clinical symptoms caused by E30, which should be noticed more [15, 16].
In this study, we reported an AM outbreak caused by E30 in Tongliao City, Inner Mongolia, China, in 2016 and evaluated the epidemiological characteristics. We explored the genetic diversity and molecular characteristics of the isolates by sequence analysis of the VP1 coding region and the full-length genome. Additionally, we investigated the phylodynamic diffusion patterns and recombination events of E30, which revealed the evolutionary features of E30 in China. This study offers insight into the outbreak characteristics and evolutionary dynamics of E30 in China.
Methods
Investigation of aseptic meningitis outbreaks and sample collection
Based on the HFMD pathogen-surveillance system of the Inner Mongolia Provincial Center for Disease Control and Prevention, an epidemic of AM in Tongliao city from June to August 2016 was recorded, and representative samples were collected for pathogen detection. The local Center for Disease Control and Prevention (CDC) staff collected the clinical samples from several local hospitals having patients with viral meningitis. Use of their clinical samples was explained to the guardians of children, and written consent was provided by guardians of children to permit analysis of their clinical samples. Patients were classified as having a viral meningitis if they presented meningeal inflammation, such as fever, headache, and vomiting. In total, 363 cases were identified during this stage. Overall, 25 stool and cerebrospinal fluid (CSF) specimens were collected from children (age ≤ 15 years) at the peak and the end of the outbreak, which covered three counties of Inner Mongolia (Fig. 1a). The laboratory-confirmed evidence of AM comprised infection with EV-A71, CVA16, or other enteroviruses. Real-time reverse transcription polymerase chain reaction (RT-PCR) was the diagnostic method used for enterovirus detection, as described previously [17, 18].

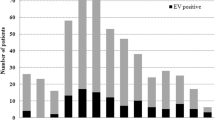
Epidemiology information of an outbreak of aseptic meningitis in Tongliao City, Inner Mongolia, China. a Cases of aseptic meningitis in townships of Tongliao city (Kailu, Naimanqi, and Kequ), Inner Mongolia, China, from June to August 2016. b Age distribution of cases reported during this outbreak
Virus isolation and molecular typing
For further etiological study, virus isolation from positive samples was performed in human rhabdomyosarcoma (RD) cells and human epithelium larynx (HEp-2) cells. Infected cell cultures were harvested after complete EV-like cytopathic effect (CPE) was observed. All experimental protocols were performed in accordance with the guidelines approved by the World Health Organization, as reported previously [19,20,21]. We ultimately obtained 12 isolates with complete EV-like CPE. Molecular typing of enterovirus isolates was performed by one-step RT-PCR amplification of the VP1 region using E30-specific primers described previously [13]. Unfortunately, we only harvested seven full-length VP1-coding-region sequences of E30 due to RT-PCR failure. The primers used for full-length genome sequencing of one E30 isolate were designed using a primer walking strategy.
Bioinformatics analysis
Seven genomic sequences sampled from different patients, including two full-length genomes, were incorporated into the molecular epidemiological analysis. A phylogenetic tree for E30 genotyping was computed using the neighbor-joining method with 1000 bootstrap replicates in MEGA 7.0 software (https://www.megasoftware.net/), which implemented the same genotyping criteria described previously [9]. Genomic dataset mining for E30 was performed and included genomic sequences from GenBank and this study. To better represent E30 genetic diversity, we removed redundant E30 sequences, resulting in 354 E30 genomic sequences derived from various locations worldwide. Genome sequences were aligned using the MUSCLE method implemented in MEGA 7.0 software [22]. We constructed a maximum-likelihood phylogenetic tree using IQ-TREE software (http://www.iqtree.org/) with 1000 bootstrap replicates, and nucleotide acid-substitution models were inferred using ModelFinder with Bayesian information criteria [23, 24].
We implemented the Bayesian inference method in BEAST software (v.1.10.4) to investigate the phylodynamics of the genomes [25]. The maximum clade credibility (MCC) tree and coalescent-based Gaussian Markov random field (GMRF) skyride plots were explored using the SYM + G4 nucleotide-substitution model. We used the sampling times of genomic sequences to calibrate the molecular clock during each run. The time signals of datasets were assessed by the Bayesian evaluation of temporal signal (BETS) method and the root-to-tip method implemented in TempEst (v.1.5.3) [26, 27]. The results supported the sufficient temporal signals of the datasets in this study (See Additional file 1: Fig. S1). We implemented 15 dataset runs combined with one genome substitution model, three different clock models, and five different coalescent tree priors. All genome sequences for Bayesian inference and acquired from different provinces of China were coded as discrete states. The asymmetric substitution model was used to infer the asymmetrical transmission rates between any pairwise region state, including the Bayesian stochastic search variable selection option [28, 29]. Path sampling and stepping stone sampling analyses in BEAST were used to choose the best parameters of Bayesian phylogenetic models [30]. We checked the convergence and effective sample size (> 200) of the parameters using Tracer software (v.1.7) [31]. We summarized the MCC tree using TreeAnnotator software (v.1.10.4), with a burn-in of the first 10% of the sampled trees. The demographic dynamics of E30 in mainland of China were assessed using the GMRF method with a time-aware smoothing parameter [32]. The GMRF skyride plots were summarized and visualized using Tracer software (v.1.7.1), and ggtree (v.1.16.3) was used to manipulate the phylogenetic tree for the best performance [33, 34].
Investigation of recombination signals
The Recombinant Detection Program (v.4.46; RDP4) was used to screen recombination signals in our entire set of genomic sequences using seven methods (RDP4, GENECONV, MaxChi, Bootscan, Chimaera, SiScan, and 3Seq) [35]. Briefly, the P2 and P3 coding-region sequences of the four strains were analyzed using the BLAST server (https://blast.ncbi.nlm.nih.gov/Blast.cgi) to compare their identity with sequences from GenBank. According to sequence similarities > 85%, these sequences were considered potential parents of the four strains and downloaded from GenBank. Phylogenetic incongruence between different regions with a p < 0.05 was considered strong evidence for recombination. We only considered recombination events that were identified by at least three methods. To confirm these putative recombination events, we utilized a smaller dataset that included the recombinant and parental strains for multiple screenings. The SimPlot program (v.3.5.1) was used for similarity plots and bootscanning analysis, with a 200-nucleotide window moving in 20-nucleotide steps [36].
Results
Profile of the AM outbreak in inner Mongolia, China
From June to August 2016, hospitalization for AM increased markedly at Tongliao People's Hospital in Inner Mongolia, China, which attracted the attention of the local CDC staff. An epidemiological curve of AM based on surveillance data from this time period in Tongliao city, Inner Mongolia, was thus generated (Fig. 1a). A total of 363 patients were clinically diagnosed from June 1st to August 31st 2016 and the epidemic spread over three townships of Tongliao city (Kailu, Naimanqi, and Kequ). Of the total number of cases, 63.1% (n = 229) were identified between June 22nd and July 17th, after which the cases gradually decreased, with certain turnovers during this progression. According to geographical distribution, Kailu and Naimanqi townships accounted for 58.4% (n = 212) and 32.8% (n = 119) of all cases, respectively, with children aged 0 to 5 years and 6 to 12 years constituting 15.7% (n = 57) and 68.9% (n = 250) of the cases, respectively (Fig. 1b). Children aged > 12 years accounted for only 4.1% (n = 15) of cases.
Pathogen and genotype identification
Twenty-five stool and CSF samples were inoculated into RD and HEp-2 cell lines for virus isolation, with complete EV-like CPE observed in 12 samples (48%). The full-length VP1 coding region of seven isolates was obtained and revealed ~ 98% nucleotide identity with E30 genomes from GenBank (accession no. KP985773). To confirm the serotype of the isolates, we constructed a neighbor-joining phylogenetic tree based on the EV-B prototypes (See Additional file 1: Fig. S1). The results showed that all isolates in this study clustered with the E30 prototype, confirming the BLAST results. The VP1 coding region of isolates in this study shared 99% nucleotide similarity between themselves, revealing a single origin of the collective outbreak. All identified enterovirus isolates met the strain-identification criteria with the homologous serotypes, including at least 75% nucleotide or 85% amino acid homology between the enterovirus isolates and the prototype enterovirus strain [37]. The genotype of the isolates in this study was investigated based on the genotyping criteria for E30 and previous reports [9, 38], with the results revealing that all isolates belonged to genotype C of E30 (Fig. 2). Moreover, genotype C showed 21.2% and 16.5% genetic divergence from genotypes A and B, respectively, with the mean genomic distance within genotypes B and C at 10.1% and 6.2%, respectively.

Generation of a phylogenetic tree. Tree based on the entire VP1 genome of 36 representative strains isolated from 1958 (Bastianni strain of the E30 prototype) to 2016, with the E-21 strain used as an outgroup and constructed using the neighbor-joining method with 1000 bootstrap replicates. Numbers at each node indicate the bootstrap support, and the letters (right) represent the E30 genotypes (A–C)
Phylodynamics of E30 in China
Two transmission lineages of E30 had been previously identified in China according to a non-redundant genome dataset from China (See Additional file 2: Fig. S2). The maximum-likelihood tree based on this dataset showed that Lineage 1 circulated from 1988 to 2010 in China and tended to have recently disappeared. Strains of Lineage 2 were detected from 2003 to the present time and dominated the spread and occurrence of the collective outbreaks. The AM outbreak in this study was caused by Lineage 2 of E30, which showed a close phylogenetic relationship with the available strains isolated from Zhejiang, Jiangsu, Shandong, Sichuan, and Yunnan provinces of China (See Additional file 2: Fig. S2a).
We implemented the Bayesian method to infer the phylogenetic relationships and transmission tendencies of E30 in China, and the time signals of several datasets were checked using root-to-tip regression and BETS (See Additional file 3: Fig. S3). All results supported the existence of time signals in the datasets, with R2 values of 0.86 and high Bayes factors (BFs). The MCC tree showed the turnovers of E30 diffusion in China, revealing phylogenetic associations among different strains (Fig. 3). Consistent with the results of the maximum-likelihood tree, we observed two distant lineages in the MCC tree, which comprised a large number of genomes from outbreaks and surveillance reports [6, 11,12,13, 39]. The results showed that the branches of E30 isolated from different provinces aggregated together, and that E30 spread simultaneously in several provinces (Fig. 3b). For example, strains from Fujian province, China, were located in different clusters of Lineage 2, revealing the existence of multiple E30 variants in that outbreak. Moreover, we observed the evolution of E30 along a date clue, where strains from several provinces intersected, indicating the complicated diffusion dynamic of E30. Several AM-related outbreaks caused by E30 were recorded and annotated in the MCC tree, indicating that E30 frequently induced AM outbreaks in China following its evolution and transmission and revealing the active status of E30 in China.

Temporal phylogenies and epidemic characteristics of E30. a Histogram of the average number of state counts based on different provinces. b The MCC phylogenetic tree based on the entire VP1 coding region in China and colored according to different provinces
We observed that outward migration from Sichuan and Gansu provinces was dominant, whereas inward migration was dominant in Fujian, Henan, Guangxi, and Inner Mongolia (Fig. 3a). The outward-migration events of Sichuan and Gansu were observed through high BF and posterior probability (PP) values (PP = 0.5 and BF > 3) (See Additional file 7: Table S1). Further, the state counts were similar according to the outward and inward migrations in Shandong and Zhejiang, respectively. Other provinces of China showed more or less equal numbers of inward and outward migrations among different provinces. The Markov rewards values of Fujian, Shandong, Taiwan, and Zhejiang provinces were significantly higher than those of other provinces, indicating that these four provinces played a significant role in the evolution and persistence of E30 over time in China (Fig. 4b). Notably, Guangdong, Yunnan, Inner Mongolia, and Henan showed relatively higher Markov rewards values compared with those of Gansu, Guangxi, Jiangsu, and Sichuan provinces.

Relative genetic diversity of E30 genomes in China. a The X-axis represents the years, and the Y-axis shows the measure of genetic diversity (logarithmic scale of Neτ). Orange shading represents 95% credibility internally; and red, green, and blue lines show the median values of the E30 population size of three datasets (all data, Lineage1, and Lineage2), respectively. b Box plot of Markov reward values. The Y-axis shows the density distribution of the total time spent in a particular location
The genetic diversity of E30 showed a fluctuant progression during evolution following the date clue (Fig. 4a). We identified two top values of E30 genetic diversity during transmission when all genomes were used (Fig. 4a, red line). The genetic diversity peaked in 2001 and 2008, whereas it showed dynamic fluctuation between 2010 and 2014, and weaker genetic diversity was observed in the most recent outbreaks. Comparison of genetic diversity between the two lineages and all genomes indicated that the relative genetic diversity of Lineage 1 peaked near 2001 and dramatically reduced after 2002. The variable tendency of Lineage 2 showed similar characteristics relative that of all of the genomes (Fig. 4a, blue line). Lineage 2 contributed to the enhanced genetic diversity of E30 after 2002, whereas Lineage 1 played a significant role in the genetic diversity of E30 before 2002. The corresponding increased tendency between the two lineages and all genomes reflected the fluctuant switches of E30 genetic diversity.
Investigation of recombination
To estimate the full-length genomic characteristics of E30, we obtained two full-length genomes. Similarity plots and bootscanning analyses revealed recombination events between the Inner Mongolia E30 isolate and the EV-B prototype at the 2A–2B junction region, implying the existence of potential recombination (See Additional file 4: Fig. S4). However, to locate the exact recombination activities between the two strains and the circulating enteroviruses, we scanned public databases to search for the recombination donor. Recombination analysis revealed overt evidence of inter-serotype recombination, as the recombination patterns of the two E30 strains in this study were similar. For the strain TL12C-NM-CHN-2016-E30 genome, breakpoint positions were identified at 3126–6627, 4066–4725, 4728–4920, and 4810–6621 (Table 1). Several EV-B strains, such as E-18, CV-B4, E-1, and CV-A9, were identified as minor putative parents using the RDP4 package. For the strain TL7C-NM-CHN-2016-E30 genome, we observed similar minor putative parents and breakpoint positions. We then constructed maximum-likelihood phylogenetic trees of the strains in this study based on the P1, P2, and P3 coding region along with the potential recombination donors, which confirmed the recombination events between the strains of this study and the E-18 strain LJ/0601/2019 (MN337405.1) (See Additional file 5: Fig. S5). Strain LJ/0601/2019 was isolated from the CSF of an adult with severe meningitis in 2019 in China. These results suggested that the role of recombination events in the evolutionary process of E30 needs to be assessed further.
Discussion
Echoviruses are currently associated with a variety of human diseases, including asymptomatic infections, febrile illness, AM, and severe diseases in newborns. E30 is one of the main pathogens that cause AM, and acute myocarditis and exhibits strong infectivity [12, 38, 40]. E30 has shown wide circulation, including to the United States, Canada, France, Italy, Germany, England, Japan, South Korea, India, and China, and has demonstrated a highly epidemic trend in the previous 10 years [12, 40,41,42,43,44]. Therefore, understanding its pathogenesis and molecular epidemiology has important public health implications. During the period from June to August 2016, the number of hospitalized patients with AM increased sharply at Tongliao People’s Hospital, thereby surpassing the baseline of previous cases with AM in Tongliao city, Inner Mongolia.
We identified 363 patients during this time period in Tongliao city, Inner Mongolia, belonging to three townships that were affected by this outbreak, with Kailu and Naimanqi townships accounting for the greatest proportion of cases (n = 331, 91.2%). Compared with the homochromous numbers of AM cases in 2015, the number of AM patients increased significantly from June to August 2016 accompanied by a higher morbidity relative to other months. Children aged 6 to 12 years constituted the greatest proportion of all cases, consistent with previous reports [9, 39]. According to previous studies, viral meningitis outbreaks caused by E30 usually occurred from June to August in local regions and at the peak of the local enterovirus infection [6, 11, 13, 39, 44]. The epidemic then spread to other villages and towns, resulting in an outbreak within a short time period [6, 9, 13, 39, 45].
Outbreaks of aseptic meningitis caused by E30 occur mostly in densely populated eastern coastal areas, such as Jiangsu, Zhejiang, Shandong, Fujian, and Guangdong [6, 7, 9, 10, 12, 13, 39]. The outbreak in the present study was caused by Lineage 2, which showed a close phylogenetic relationship with the strains isolated from Zhejiang, Jiangsu, Shandong, Sichuan, and Yunnan provinces of China. Using the Bayesian method, we observed the turnover of E30 diffusion in China, implying its complex diffusion dynamic. The branches of E30 isolated from the different provinces aggregated together, and E30 spread simultaneously in several provinces simultaneously. E30 is widespread in China and has caused a large number of AM outbreaks. Moreover, we observed the evolution of E30 along the date clue, indicating gradual break outs in different provinces. The active activity status of E30 in China promoted its evolution and transmission, and the accumulation of genomic variants might play a significant role in local outbreaks of AM.
Mapping of transmission links through Bayesian inference showed that Sichuan and Gansu provided more outward migrations, whereas Fujian, Henan, Guangxi, and Inner Mongolia had more inward migrations. Additionally, more emigration events were found in Gansu, Sichuan, Guangxi, and Yunnan provinces, with high PP and BF support (See Additional file 7: Table S1). The outbreak in the present study possibly originated from these E30-migration events; however, we were unable to directly locate the accurate source of E30 due to low PP and BF support for Inner Mongolia (data not shown). However, the strains isolated from Sichuan province shared the closest phylogenetic relationships with those from Inner Mongolia (this study). Further, the high PP and BF support verified the transmission events from Gansu to Sichuan provinces, suggesting that the two outbreaks (Sichuan and Inner Mongolia) possibly possessed the same origin (See Additional file 6: Fig. S6, Additional file 7). Moreover, Gansu province could play an important role in E30 outbreaks and spread in Sichuan and Inner Mongolia based on the highest PP and BF support (See Additional file 7: Table S1). Furthermore, Gansu, Sichuan, and Inner Mongolia are neighbors in terms of their geographic distribution in China, suggesting local outbreaks and spread events among different provinces of China. We also observed that Fujian, Shandong, Taiwan, and Zhejiang provinces showed higher Markov rewards values as compared with other provinces, indicating that they played significant roles in E30 evolution and circulation over time in China. The eastern provinces of China were primary regions experiencing E30 infection and played an important role for further nationwide diffusion.
We then assessed the relative genetic diversity following the date clue, which showed fluctuant progression. Thus, the genetic diversity peaked ~ 2001 and ~ 2008, and different lineages showed polymorphic characteristics. The relative genetic diversity of Lineage 1 showed a similar fluctuant progression with that of all genomes before 2002, whereas that of Lineage 2 showed a similar fluctuant progression with that of all genomes after 2002. Therefore, Lineage 2 contributed to the enhanced genetic diversity of E30 after 2002, whereas Lineage 1 was important for the genetic diversity of E30 before 2002. This explains the contribution of fluctuant genetic diversity through the switch between different lineages.
The nucleotide and amino acid sequences in the P2 and P3 regions are highly conserved within an enterovirus species, and the P2 and P3 sequences do not correlate with EV serotypes due to frequent recombination; however, these sequences clearly distinguish different EV species [46]. In the present study, we identified overt evidence of inter-serotype recombination events. After screening several recombination signals, strain MN337405.1_Echovirus_E18_LJ/0601/2019 was detected as the putative recombination donor. This strain provided the raw recombination materials in the P2 and P3 coding regions for other recombinants. Frequent recombination and mutations in enteroviruses are recognized as the main mechanisms associated with their evolution, enabling their rapid response and adaption to new environments [46]. Accumulation of inter-species and intra-species recombination events is regarded as a strong driver for emergence and disappearance of certain enterovirus serotypes. Some studies have confirmed the ease of intra-species recombination events, and that EV-B is more susceptible to recombination [47,48,49]. The recombination events identified in the present study (i.e., the recombination donor was isolated from the CSF of an adult with severe meningitis in 2019 in China) verifies this. The recombination signals imply a vital role for E30 evolution and might be related to E30 pathogenicity and transmission.
Conclusion
In this study, clinical specimens were collected from patients with AM, and the pathogens causing viral meningitis were screened. This process and subsequent investigation of the phylodynamic characteristics of the pathogens enhanced our understanding of the etiology of this outbreak of meningitis syndromes in Inner Mongolia. Investigation of the epidemiological and genetic characteristics of the E30 strains in Inner Mongolia provided a solid foundation for future detailed molecular epidemiological studies in China. Due to the recent increases in viral meningitis outbreaks in China, it is necessary to establish a pathogen-surveillance system in China that targets enterovirus-related syndromes in order to control enterovirus transmission and outbreaks [50, 51].
Availability of data and materials
The data generated in this study are available at GenBank (accession nos. MW080371–MW080377) and China Virus Identification Net (accession nos. CVIN_AA002571-CVIN_AA002577).
Abbreviations
- AFP:
-
Acute flaccid paralysis
- AM:
-
Aseptic meningitis
- BETS:
-
Bayesian evaluation of temporal signal
- BF:
-
Bayes factor
- CDC:
-
Center for Disease Control and Prevention
- CPE:
-
Cytopathic effect
- CSF:
-
Cerebrospinal fluid
- E30:
-
Echovirus 30
- EV-B:
-
Enterovirus species B
- GMRF:
-
Gaussian Markov random field
- HEp-2:
-
Human epithelium larynx cell line
- HFMD:
-
Hand, foot, and mouth disease
- MCC:
-
Maximum clade credibility
- PP:
-
Posterior probability
- RD:
-
Rhabdomyosarcoma cell line
- RDP4:
-
Recombinant Detection Program
- RT-PCR:
-
Reverse transcription polymerase chain reaction
References
Sun Y, Miao Z, Yan J, Gong L, Chen Y, Chen Y, Mao H, Zhang Y. Sero-molecular epidemiology of enterovirus-associated encephalitis in Zhejiang Province, China, from 2014 to 2017. Int J Infect Dis. 2019;79:58–64.
Zhang Y, Tan XJ, Wang HY, Yan DM, Zhu SL, Wang DY, Ji F, Wang XJ, Gao YJ, Chen L, et al. An outbreak of hand, foot, and mouth disease associated with subgenotype C4 of human enterovirus 71 in Shandong, China. J Clin Virol. 2009;44:262–7.
Prill MM, Dahl RM, Midgley CM, Chern SW, Lu X, Feikin DR, Sakthivel SK, Nix WA, Watson JT, Gerber SI, Oberste MS. Severe respiratory illness associated with rhinovirus during the enterovirus D68 outbreak in the United States, August 2014-November 2014. Clin Infect Dis. 2018;66:1528–34.
Zell R, Delwart E, Gorbalenya AE, Hovi T, King AMQ, Knowles NJ, Lindberg AM, Pallansch MA, Palmenberg AC, Reuter G, et al. ICTV virus taxonomy profile: picornaviridae. J Gen Virol. 2017;98:2421–2.
Bubba L, Broberg EK, Jasir A, Simmonds P, Harvala H, Redlberger-Fritz M, Nikolaeva-Glomb L, Havlíčková M, Rainetova P, Fischer TK, et al. Circulation of non-polio enteroviruses in 24 EU and EEA countries between 2015 and 2017: a retrospective surveillance study. Lancet Infect Dis. 2020;20:350–61.
Yang XH, Yan YS, Weng YW, He AH, Zhang HR, Chen W, Zhou Y. Molecular epidemiology of Echovirus 30 in Fujian, China between 2001 and 2011. J Med Virol. 2013;85:696–702.
Tao Z, Wang H, Li Y, Liu G, Xu A, Lin X, Song L, Ji F, Wang S, Cui N, Song Y: Molecular epidemiology of human enterovirus associated with aseptic meningitis in Shandong Province, China, 2006–2012. PLoS One 2014, 9:e89766.
Lema C, Torres C, Van der Sanden S, Cisterna D, Freire MC, Gomez RM. Global phylodynamics of Echovirus 30 revealed differential behavior among viral lineages. Virology. 2019;531:79–92.
Chen J, Han Z, Wu H, Xu W, Yu D, Zhang Y. A large-scale outbreak of echovirus 30 in Gansu Province of China in 2015 and Its phylodynamic characterization. Front Microbiol. 2020;11:1137.
Zhao YN, Jiang QW, Jiang RJ, Chen L, Perlin DS. Echovirus 30, Jiangsu Province, China . Emerg Infect Dis. 2005;11:562–7.
Chen P, Lin X, Liu G, Wang S, Song L, Tao Z, Xu A. Analysis of enterovirus types in patients with symptoms of aseptic meningitis in 2014 in Shandong, China . Virology. 2018;516:196–201.
Wang JR, Tsai HP, Huang SW, Kuo PH, Kiang D, Liu CC. Laboratory diagnosis and genetic analysis of an echovirus 30-associated outbreak of aseptic meningitis in Taiwan in 2001. J Clin Microbiol. 2002;40:4439–44.
Chen Y, Sun Y, Yan J, Miao Z, Xu C, Zhang Y, Mao H, Gong L. Molecular epidemiology and prevalence of echovirus 30 in Zhejiang Province, China, from 2002 to 2015. J Microbiol Biotechnol. 2017;27:2221–7.
Ivanova OE, Yarmolskaya MS, Eremeeva TP, Babkina GM, Baykova OY, Akhmadishina LV, Krasota AY, Kozlovskaya LI, Lukashev AN: Environmental Surveillance for Poliovirus and Other Enteroviruses: Long-Term Experience in Moscow, Russian Federation, 2004(-)2017. Viruses 2019, 11.
Sousa IP, Jr., Burlandy FM, Lima STS, Maximo ACB, Figueiredo MAA, Maia Z, da Silva EE: Echovirus 30 detection in an outbreak of acute myalgia and rhabdomyolysis, Brazil 2016–2017. Clin Microbiol Infect 2019, 25:252 e255–252 e258.
Mauri E, Mastrangelo A, Testa S, Pellegrinelli L, Pariani E, Binda S, Triulzi F, Barbieri S, Bana C, Montini G, Dilena R: Acute flaccid paralysis due to Echovirus 30 in an immunosuppressed transplant recipient. J Neurovirol 2019.
Cui A, Xu C, Tan X, Zhang Y, Zhu Z, Mao N, Lu Y, Xu W: The development and application of the two real-time RT-PCR assays to detect the pathogen of HFMD. PLoS One 2013, 8:e61451.
Zhang S, Wang J, Yan Q, He S, Zhou W, Ge S, Xia N: A one-step, triplex, real-time RT-PCR assay for the simultaneous detection of enterovirus 71, coxsackie A16 and pan-enterovirus in a single tube. PLoS One 2014, 9:e102724.
Han Z, Zhang Y, Huang K, Cui H, Hong M, Tang H, Song Y, Yang Q, Zhu S, Yan D, Xu W. Genetic characterization and molecular epidemiological analysis of novel enterovirus EV-B80 in China. Emerg Microbes Infect. 2018;7:193.
Luo HM, Zhang Y, Wang XQ, Yu WZ, Wen N, Yan DM, Wang HQ, Wushouer F, Wang HB, Xu AQ, et al. Identification and control of a poliomyelitis outbreak in Xinjiang, China . N Engl J Med. 2013;369:1981–90.
Blake IM, Pons-Salort M, Molodecky NA, Diop OM, Chenoweth P, Bandyopadhyay AS, Zaffran M, Sutter RW, Grassly NC. Type 2 poliovirus detection after global withdrawal of trivalent oral vaccine. N Engl J Med. 2018;379:834–45.
Kumar S, Stecher G, Tamura K: MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 2016, 33:1870–1874.
Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32:268–74.
Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 2017;14:587–9.
Bouckaert R, Vaughan TG, Barido-Sottani J, Duchene S, Fourment M, Gavryushkina A, Heled J, Jones G, Kuhnert D, De Maio N, et al: BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. PLoS Comput Biol 2019, 15:e1006650.
Baele G, Lemey P, Suchard MA. Genealogical working distributions for bayesian model testing with phylogenetic uncertainty. Syst Biol. 2016;65:250–64.
Rambaut A, Lam TT, Max Carvalho L, Pybus OG: Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol 2016, 2:vew007.
Lemey P, Rambaut A, Bedford T, Faria N, Bielejec F, Baele G, Russell CA, Smith DJ, Pybus OG, Brockmann D, Suchard MA: Unifying viral genetics and human transportation data to predict the global transmission dynamics of human influenza H3N2. PLoS Pathog 2014, 10:e1003932.
Lemey P, Rambaut A, Drummond AJ, Suchard MA: Bayesian phylogeography finds its roots. PLoS Comput Biol 2009, 5:e1000520.
Baele G, Lemey P, Bedford T, Rambaut A, Suchard MA, Alekseyenko AV. Improving the accuracy of demographic and molecular clock model comparison while accommodating phylogenetic uncertainty. Mol Biol Evol. 2012;29:2157–67.
Rambaut A, Drummond AJ, Xie D, Baele G, Suchard MA: Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst Biol 2018, 67:901–904.
Minin VN, Bloomquist EW, Suchard MA. Smooth skyride through a rough skyline: Bayesian coalescent-based inference of population dynamics. Mol Biol Evol. 2008;25:1459–71.
Yu G, Smith DK, Zhu H, Guan Y, Lam TTY, McInerny G. ggtree: an r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol Evol. 2016;8:28–36.
Yu G, Lam TT, Zhu H, Guan Y. Two Methods for Mapping and Visualizing Associated Data on Phylogeny Using Ggtree. Mol Biol Evol. 2018;35:3041–3.
Martin DP, Murrell B, Golden M, Khoosal A, Muhire B: RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol 2015, 1:vev003.
Salminen MO, Carr JK, Burke DS, McCutchan FE. Identification of breakpoints in intergenotypic recombinants of HIV type 1 by bootscanning. AIDS Res Hum Retroviruses. 1995;11:1423–5.
Oberste MS, Maher K, Kilpatrick DR, Pallansch MA. Molecular evolution of the human enteroviruses: correlation of serotype with VP1 sequence and application to picornavirus classification. J Virol. 1999;73:1941–8.
Oberste MS, Maher K, Kennett ML, Campbell JJ, Carpenter MS, Schnurr D, Pallansch MA. Molecular epidemiology and genetic diversity of echovirus type 30 (E30): genotypes correlate with temporal dynamics of E30 isolation. J Clin Microbiol. 1999;37:3928–33.
Xiao H, Guan D, Chen R, Chen P, Monagin C, Li W, Su J, Ma C, Zhang W, Ke C. Molecular characterization of echovirus 30-associated outbreak of aseptic meningitis in Guangdong in 2012. Virol J. 2013;10:263.
Bailly JL, Beguet A, Chambon M, Henquell C, Peigue-Lafeuille H. Nosocomial transmission of echovirus 30: molecular evidence by phylogenetic analysis of the VP1 encoding sequence. J Clin Microbiol. 2000;38:2889–92.
dos Santos GP, da Costa EV, Tavares FN, da Costa LJ, da Silva EE. Genetic diversity of Echovirus 30 involved in aseptic meningitis cases in Brazil (1998–2008). J Med Virol. 2011;83:2164–71.
Holmes CW, Koo SS, Osman H, Wilson S, Xerry J, Gallimore CI, Allen DJ, Tang JW. Predominance of enterovirus B and echovirus 30 as cause of viral meningitis in a UK population. J Clin Virol. 2016;81:90–3.
Mladenova Z, Buttinelli G, Dikova A, Stoyanova A, Troyancheva M, Komitova R, Stoycheva M, Pekova L, Parmakova K, Fiore L. Aseptic meningitis outbreak caused by echovirus 30 in two regions in Bulgaria, May-August 2012. Epidemiol Infect. 2014;142:2159–65.
Maruo Y, Nakanishi M, Suzuki Y, Kaneshi Y, Terashita Y, Narugami M, Takahashi M, Kato S, Suzuki R, Goto A, et al. Outbreak of aseptic meningitis caused by echovirus 30 in Kushiro, Japan in 2017. J Clin Virol. 2019;116:34–8.
Kim HJ, Kang B, Hwang S, Hong J, Kim K, Cheon DS. Epidemics of viral meningitis caused by echovirus 6 and 30 in Korea in 2008. Virol J. 2012;9:38.
Lukashev AN. Role of recombination in evolution of enteroviruses. Rev Med Virol. 2005;15:157–67.
McWilliam Leitch EC, Cabrerizo M, Cardosa J, Harvala H, Ivanova OE, Kroes AC, Lukashev A, Muir P, Odoom J, Roivainen M, et al. Evolutionary dynamics and temporal/geographical correlates of recombination in the human enterovirus echovirus types 9, 11, and 30. J Virol. 2010;84:9292–300.
Kyriakopoulou Z, Pliaka V, Amoutzias GD, Markoulatos P. Recombination among human non-polio enteroviruses: implications for epidemiology and evolution. Virus Genes. 2015;50:177–88.
Oberste MS, Maher K, Pallansch MA. Evidence for frequent recombination within species human enterovirus B based on complete genomic sequences of all thirty-seven serotypes. J Virol. 2004;78:855–67.
Gao GF. From “A”IV to “Z”IKV: Attacks from Emerging and Re-emerging Pathogens. Cell. 2018;172:1157–9.
Harvala H, Broberg E, Benschop K, Berginc N, Ladhani S, Susi P, Christiansen C, McKenna J, Allen D, Makiello P, et al. Recommendations for enterovirus diagnostics and characterisation within and beyond Europe. J Clin Virol. 2018;101:11–7.
Acknowledgements
We would like to acknowledge the staff of the local Center for Disease Control and Prevention (CDC) in Inner Mongolia for collecting the clinical samples and conducting the epidemiological investigations.
Funding
This study was supported by the Scientific and Technological Innovation Guidance Project of the Department of Finance of Inner Mongolia Autonomous region (2016–5); the Inner Mongolia Autonomous region "Grassland Talent" team Rolling Project (2016–10)-Disease prevention and control innovation team; and the National Key Research and Development Program of China (Project No. 2021YFC0863000), the National Science and Technology Major project of China (project nos. 2018ZX10711001, 2017ZX10104001, 2018ZX10713002, 2018ZX10101002-001–003, and 2018ZX10101002-004–006), and Beijing Natural Science Foundation (Project No. L192014). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
YZ, HL, WW, and WX conceived and designed the experiments; XT and YH performed the experiments; XT, ZH, and YZ analyzed the data; ZH and YZ wrote the main manuscript and prepared all the tables and figures; and all authors reviewed the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Written informed consent for the use of their clinical samples was obtained from all individuals included in the study or from their guardians. The study was approved by the Second Ethics Review Committee of the National Institute for Viral Disease Control and Prevention (IVDC), Chinese Center for Disease Control and Prevention; all experimental protocols were approved by the IVDC; and the methods were carried out in accordance with the approved guidelines.
Consent to publication
Not applicable.
Competing interest
The authors declare that no competing interests exist.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
. Fig. S1: The neighbour-joining phylogenetic tree, based on the VP1 coding region, for serotyping. The EV-B prototype sequences of the VP1 coding region and the genome sequences in this study were used.
Additional file 2
. Fig. S2: (A). Magnification based on the red box in Figure S2 (B) and the branches coloured in red represent the E30 isolates from the collective outbreak in this study. (B). The midpoint-rooted maximum likelihood phylogenetic tree of E30 strains isolated from China. Scale bars represent the substitutions per site per year.
Additional file 3
. Fig. S3: (A) Linear regression of root-to-tip divergence and sample dates. (B) Results of the Bayesian evaluation of temporal signal (BETS). The temporal signal of E-30 datasets was estimated using these two methods to assure sufficient temporal signals.
Additional file 4
. Fig. S4: Similarity and bootscanning plot of EV-B prototypes and genomes isolated from this study, which was used to scan the recombination signals.
Additional file 5
. Fig. S5: Maximum likelihood phylogenetic tree of E30 strains isolated from this study and the potential recombination donors. Scale bars represent the substitutions per site per year. (A), (B), and (C) represent the phylogenetic tree based on the P1, P2 and P3 coding regions.
Additional file 6
. Fig. S6: Spatial transmission pathways of E30 inferred in this study using the Bayesian Stochastic Search Variable Selection (BSSVS) method. The solid black arrow shows the possible origin pathway of the outbreak in this study.
Additional file 7
. Table S1: The migration events of E30 based on the VP1 coding region.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Tian, X., Han, Z., He, Y. et al. Temporal phylogeny and molecular characterization of echovirus 30 associated with aseptic meningitis outbreaks in China. Virol J 18, 118 (2021). https://doi.org/10.1186/s12985-021-01590-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12985-021-01590-4