
Abstract
Background
Human pegivirus (HPgV)—formerly known as GBV-C—is a member of the Flaviviridae family and belongs to the species Pegivirus C. It is a non-pathogenic virus and is transmitted among humans mainly through the exposure to contaminated blood and is often associated with human immunodeficiency virus (HIV) infection, among other viruses. This study aimed to determine the prevalence of HPgV viremia, its association with HIV and clinical epidemiological factors, as well as the full-length sequencing and genome characterization of HPgV recovered from blood donors of the HEMOPA Foundation in Belém-PA-Brazil.
Methods
Plasma samples were obtained from 459 donors, tested for the presence of HPgV RNA by the RT-qPCR. From these, a total of 26 RT-qPCR positive samples were submitted to the NGS sequencing approach in order to obtain the full genome. Genome characterization and phylogenetic analysis were conducted.
Results
The prevalence of HPgV was 12.42%. We observed the highest prevalences among donors aged between 18 and 30 years old (16.5%), with brown skin color (13.2%) and men (15.8%). The newly diagnosed HIV-1 prevalence was 26.67%. The HPgV genotype 2 (2a and 2b) was identified. No data on viral load value was found to corroborate the protective effect of HPgV on HIV evolution.
Conclusions
This study provided information regarding the HPgV infection among blood donors from HEMOPA Foundation. Furthermore, we genetically characterized the HPgV circulating strains and described by the first time nearly complete genomes of genotype 2 in Brazilian Amazon.
Similar content being viewed by others


Background
Human pegivirus (HPgV), formerly known as GBV-C or hepatitis G virus (HGV), is a member of the Flaviviridae family, belongs to the species Pegivirus C [1]. HPgV is an enveloped virus with a single-stranded, positively polarized RNA genome comprising approximately 9,400 nucleotides. The viral genome is similar to the genome of the hepatitis C virus and contains a single open reading frame (ORF) located between the untranslated regions (UTRs) at the 5′ and 3′ ends of the viral genome. The 5′-NTR region is highly conserved with an internal ribosome entry site (IRES) and is responsible for the initiation of the translation of the viral RNA, resulting in the synthesis of a polyprotein of approximately 3,000 amino acid residues. Through the action of cellular peptidases and viral proteases, the polyprotein is cleaved to produce eight mature yet incompletely characterized proteins, including the two structural (E1 and E2) and seven non-structural (NS) proteins [2,3,4].
HPgV is transmitted among humans mainly through exposure to contaminated blood. This transmission profile deems HPgV as a common coinfection with other viruses such as HIV-1, hepatitis C virus (HCV), and Ebola virus [5,6,7]. Up to 40% of the individuals infected with HIV and/or HCV are positive for HPgV infection [8, 9]
People HIV-1 co-infected with HPgV experience slower disease progression that may be influenced by the interference of HPgV on the pathogenicity of HIV-1 [10, 11]. However, the mechanism by which HPgV mediates this protective effect remains inconclusive [12, 13].
Several studies carried out in different populations in the last decades in Brazil have shown varying prevalence rates of HPgV infections [14, 15]. Studies among healthy blood donors conducted in Brazil revealed prevalence rates of 19.5% and 9.7% among individuals with prior exposure and active infection, respectively. [16]. However, the most significant prevalence reported was among patients with HIV, with a value reaching up to 34% [17].
The prevalence of the virus is lower in the developed countries (1–5%) than in the developing countries -(approximately 20%), with South America exhibiting a prevalence rate of up to 14.6% among blood donors [4]. Seroprevalence studies revealed the presence of anti-E2 antibodies in 19.5% of healthy blood donors [18]. However, data about the soroprevalence of HPgV viremia and its circulating strains in the Northern Brazilian population are scarce, particularly among blood donors.
This study aimed to determine the prevalence of HPgV viremia and its association with clinical epidemiological factors and the presence of HIV, as well as the complete genome characterization of HPgV strains in volunteer blood donors from a public hemotherapy service in Northern Brazil.
Methods
Blood donors and the collection of serum samples
A cross-sectional study was performed to determine the prevalence of HPgV infection among blood donors from the HEMOPA Foundation between March 2017 and April 2018. Epidemiological data were obtained through access to the HEMOPA Foundation donor registry. The sample size was calculated using EpiInfo™ software [19] based on the presumed prevalence of 5% to 10% of HPgV in Brazil [17, 20]. For this calculation, the number of blood donors registered in 2016 at the HEMOPA Foundation (63,501), 95% confidence level, and 20% margin adjustment was used to obtain a total of 366 individuals. A total of 459 serum samples (400 μL) from the blood donors from the HEMOPA Foundation were tested.
Extraction and detection of HIV, HCV, and HPgV nucleic acids
The extraction of nucleic acids was performed using the QIAmp RNA mini Kit (Qiagen®, Hilden, Germany) according to the manufacturer’s recommendations. HIV and HCV detection were performed with Hemocenter’s Nuclear Acid Test Platform (NAT) using the HIV/HCV NAT kit (Bio Manguinhos®, Rio de Janeiro, Brazil), according to the manufacturer’s recommendations.
The presence of HPgV nucleic acid was evaluated by the RT-qPCR, using the custom Assay TaqMan® Fast Virus 1-Step, developed by AB Applied Biosystems (Foster City, California, EUA), following the manufacturer’s Fast protocol as follows: 1 cycle of reverse transcription (RT) for 2 min at 50 °C; inactivation of Reverse Transcription (RT)/start of denaturation (1 cycle) for 20 s at 95 °C; amplification for 40 cycles of 95 °C for 3 s and 60 °C for 30 s. The selected primers corresponded to the 5′-UTR of the viral genome according to GenBank NC_001710 and were as follows: RTG1 (GTGGTGGATGGGTGATGACA; sense), RTG2 (GACCCACCTATAGTGGCTACCA; antisense), and NFQ (5′-FAM-CCGGGATTTACGACCTACC3′; probe) [17].
Quantification of HIV-1 and HPgV plasma viral load
HIV-1 viral load was measured in a Real-Time Rotor-Gene® Q platform using artus HI Virus-1 RG RT-PCR (QIAGEN Hilden, Germany) and HPgV viral load was measured in a Real-Time LightCycler® 480 Instrument II (Roche Applied Science, Penzberg, Germany) using TaqMan® Fast Virus 1-Step Master Mix (Foster City, California, USA). Both methods strictly followed the manufacture´s recommendation.
High-throughput sequencing
The RNA, obtained in the nucleic acid extraction step, was quantified in Qubit 2.0 fluorometer (Thermo Fisher Scientific), using the QubitTM RNA HS Assay Kit 500 assays (Invitrogen by Thermo Fisher Scientific). Then, cDNA was synthesized using the cDNA Synthesis System Roche® kit (Roche Applied Science), as described by the manufacturer. The subsequent step was the quantification of cDNA using the Qubit 2.0 fluorometer (Thermo Fisher Scientific), using the QubitTM dsDNA HS Assay Kit (Invitrogen by Thermo Fisher Scientific) and analysis of cDNA integrity in the equipment 2100 Bioanalyzer (Agilent Technologies) using the high sensitivity DNA reagents kit (Agilent Technologies). Genome sequencing was performed using the HiSeq 2500 platform (Illumina) as previously described [21].
Bioinformatics analysis
Generated reads were filtered, adapters and reads with Phred quality scores below 20 and size less than 50 nt, were removed using Trim Galore 0.4.4, Cutadapt and Prinseq-lite 0.20.4 software [22,23,24]. The filtered reads were used in de novo assembly strategy applying two software: IDBA-UD v.1.1.3 [25] and MEGAHIT v.1.1.3 [26], both set to a k-mer range of 21 to 91, varying every 10 k-mer. For the removal of redundant data, generated contigs were processed using CD-Hit-Est v.4.7 [27] set to a threshold of 90% identity. Then, the non-redundant contigs were aligned against the NCBI non-redundant protein database using the Blastx (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=blastx&PAGE_TYPE=BlastSearch&LINK_LOC=blasthome) algorithm and the software Diamond v.0.9.22 [28].
Genome characterization and phylogenetic inference
The HPgV genomes sequences, identified by the Blastx algorithm, were used for predicting the coding region (Open Reading Frame; ORF), as well as the 5′-UTR and 3′-UTR regions using the Geneious v9 tool. Viral genomes were aligned with other HPgV complete genomes available in GenBank database using the MAFFT v7 software [29]. For phylogenetic analyses, complete ORFs from aligned genomes were used to perform the phylogenetic reconstructions using the maximum-likelihood method, generated by RAxML v.8.2.12 [30], applying 1000 bootstrap replicates [31] and the best nucleotide replacement model calculated by JModelTest [32]. Complete genome sequences of HPgV obtained in this study have been submitted to GenBank (accession numbers MN215894–MN21591).
Statistical analysis
The differences between the groups were analyzed with the chi-square test, G test of independence, Student’s t-test, and odds ratio. The level of significance of α = 0.05 was adopted for the rejection of the null hypothesis. Statistical analyses were performed using the BioEstat program version 5.3 and GraphPad Prism version 8; Microsoft Excel Professional 2007 program was used for data processing and to prepare tables and databases.
Results
Plasma samples were obtained from 459 donors at the time of screening. The prevalence of HPgV in the samples was 12.42% (n = 57) and a total of 26.14% (n = 120) of the donors were HIV positive, diagnosed through routine tests carried out at the HEMOPA Foundation. The HPgV prevalence among HIV donors was 26.67% (n = 32, p < 0 0.0001). HIV positive donors were four times more likely to have HPgV infection than those HIV negative (odds ratio = 4.56, p < 0.0001, 95% confidence interval [CI]:2.57–8.10).
The highest prevalences of HPgV were observed among donors with ages ranging from 18 to 30 years old (16.5%, p = 0.024), men (15,8%, p = 0.005) with brown skin color (13.2%, p = 0,462) and 12 or more years of education (24.0%, p < 0.0001) (Table 1).
In 18 of the 57 HPgV positive samples (31.6%) near-complete genomes sequences were obtained. The BlastX result of the 18 genomes obtained showed 91% to 93.24% identity with Human pegivirus sequences from the United Kingdom (LT009489 and LT009494), France (MH053115) and Japan (D87255), available from GenBank / NCBI (Table 2). The pairwise alignment of the polyprotein amino acid sequences of these four strains of the bank along with the 18 described sequences showed an identity of 98.6%.
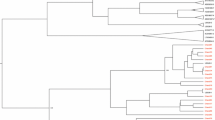
All nearly complete genome sequences showed the common genome organization related to members of the HPgV: unique and large ORF flanked by 5′and 3′ UTRs. The phylogenetic tree represented the genotypes (1, 2a, 2b, 3, 4, 6 and 7), including two subgenotypes (2a and 2b). All the sequences of the HPgV genome obtained clustered with sequences of the HPgV genotype 2, subgenotypes 2a and 2b, with high bootstrap values (> 90%) (Fig. 1).

Phylogenetic tree of Human pegivirus (HPgV) generated with complete polyprotein ORF, using RAxML with the GTR + I + G + F nucleotide substitution model using 1000 bootstrap replicas displaying only values greater than 70
Viral load was compared in two groups. First we compared HIV viral load in monoinfected (HIV-1) and coinfected (HIV-1/HPgV). Then, we compared HPgV viral load in monoinfected (HPgV) and coinfected group (HPgV/HIV-1). We found a higher HIV-1 viral load in the coinfected (2.72 Log10) than in the monoinfected group (2.00 Log10; Fig. 2a). While a higher HPgV viral load (4.28 Log10) was observed in the monoinfected group in comparison with coinfected group (HPgV, Fig. 2b).

Virological profiles of newly diagnosed blood donors with HIV-1 and HPgV. a Comparison of plasma HIV-1 viral load between HIV-1 monoinfected group and HIV-1/HPgV coinfected group. b Comparison of plasma HPgV viral load between HPgV monoinfected group and HPgV/HIV-1 coinfected group
Discussion
The prevalence of HPgV-1 among blood donors was 12,4%, which is consistent and not significantly different from the expected prevalence in developing countries (up to 20%) [4, 33]. The prevalence calculated in this study was 2,8% higher than reported Slavov et al. [34] in a study among blood donors from the city of Macapá (northern Brazil). Previous studies have shown that the prevalence of HPgV among blood donors in most regions of Brazil varies from 5 to 10% [16, 35,36,37,38], although Da Mota et al. [39] have found a high prevalence of 21.7% in the southernmost region of Brazil.
In our findings, the highest prevalence of HPgV occurred among subjects between 18 and 30 years of age (16.5%), males (15.8%), and brown individuals (13.2%). It is important to highlight that the epidemiological profile of the donors was similar to that observed in the epidemiology of HIV/AIDS in Brazil, wherein the majority of the infected individuals were male and young subjects (15 to 39 years) with up to 11 years of study (completed high school) [40].
The prevalence of HPgV among the individuals diagnosed with HIV-1 reported in this study was 26,7%, which is 9,7% higher than that reported by Miranda et al. [41]. [38] The high prevalence of HPgV among HIV-1 individuals has been reported in several studies in Brazil and the world [42,43,44]. The association between the presence of HPgV and HIV is owing to the fact that HPgV likely acts as a protective factor for the development of HIV [44,45,46].
As was seen in some studies, HIV-1 infected people have reduced mortality when co-infected with HPgV [47, 48]. This protective effect may be due to the inducing effect of HPgV on CD4 and CD8 T lymphocytes, leading to antiretroviral factors secretion and also to the reduction of the expression of the HIV-1 co-receptor CCR5, as reported by Jung et al. and Xiang et al., on in vitro experiments [49, 50]. Nevertheless, the present study showed no evidence of viral load value that corroborated with the protective effect of HPgV in the evolution of HIV-1, instead, HIV-1 viral load in the coinfected group (HIV-1/HPgV) was 0.72 Log10 (p = 0,002) higher than in a monoinfected group (HIV-1 positive). Another consideration is that all individuals in our sample were newly diagnosed with HIV-1 during the acute phase, suggesting that HPgV does not exert a protective effect on the pathogenesis of HIV-1 during the acute phase as suggested Bailey et al. [51].
On trials investigating the interaction of SPgV (Simian Pegivirus) and SIV (Simian immunodeficiency virus) infection, Bailey et al. found no evidence of a protective effect of SPgV on the evolution of SIV in the acute phase of infection. The protective immunomodulatory effect of SPgV was observed only in the chronic phase of SIV infection [51]. Extending this observation, our findings corroborate the hypothesis suggested that HPgV does not exert a protective effect during the acute phase of HIV infection, since the HIV positive individuals in this study were all newly diagnosed. Otherwise, as seen in several other studies, there is a likely beneficial relationship between HPgV and the chronic phase of HIV infection [4, 47, 52, 53].
The phylogenetic analysis revealed the presence of genotype 2 and the subtypes 2a and 2b in the studied population. These findings corroborate previous studies that identified these same genotypes in other regions of Brazil [20, 35, 42] and in Brazilian Amazon [34].
HPgV is known as a non-pathogenic virus and is not part of the routine diagnosis in the HEMOPA Foundation, but further studies are necessary to evaluate the unclear aspects related to HPgV infection especially those related to viral biology and interaction with HIV-1. This study genetically characterized and identified, by the first time, the circulating strains of HPgV among blood donors from HEMOPA Foundation and described by the first time nearly complete genomes of genotype 2 in Brazilian Amazon.
Conclusions
This study provided information regarding the HPgV infection among blood donors from HEMOPA Foundation. Furthermore, we genetically characterized the HPgV circulating strains and described by the first time the genotype 2 genomes in the Brazilian Amazon region.
Availability of data and materials
Not applicable.
Abbreviations
- cDNA:
-
Complementary deoxyribonucleic acid
- dsDNA:
-
Double-stranded deoxyribonucleic acid
- GBV-C:
-
GB virus C
- HCV:
-
Hepatitis C virus
- HEMOPA Foundation:
-
Foundation Center for Hemotherapy and Hematology of Pará
- HIV:
-
Human immunodeficiency virus
- HPgV:
-
Human Pegivirus
- IRES:
-
Internal ribosome entry site
- ORF:
-
Open reading frame
- RNA:
-
Ribonucleic acid
- RT-qPCR:
-
Reverse transcription real-time polymerase chain reaction
- SIV:
-
Simian immunodeficiency virus.
- SPgV:
-
Simian Pegivirus
- UTRs:
-
Untranslated regions
References
Simmonds P, Becher P, Bukh J, Gould EA, Meyers G, Monath T, et al. ICTV virus taxonomy profile: dicistroviridae. J Gen Virol. 2017;98:355–6.
Chowdhury AY, Tavis JE, George SL. Human pegivirus (GB virus C) NS3 protease activity inhibits induction of the type I interferon response and is not inhibited by HCV NS3 protease inhibitors. Virology. 2014;456–457:300–9. https://doi.org/10.1016/j.virol.2014.03.018.
Stapleton JT, Foung S, Muerhoff AS, Bukh J, Simmonds P. The GB viruses: a review and proposed classification of GBV-A, GBV-C (HGV), and GBV-D in genus Pegivirus within the family Flaviviridae. J Gen Virol. 2011;92:233–46.
Mohr EL, Stapleton JT. GB virus type C interactions with HIV: the role of envelope glycoproteins. J Viral Hepat. 2009;16:757–68.
Lauck M, Bailey AL, Andersen KG, Goldberg TL, Sabeti PC, O’Connor DH. GB Virus C coinfections in West African Ebola patients. J Virol. 2015;89:2425–9.
Berg MG, Lee D, Coller K, Frankel M, Aronsohn A, Cheng K, et al. Discovery of a novel human pegivirus in blood associated with hepatitis C virus co-infection. PLOS Pathog. 2015;11:e1005325.
Ng KT, Takebe Y, Chook JB, Chow WZ, Chan KG, Abed Al-Darraji HA, et al. Co-infections and transmission networks of HCV, HIV-1 and HPgV among people who inject drugs. Sci Rep. 2015;5:15198.
Tenckhoff S, Kaiser T, Bredeek F, Donfield S, Menius E, Lail A, et al. Role of GB virus C in HIV-1-infected and hepatitis C virus-infected hemophiliac children and adolescents. J Acquir Immune Defic Syndr. 2012;61:243–8.
Feng Y, Zhao W, Feng Y, Dai J, Li Z, Zhang X, et al. A novel genotype of GB virus C: Its identification and predominance among injecting drug users in Yunnan, China. PLoS ONE. 2011;6:e21151.
Williams CF, Klinzman D, Yamashita TE, Xiang J, Polgreen PM, Rinaldo C, et al. Persistent GB virus C infection and survival in HIV-infected men. N Engl J Med. 2004;350:981–90.
Xiang J, Wünschmann S, Diekema DJ, Klinzman D, Patrick KD, George SL, et al. Effect of coinfection with GB virus C on survival among patients with HIV infection. N Engl J Med. 2001;345:707–14.
Maidana-Giret MT, Silva TM, Sauer MM, Tomiyama H, Levi JE, Bassichetto KC, et al. GB virus type C infection modulates T-cell activation independently of HIV-1 viral load. AIDS. 2009;23:2277–87.
Stapleton JT, Chaloner K, Martenson JA, Zhang J, Klinzman D, Xiang J, et al. GB virus C infection is associated with altered lymphocyte subset distribution and reduced T cell activation and proliferation in HIV-infected individuals. PLoS ONE. 2012;7:e50563.
Ramos Filho R, Carneiro MAS, Teles S, a, Dias M a, Cardoso DDP, Lampe E, et al. GB virus C/hepatitis G virus infection in dialysis patients and kidney transplant recipients in Central Brazil. Mem Inst Oswaldo Cruz. 2004;99:639–43.
Gallian P, Rodrigues V, Cantaloube JF, Dessein H, de Micco P, Dessein AJ, et al. High prevalence of GB-C/hepatitis G virus in a Brazilian population with helminth infection. J Med Virol. 1998;56:310–5.
Levi JE, Contri DG, Lima LP, Takaoka DT, Garrini RH, Santos W, et al. High prevalence of GB virus c/hepatitis G virus RNA among Brazilian blood donors. Rev Inst Med Trop Sao Paulo. 2003;45:75–8.
Da Mota LD, Nishiya AS, Finger-Jardim F, Barral MFM, Silva CM, Nader MM, et al. Prevalence of human pegivirus (HPgV) infection in patients carrying HIV-1C or non-C in southern Brazil. J Med Virol. 2016;88:2106–14.
Ross RS, Viazov S, Schmitt U, Schmolke S, Tacke M, Ofenloch-Haehnle B, et al. Distinct prevalence of antibodies to the E2 protein of GB virus C/hepatitis G virus in different parts of the world. J Med Virol. 1998;54:103–6.
Centers for Disease Control and Prevention. Epi InfoTM. Epi Info. 2013.
Giret MTM, Miraglia JL, Sucupira MCA, Nishiya A, Levi JE, Diaz RS, et al. Prevalence, incidence density, and genotype distribution of GB virus C infection in a cohort of recently HIV-1-infected subjects in Sao Paulo, Brazil. PLoS ONE. 2011;6:e18407.
Pease J, Sooknanan R. A rapid, directional RNA-seq library preparation workflow for Illumina® sequencing. Nat Methods. 2012;9:i–ii.
Felix K. Trim Galore! https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/; 2017.
Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17:10.
Schmieder R, Edwards R. Quality control and preprocessing of metagenomic datasets. Bioinformatics. 2011;27:863–4.
Peng Y, Leung HCM, Yiu SM, Chin FYL. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics. 2012;28:1420–8.
Li D, Liu C-M, Luo R, Sadakane K, Lam T-W. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics. 2015;31:1674–6.
Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–9.
Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nat Methods. 2015;12:59–60.
Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–80.
Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–3.
Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution (N Y). 1985;39:783.
Posada D. jModelTest: phylogenetic model averaging. Mol Biol Evol. 2008;25:1253–6.
AbuOdeh RO, Al-Absi E, Ali NH, Khalili M, Al-Mawlawi N, Hadwan TA, et al. Detection and phylogenetic analysis of human pegivirus (GBV-C) among blood donors and patients infected with hepatitis B virus (HBV) in Qatar. J Med Virol. 2015;87:2074–81.
Slavov SN, Maraninchi Silveira R, Hespanhol MR, Sauvage V, Rodrigues ES, Fontanari Krause L, et al. Human pegivirus-1 (HPgV-1) RNA prevalence and genotypes in volunteer blood donors from the Brazilian Amazon. Transfus Clin Biol. Société française de transfusion sanguine (SFTS). 2019;1:1–6.
Slavov SN, Silveira RM, Rodrigues ES, Diefenbach CF, Zimmermann AM, Covas DT, et al. Human pegivirus-1 (HPgV-1, GBV-C) RNA prevalence and genotype diversity among volunteer blood donors from an intra-hospital hemotherapy service in Southern Brazil. Transfus Apher Sci. 2019;58:174–8.
Ribeiro-dos-Santos G, Nishiya AS, Nascimento CMR, Bassit L, Chamone DF, Focaccia R, et al. Prevalence of GB virus C (hepatitis G virus) and risk factors for infection in São Paulo, Brazil. Eur J Clin Microbiol Infect Dis. 2002;21:438–43.
Oliveira LA, Martins RMB, Carneiro MAS, Teles SA, Silva SA, Cardoso DDP, et al. Prevalence and genotypes of GB virus C/hepatitis G virus among blood donors in Central Brazil. Mem Inst Oswaldo Cruz. 2002;97:953–7.
Bassit L, Kleter B, Ribeiro-dos-Santos G, Maertens G, Sabino E, Chamone D, et al. Hepatitis G virus: prevalence and sequence analysis in blood donors of São Paulo. Brazil Vox Sang. 1998;74:83–7.
Da Mota LD, Finger-Jardim F, Silva CM, Germano FN, Nader MM, Gonçalves CV, et al. High prevalence and autochtonous transmission of human pegivirus (HPgV-1) in blood donors in the extreme southern of Brazil. J Med Virol. 2019;91:31–7.
BRASIL: MINISTÉRIO DA SAÚDE. Boletim Epidemiológico HIV/Aids | 2019. Secretaria de Vigilância em Saúde | Ministério da Saúde; 2019. Available from: file:///C:/Users/AEB-PC/Downloads/boletim_hivaids_2019.pdf
de Miranda BKB, de Sá KSG, da Silva ANR, Feitosa RNM, Cayres-Vallinoto IMV, Ishak R, et al. GBV-C/HIV-1 coinfection is associated with low HIV-1 viral load and high CD4+ T lymphocyte count. Arch Virol. 2017. https://doi.org/10.1007/s00705-017-3514-y.
Mota LDD, Finger-Jardim F, Silva CM, Germano FN, Nader MM, Gonçalves CV, et al. Molecular and clinical profiles of human pegivirus type 1 infection in individuals living with HIV-1 in the extreme South of Brazil. Biomed Res Int. 2019;2019:1–11.
Santos LM, Lobato RC, Barral MFM, Gonçalves CV, da Hora VP, Martinez AMB. Prevalence and vertical transmission of human pegivirus among pregnant women infected with HIV. Int J Gynaecol Obstet. 2017;38:42–9.
Miao Z, Gao L, Song Y, Yang M, Zhang M, Lou J, et al. Prevalence and Clinical Impact of Human Pegivirus-1 Infection in HIV-1-Infected Individuals in Yunnan, China. Viruses. 2017;9:28.
Chivero ET, Bhattarai N, McLinden JH, Xiang J, Stapleton JT. Human Pegivirus (HPgV; formerly known as GBV-C) inhibits IL-12 dependent natural killer cell function. Virology. 2015;485:116–27. https://doi.org/10.1016/j.virol.2015.07.008.
Gómara MJ, Galatola R, Gutiérrez A, Gimeno MC, Gatell JM, Sánchez-Merino V, et al. HIV-1 inhibiting capacity of novel forms of presentation of GB virus C peptide domains is enhanced by coordination to gold compounds. Curr Med Chem. 2014;21:238–50.
Vahidnia F, Petersen M, Stapleton JT, Rutherford GW, Busch M, Custer B. Acquisition of GB virus type C and lower mortality in patients with advanced HIV disease. Clin Infect Dis. 2012;55:1012–9.
Lanteri MC, Vahidnia F, Tan S, Stapleton JT, Norris PJ, Heitman J, et al. Downregulation of cytokines and chemokines by GB virus C after transmission via blood transfusion in HIV-positive blood recipients. J Infect Dis. 2015;211:1585–96.
Jung S, Knauer O, Donhauser N, Eichenmüller M, Helm M, Fleckenstein B, et al. Inhibition of HIV strains by GB virus C in cell culture can be mediated by CD4 and CD8 T-lymphocyte derived soluble factors. AIDS. 2005;19:1267–72.
Xiang J, George SL, Wünschmann S, Chang Q, Klinzman D, Stapleton JT. Inhibition of HIV-1 replication by GB virus C infection through increases in RANTES, MIP-1alpha, MIP-1beta, and SDF-1. Lancet (London, England). 2004;363:2040–6.
Bailey AL, Buechler CR, Matson DR, Peterson EJ, Brunner KG, Mohns MS, et al. Pegivirus avoids immune recognition but does not attenuate acute-phase disease in a macaque model of HIV infection. PLOS Pathog. 2017;13:e1006692.
Horemheb-Rubio G, Ramos-Cervantes P, Arroyo-Figueroa H, Ávila-Ríos S, García-Morales C, Reyes-Terán G, et al. High HPgV replication is associated with improved surrogate markers of HIV progression. PLoS ONE. 2017;12:e0184494.
Vahidnia F, Petersen M, Rutherford G, Busch M, Assmann S, Stapleton JT, et al. Transmission of GB virus type C via transfusion in a cohort of HIV-infected patients. J Infect Dis. 2012;205:1436–42.
Acknowledgements
Not applicable
Funding
The work was supported by the HEMOPA foundation, Evandro Chagas Institute, Foundation for Scientific and Technological Development in Health (FIOTEC; Project PRES-012-FIO-16) and funded in part by the research productivity project CNPq (302584/2015–3).
Author information
Authors and Affiliations
Contributions
AS contributed to the conception of the work; acquisition, analysis, and interpretation of data for the manuscript; revising the work critically for intellectual content. CS worked on the acquisition, analysis, and interpretation of data. RB contributed to the acquisition, analysis, and interpretation of the data. PS and PM contributed to the acquisition of data. LL and RB contributed to the conception of the work; acquisition, analysis, and interpretation of data for the manuscript; revising it critically for intellectual content. MN and PM contributed to the conception of the work; acquisition, analysis, and interpretation of data for the manuscript; revising it critically for intellectual content. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All procedures performed in this study involving human participants were in accordance with the ethical standards of the institutional research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. This work was approved by the Human Research Ethics Committee of the State University of Pará (Opinion No. 1.868.004/CAAE: 62348616.4.0000.5174) and all donors were informed of their participation in this research project by signing the Term Free and Informed Consent (EHIC) and all the privacy rights were observed.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Silva, A.S.N., Silva, C.P., Barata, R.R. et al. Human pegivirus (HPgV, GBV-C) RNA in volunteer blood donors from a public hemotherapy service in Northern Brazil. Virol J 17, 153 (2020). https://doi.org/10.1186/s12985-020-01427-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12985-020-01427-6