
Abstract
Background
We previously reported the discovery of a novel, putative flavivirus designated T’Ho virus in Culex quinquefasciatus mosquitoes in the Yucatan Peninsula of Mexico. A 1358-nt region of the NS5 gene was amplified and sequenced but an isolate was not recovered.
Results
The complete genome of T’Ho virus was sequenced using a combination of unbiased high-throughput sequencing, 5′ and 3′ rapid amplification of cDNA ends, reverse transcription-polymerase chain reaction and Sanger sequencing. The genome contains a single open reading frame of 10,284 nt which is flanked by 5′ and 3′ untranslated regions of 97 and 556-nt, respectively. Genome sequence alignments revealed that T’Ho virus is most closely related to Rocio virus (67.4% nucleotide identity) and Ilheus virus (65.9%), both of which belong to the Ntaya group, followed by other Ntaya group viruses (58.8–63.3%) and Japanese encephalitis group viruses (62.0–63.7%). Phylogenetic inference is in agreement with these findings.
Conclusions
This study furthers our understanding of flavivirus genetics, phylogeny and diagnostics. Because the two closest known relatives of T’Ho virus are human pathogens, T’Ho virus could be an unrecognized cause of human disease. It is therefore important that future studies investigate the public health significance of this virus.
Similar content being viewed by others


Background
The genus Flavivirus (family Flaviviridae) contains more than 70 viruses, most of which are transmitted to vertebrates by arthropod vectors such as mosquitoes and ticks [23]. The genus is divided into at least 14 groups on the basis of nucleotide (nt) and deduced amino acid sequence data, antigenic relatedness and other characteristics. Two groups within the genus are the Ntaya and Japanese encephalitis (JE) groups. According to the Ninth Report of the International Committee on Taxonomy of Viruses, the Ntaya group consists of six viruses: Bagaza virus (BAGV), Ilheus virus (ILHV), Israel turkey meningoencephalitis virus (ITV), Ntaya virus (NTAV), Tembusu virus (TMUV) and Zika virus (ZIKV). Rocio virus (ROCV) is considered to represent a subtype of ILHV. The JE group consists of eight viruses: Cacipacore virus (CPCV), Japanese encephalitis virus (JEV), Koutango virus (KOUV), Murray Valley encephalitis virus (MVEV), St. Louis encephalitis virus (SLEV), Usutu virus (USUV), West Nile virus (WNV) and Yaounde virus (YAOV), in addition to Alfuy virus (ALFV) and Kunjin virus (KUNV) which are considered to represent subtypes of MVEV and WNV, respectively.
All flaviviruses possess a single-stranded, positive-sense RNA genome of approximately 11 kb [29]. The genome encodes a major open reading frame (ORF) that is flanked by 5′ and 3′ untranslated regions (UTRs) of ~100 and ~400–700 nt, respectively. The ORF encodes a polyprotein that is co- and post-translationally cleaved to generate three structural proteins, designated the capsid (C), premembrane/membrane (prM/M) and envelope (E) proteins, and at least seven nonstructural (NS) proteins in the gene order: 5′–C–prM (M)–E–NS1–NS2A–NS2B–NS3–NS4A–2K–NS4B–NS5–3′. Some viruses in the JE group utilize efficient −1 ribosomal frameshifting to produce a larger NS1-related protein (NS1’) [18, 33].
Previously, we provided evidence that a novel flavivirus (designated T’Ho virus) occurs in the Yucatan Peninsula of Mexico [16]. The putative virus was identified in a pool of Culex quinquefasciatus mosquitoes collected in 2007 at the Merida zoo, Yucatan State. A 1358-nt region of the NS5 gene was amplified and sequenced by reverse transcription-polymerase chain reaction (RT-PCR) and Sanger sequencing using flavivirus-specific primers. Application of BLASTn analysis revealed that the sequence is genetically equidistant to the corresponding regions of SLEV (72.6% identical), ILHV (72.2%), JEV (72.1%), USUV (71.8%), ROCV (71.4%), MVEV (71.3%), WNV (71.1%) and BAGV (70.1%). Although we successfully amplified T’Ho virus RNA, we were not able to obtain an isolate by virus isolation in African Green Monkey kidney (Vero) or Aedes albopictus (C6/36) mosquito cells or suckling mouse brain inoculation. In this study, the complete genome sequence of T’Ho virus was determined and its genetic relatedness to other flaviviruses was assessed.
Methods
High-throughput sequencing
Trizol Reagent (Invitrogen, Carlsbad, CA, USA) was used to extract total RNA from the pool of Cx. quinquefasciatus previously shown to contain T’Ho virus RNA. Protocols used for the collection, identification and homogenization of mosquitoes have been described elsewhere [16]. RNA extracts were reverse transcribed using SuperScript III (Thermo Fisher, Waltham, MA, USA) with random hexamers. The complementary DNA (cDNA) was RNase-H treated prior to second strand synthesis with Klenow Fragment (NEB, Ipswich, MA, USA). The generated double stranded (ds) DNA was sheared to an average fragment size of 200 bp using manufacturer’s standard settings (Covaris focused-ultrasonicator E210; Woburn, MA, USA). Sheared products were purified (Agencourt Ampure DNA purification beads, Beckman Coulter, Brea, CA, USA) and libraries constructed. Sheared nucleic acid was end-repaired, dA-tailed, ligated to sequencing adapters (NEBNext modules, NEB), PCR amplified (Phusion High-Fidelity DNA polymerase, NEB) and quantitated by Bioanalyzer (Agilent, Santa Clara, CA, USA) for sequencing. Sequencing on the Illumina HiSeq 2500 platform (Illumina, San Diego, CA, USA) resulted in an average of 180 million reads per lane. Samples were de-multiplexed using Illumina software and FastQ files generated. Data were quality filtered and trimmed (Slim-Filter) and de novo assembled using Dwight assembler at custom settings [20]. The generated contiguous sequences (contigs) and unique singleton reads were subjected to homology search using BLASTn and BLASTx against the GenBank database.
5′ and 3′ rapid amplification of cDNA ends
The extreme 5′ and 3′ ends of the T’Ho virus genome were determined by 5′ rapid amplification of cDNA ends (RACE) and 3′ RACE, respectively. In the 5′ RACE reactions, total RNA was reversed transcribed using a T’Ho virus-specific primer. Complementary DNAs were purified by ethanol precipitation and oligo (dC) tails were added to the 3′ ends using 15 units of terminal deoxynucleotidyl transferase (Invitrogen, Carlsbad, CA, USA). Tailing reactions were performed at 37 °C for 30 min and then terminated by heat-inactivation (65 °C for 10 min). Oligo dC-tailed cDNAs were purified by ethanol precipitation then PCR-amplified using a consensus forward primer specific to the C-tailed termini (5′-GACATCGAAAGGGGGGGGGGG-3′) and a reverse primer specific to the T’Ho virus cDNA sequence. In the 3′ RACE reactions, polyadenylate [poly (A)] tails were added to the 3′ ends of the T’Ho virus genomic RNA using 6 units of poly (A) polymerase (Ambion, Austin, TX, USA). Tailing reactions were performed at 37 °C for 1 h and then terminated by heat-inactivation (65 °C for 10 min). Poly (A)-tailed RNA was reverse transcribed using a poly (A) tail-specific primer (5′-GGCCACGCGTCGACTAGTACTTTTTTTTTTTTTTTTT-3′). Complementary DNAs were PCR amplified using a forward primer specific to the T’Ho virus cDNA sequence and a reverse primer that matched the 5′ half of the poly (A)-specific reverse transcription primer (5′-GGCCACGCGTCGACTAGTAC-3′). PCR products generated from the 5′ and 3′ RACE reactions were inserted into the pCR4-TOPO cloning vector (Invitrogen, Carlsbad, CA, USA) and ligated plasmids were transformed into competent TOPO10 Escherichia coli cells (Invitrogen, Carlsbad, CA). Cells were grown on Lysogeny broth (LB) agar containing ampicillin (50 μg/ml) and kanamycin (50 μg/ml), and colonies were screened for inserts by PCR amplification. An aliquot of each PCR product was examined by 1% agarose gel electrophoresis and several PCR products were purified using a QIAquick spin column (Qiagen, Valencia, CA, USA) and sequenced using a 3730 × 1 DNA sequencer (Applied Biosystems, Foster City, CA, USA).
Nucleotide and amino acid sequence alignments
The genomic and predicted amino acid sequences of T’Ho virus were aligned to all other sequences in the Genbank database by application of BLASTn and BLASTp, respectively [1]. Sequence alignments using Clustal Omega (available at http://www.ebi.ac.uk/Tools/msa/clustalo/) were performed to calculate percent nucleotide and amino acid identities between select sequences.
Phylogenetic analysis
The complete ORF sequences of T’Ho virus and other flaviviruses were aligned using Muscle v3.8.31 [14]. Phylogenetic trees were constructed by MrBayes v3.2.6 (evolutionary model GTR with gamma-distributed rates in six categories plus invariant sites, two chains run for 3 × 106 generations with 25% burn-in, resulting in maximum PSRF 1.005 among all continuous parameters) [38], neighbor-joining (NJ, Mega v7.0.20 using Maximum Composite Likelihood distances with Gamma-distributed rates, alpha set to 0.58, pairwise gap deletion and 500 bootstraps), maximum parsimony (MP, Mega v7.0.20 using complete gap deletion and 500 bootstraps) [26] and maximum likelihood (ML, RaxML v8.2.8, using GTRGAMMAIX model and bootstopping criterion autoMRE) [42]. The GTR + G + I model was selected by sample size corrected AICc in jModelTest v2.1.6, and this finding motivated the models chosen for the NJ and ML analyses [36].
Results
Genomic organization and BLAST analysis
The T’Ho virus genome consists of 10,937 nt (Genbank Accession No. EU879061.2) and contains a single 10,284-nt ORF flanked by 5′ and 3′ UTRs of 97 and 556-nt, respectively. The ORF encodes the three structural and seven nonstructural proteins common to all known flaviviruses. The position and length of each gene and untranslated region are shown in Table 1. The complete genome and deduced polyprotein amino acid sequences of T’Ho virus were aligned to all other flaviviruses sequences for which complete genome data are available. The nucleotide sequence alignments indicated that T’Ho virus is most closely related to ROCV (67.4% identity) and ILHV (65.9%) followed by other Ntaya group viruses (58.8–63.3%) and JE group viruses (62.0–63.7%) (Table 2). The polyprotein of T’Ho virus has greatest amino acid identity to ROCV (72.2%) and ILHV (70.6%) followed by other Ntaya group viruses (57.1–66.3%) and JE group viruses (63.2–66.3%). The genome sequence of T’Ho virus was inspected for potential overlapping genes but none were identified.
Organization of the polyprotein
Cleavage sites in the T’Ho virus polyprotein are shown in Table 3. These sites were predicted by aligning the polyprotein sequence of T’Ho virus to those of select other flaviviruses for which cleavage sites have previously been predicted and in some instances experimentally verified. The predicted cleavage sites for T’Ho virus are in agreement with the rules previously established for flaviviruses. The C/prM, prM/E, E/NS1 and 2K/NS4B polyprotein junctions of most known flaviviruses conform to predicted signalase cleavage sites [6] and similar sites were identified at these locations for T’Ho virus (Table 3). The NS1/NS2A junctions of most known flaviviruses occur after a Val-X-Ala site that fulfills the ‘-1, −3’ rule for a signalase site but lacks an upstream hydrophobic domain [6]. The predicted NS1/NS2A junction of T’Ho virus fulfills these requirements. For most flaviviruses, the predicted pr/M junction occurs after an Arg-X-Lys/Arg-Arg or Arg-X-X-Arg motif. T’Ho virus adheres to this rule. The flavivirus virion C/anchor, NS2A/NS2B, NS2B/NS3, NS3/NS4A, NS4A/2K and NS4B/NS5 junctions are commonly cleaved after two basic amino acid residues (KR, RR or RK) [5,6,7] and sites in T’Ho virus are consistent with that rule.
Potential N-linked glycosylation sites were identified using the NetNGlyc 1.0 Server (available at http://www.cbs.dtu.dk/services/NetNGlyc/) with the consensus sequence defined as Asn-X-Ser/Thr (where X is not Pro) in the context of specific surrounding sequences. Fourteen Asn-X-Ser/Thr motifs were identified in the T’Ho virus polyprotein sequence. Eight motifs are predicted to be utilized by N-linked glycans: these are located at prM15, prM148, E154, NS1130, NS1207, NS2A122, NS5234 and NS5655.
Phylogenetic analysis
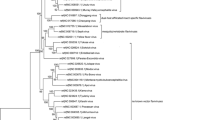
A ML phylogenetic tree was constructed by RAxML v8.2.8 using the complete ORF sequences of T’Ho virus and all 30 mosquito-borne flaviviruses (species and subtypes) listed in the Ninth Report of the International Committee on Taxonomy of Viruses (Fig. 1). Cell fusing agent virus (an insect-specific flavivirus) was used as the outgroup. Phylogenetic trees were also constructed using Bayesian, NJ and MP methods (data not shown). In all trees, T’Ho virus was most closely related to ROCV and ILHV. The Bayesian posterior support for this grouping was strong (100%), in agreement with similarly strong bootstrap support (100% for ML, 100% for MP, 94% for NJ). These viruses cluster within a larger clade that contains the Ntaya group viruses with the exception of ZIKV, although in phylogenies based on short conserved polymerase motifs used in taxonomic classification ZIKV appears more closely related to the Ntaya group viruses [24]. The Bayesian posterior support for this topological arrangement is 100%, but bootstrap support is weaker ranging from 49% for MP to 79% for NJ. Trees generated by all methods agree on the composition of all clades shown in Fig. 1 with 98% or more bootstrap support, but the relative placement of these clades is uncertain (data not shown).

Unrooted maximum likelihood phylogeny of T'Ho virus and other relevant flaviviruses estimated by RAxML. The support out of 100 bootstraps is indicated on each branch. The branch length of cell fusing agent virus (CFAV; the outgroup) is not shown to scale: the actual estimated branch length is 15.60. Although T’Ho virus clusters with Ntaya and JE group viruses with 88% bootstrap support in this phylogeny and 98% posterior support in the MrBayes tree, the CFAV outgroup splits these two groups in around 50% of neighbor-joining bootstrap trees; there is considerable uncertainty in the deep branches of the phylogeny. Genbank Accession numbers for sequences used in the analysis are as follows: T’Ho virus, EU879061.2; Alfuy virus, AY898809.1; Aroa virus, KF917535.1; Bagaza virus, NC_012534.1; Bussuquara virus, AY632536.4; Cacipacore virus, LN849009.1; Cell fusing agent virus, NC_001564.1; Dengue virus type 1, AY277665.2; Dengue virus type 4, KF041260.1; Dengue virus type 2, U87411.1; Dengue virus type 3, AY099336.1; Iguape virus, AY632538.4; Ilheus virus, NC_009028.2; Israel turkey meningoencephalomyelitis virus, KC734552.1; Japanese encephalitis virus, NC_001437.1; Kokobera virus, NC_009029.2; Koutango virus, EU082200.2; Kunjin virus, KX394383.1; Murray Valley encephalitis virus, AF161266.1; Naranjal virus, KF917538.1; Ntaya virus, NC_018705.3; Rocio virus, AY632542.4; Sepik virus, NC_008719.1; St. Louis encephalitis virus, DQ525916.1; Stratford virus, KM225263.1; Tembusu virus, NC_015843.2; Usutu virus, NC_006551.1; Wesselsbron virus, NC_012735.1; West Nile virus, M12294.2; Yaounde virus, EU082199.2; Yellow fever virus, NC_002031.1 and Zika virus, NC_012532.1
Discussion
We report the complete genome sequence of T’Ho virus, a novel putative flavivirus discovered in Cx. quinquefasciatus from the Yucatan Peninsula of Mexico. The closest known relatives of T’Ho virus are ROCV and ILHV, that both belong to the Ntaya group of mosquito-borne flaviviruses, with 67.4% and 65.9% nt. identity, respectively. Based on genetic criteria, we propose that T’Ho virus is classified as a new species within the genus Flavivirus. It has been suggested that flaviviruses with >84% nucleotide sequence identity should be classified within the same species [28]. The genome of T’Ho virus has only 58.8–67.4% nt. identity (mean: 63.4%) to the seven viral species and subtypes currently assigned to the Ntaya group, and this amount of genetic relatedness is not dissimilar to that observed between group members (≥57.9% identity; mean: 65.9%). Phylogenetic analysis of full polyprotein sequences revealed that T’Ho virus forms a distinct clade with Ntaya group viruses, except for ZIKV. Based on previous sequence analysis of a 1358-nt region of the NS5 gene of T’Ho virus that indicated it to be genetically equidistant to ILHV, SLEV, WNV, ROCV and JEV, we speculated that T’Ho virus could also serologically resembled WNV [16]; however, the NS5 gene is one of the most conserved regions of the flavivirus genome [6] and sequence alignments and phylogenetic studies performed using relatively short, conserved sequences are not as robust as those performed with complete genomes.
The two closest known relatives of T’Ho virus are recognized human pathogens. ROCV was responsible for several epidemics of severe encephalitis in Brazil in the 1970s [10, 11, 17, 44]. A case fatality rate of 10% and long-term sequelae in 20% of the surviving patients were reported. ILHV has been sporadically isolated from humans in Central America, South America and the Caribbean, and symptomatic infections are usually characterized by fever, headache, myalgia and arthralgia although central nervous system manifestations have also been reported [25, 37, 39, 40, 45]. Because T’Ho virus is most closely related to known human pathogens, it too could be a cause of human disease. Most other Ntaya group viruses are also recognized pathogens; ZIKV is a cause of febrile illness, neonatal microcephaly and linked to Guillain-Barré syndrome [47]. BAGV, ITV and TMUV are known avian pathogens [4, 9, 30]. Most JE group viruses are also serious pathogens of humans and other vertebrates [21].
An isolate of T’Ho virus is not available; thus, experimental infection studies cannot currently be performed to assess the vector and reservoir competence of mosquito and vertebrate species likely to be involved in its amplification. However, probable vectors and reservoir hosts of T’Ho virus can be inferred from the information available for its closest known relatives. The amplification cycles of ROCV and ILHV are not well defined but most isolations have been made from Aedes and Psorophora spp. mosquitoes (particularly Ps. ferox) with birds implicated as principal reservoir hosts [2, 12, 17, 19, 31, 34]. Most other Ntaya group viruses are primarily maintained in transmission cycles between Culex spp. mosquitoes and birds [13, 22, 43], the notable exception being ZIKV which cycles between Aedes spp. mosquitoes and primates [47]. JE group viruses are primarily maintained in transmission cycles between Culex spp. mosquitoes and birds [21]. It is therefore likely that the principal amplification vectors of T’Ho virus are Culex, Aedes and/or Psorophora spp. mosquitoes and the principal reservoir hosts are birds.
Conclusion
In conclusion, we describe a novel species, T’Ho virus, of the genus Flavivirus, whose closest known relatives are human pathogens; thus, it is feasible to suggest that T’Ho virus may be an unrecognized cause of human disease in the Yucatan Peninsula of Mexico. Our report enables directly the creation of specific PCR diagnostic assays. However, serological cross-reactivity is common with flaviviruses [3], as exemplified in the ongoing ZIKV pandemic where the differential diagnosis of ZIKV and dengue virus infections is difficult [46]. Accordingly, virus isolates or a recombinant virus will be needed for serosurveillance studies in Latin America where several other flaviviruses, including dengue virus, yellow fever virus, ZIKV, WNV, ROCV and ILHV, may co-circulate in overlapping geographic areas.
Abbreviations
- ALFV:
-
Alfuy virus
- BAGV:
-
Bagaza virus
- C:
-
Capsid
- cDNA:
-
Complementary DNA
- CPCV:
-
Cacipacore virus
- ds:
-
Double stranded
- E:
-
Envelope
- ILHV:
-
Ilheus virus
- ITV:
-
Israel turkey meningoencephalitis virus
- JE:
-
Japanese encephalitis
- JEV:
-
Japanese encephalitis virus
- KOUV:
-
Koutango virus
- KUNV:
-
Kunjin virus
- LB:
-
Lysogeny broth
- ML:
-
Maximum likelihood
- MP:
-
Maximum parsimony
- MVEV:
-
Murray Valley encephalitis virus
- NJ:
-
Neighbor-joining
- NS:
-
Nonstructural
- nt:
-
Nucleotide
- NTAV:
-
Ntaya virus
- ORF:
-
Open reading frame
- Poly (A):
-
Polyadenylate
- prM/M:
-
Premembrane/membrane
- RACE:
-
Rapid amplification of cDNA ends
- ROCV:
-
Rocio virus
- RT-PCR:
-
Reverse transcription-polymerase chain reaction
- SLEV:
-
St. Louis encephalitis virus
- TMUV:
-
Tembusu virus
- USUV:
-
Usutu virus
- UTRs:
-
Untranslated regions
- WNV:
-
West Nile virus
- YAOV:
-
Yaounde virus
- ZIKV:
-
Zika virus
References
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10.
Anderson CR, Aitken THG, Downs WG. The isolation of ilheus virus from wild caught forest mosquitoes in trinidad. Am J Trop Med Hyg. 1956;5:621–5.
Calisher CH, Karabatsos N, Dalrymple JM, Shope RE, Porterfield JS, Westaway EG, et al. Antigenic relationships between flaviviruses as determined by cross-neutralization tests with polyclonal antisera. J Gen Virol. 1989;70(Pt 1):37–43.
Cao Z, Zhang C, Liu Y, Liu Y, Ye W, Han J, et al. Tembusu virus in ducks, china. Emerg Infect Dis. 2011;17:1873–5.
Chambers TJ, Grakoui A, Rice CM. Processing of the yellow fever virus nonstructural polyprotein: a catalytically active NS3 proteinase domain and NS2B are required for cleavages at dibasic sites. J Virol. 1991;65:6042–50.
Chambers TJ, Hahn CS, Galler R, Rice CM. Flavivirus genome organization, expression, and replication. Annu Rev Microbiol. 1990;44:649–88.
Chambers TJ, Weir RC, Grakoui A, McCourt DW, Bazan JF, Fletterick RJ, et al. Evidence that the N-terminal domain of nonstructural protein NS3 from yellow fever virus is a serine protease responsible for site-specific cleavages in the viral polyprotein. Proc Natl Acad Sci U S A. 1990;87:8898–902.
Coia G, Parker MD, Speight G, Byrne ME, Westaway EG. Nucleotide and complete amino acid sequences of Kunjin virus: definitive gene order and characteristics of the virus-specified proteins. J Gen Virol. 1988;69(Pt 1):1–21.
Davidson I, Grinberg R, Malkinson M, Mechani S, Pokamunski S, Weisman Y. Diagnosis of turkey meningoencephalitis virus infection in field cases by RT-PCR compared to virus isolation in embryonated eggs and suckling mice. Avian Pathol. 2000;29:35–9.
de Souza Lopes O, Coimbra TL, de Abreu Sacchetta L, Calisher CH. Emergence of a new arbovirus disease in Brazil. I. Isolation and characterization of the etiologic agent, Rocio virus. Am J Epidemiol. 1978;107:444–9.
de Souza Lopes O, de Abreu Sacchetta L, Francy DB, Jakob WL, Calisher CH. Emergence of a new arbovirus disease in Brazil. III. Isolation of Rocio virus from Psorophora ferox (Humboldt, 1819). Am J Epidemiol. 1981;113:122–5.
Derodaniche E. Isolation of the virus of Ilheus encephalitis from mosquitoes of the genus psorophora captured in Honduras. Am J Trop Med Hyg. 1956;5:797–801.
Diallo M, Nabeth P, Ba K, Sall AA, Ba Y, Mondo M, et al. Mosquito vectors of the 1998-1999 outbreak of Rift Valley fever and other arboviruses (Bagaza, Sanar, Wesselsbron and West Nile) in Mauritania and Senegal. Med Vet Entomol. 2005;19:119–26.
Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7.
Falgout B, Markoff L. Evidence that flavivirus NS1-NS2A cleavage is mediated by a membrane-bound host protease in the endoplasmic reticulum. J Virol. 1995;69:7232–43.
Farfan-Ale JA, Lorono-Pino MA, Garcia-Rejon JE, Hovav E, Powers AM, Lin M, et al. Detection of RNA from a novel West Nile-like virus and high prevalence of an insect-specific flavivirus in mosquitoes in the Yucatan Peninsula of Mexico. Am J Trop Med Hyg. 2009;80:85–95.
Figueiredo LT. Emergent arboviruses in Brazil. Rev Soc Bras Med Trop. 2007;40:224–9.
Firth AE, Atkins JF. A conserved predicted pseudoknot in the NS2A-encoding sequence of West Nile and Japanese encephalitis flaviviruses suggests NS1′ may derive from ribosomal frameshifting. Virol J. 2009;6:14.
Galindo P, Derodaniche E. Birds as hosts of Ilheus encephalitis virus in Panama. Am J Trop Med Hyg. 1961;10:395–6.
Golovko G, Khanipov K, Rojas M, Martinez-Alcantara A, Howard JJ, Ballesteros E, et al. Slim-filter: an interactive windows-based application for illumina genome analyzer data assessment and manipulation. BMC Bioinf. 2012;13:166.
Gubler DJ, Kuno G, Markoff L. Flaviviruses. In: Knipe DM, Howley PM, editors. Fields Virology. Fifth ed. Philadelphia: Lippincott Williams and Wilkins; 2007. p. 1153–252.
Hubalek Z, Rudolf I, Nowotny N. Arboviruses pathogenic for domestic and wild animals. Adv Virus Res. 2014;89(89):201–75.
ICTV. Virus taxonomy: classification and nomenclature of viruses: ninth report of the international committee on taxonomy of viruses. San Diego: Elsevier Academic Press; 2012.
ICTV, 2015. The online (10th) report of the international committee on taxonomy of viruses, https://talk.ictvonline.org/ictv-reports/ictv_online_report/positive-sense-rna-viruses/w/flaviviridae. Accessed 6 Mar 2017.
Johnson BW, Cruz C, Felices V, Espinoza WR, Manock SR, Guevara C, et al. Ilheus virus isolate from a human, Ecuador. Emerg Infect Dis. 2007;13:956–8.
Kumar S, Stecher G, Peterson D, Tamura K. MEGA-CC: computing core of molecular evolutionary genetics analysis program for automated and iterative data analysis. Bioinformatics. 2012;28:2685–6.
Kuno G, Chang GJ. Full-length sequencing and genomic characterization of Bagaza, Kedougou, and Zika viruses. Arch Virol. 2007;152:687–96.
Kuno G, Chang GJ, Tsuchiya KR, Karabatsos N, Cropp CB. Phylogeny of the genus Flavivirus. J Virol. 1998;72:73–83.
Lindenbach BD, Murray CL, Thiel H-J, Rice CM. Flaviviridae. In: Knipe DM, Howley PM, editors. Fields Virolgy. Philadelphia: Lippincott Williams and Wilkins; 2013. p. 712–46.
Llorente F, Perez-Ramirez E, Fernandez-Pinero J, Elizalde M, Figuerola J, Soriguer RC, et al. Bagaza virus is pathogenic and transmitted by direct contact in experimentally infected partridges, but is not infectious in house sparrows and adult mice. Vet Res. 2015;46:93.
Lopes OD, Sacchetta LD, Francy DB, Jakob WL, Calisher CH. Emergence of a new arbovirus disease in Brazil. 3. isolation of Rocio virus from Psorophora-Ferox (Humboldt, 1819). Am J Epidemiol. 1981;113:122–5.
Medeiros DB, Nunes MR, Vasconcelos PF, Chang GJ, Kuno G. Complete genome characterization of Rocio virus (Flavivirus: Flaviviridae), a Brazilian flavivirus isolated from a fatal case of encephalitis during an epidemic in Sao Paulo state. J Gen Virol. 2007;88:2237–46.
Melian EB, Hinzman E, Nagasaki T, Firth AE, Wills NM, Nouwens AS, et al. NS1′ of flaviviruses in the Japanese encephalitis virus serogroup is a product of ribosomal frameshifting and plays a role in viral neuroinvasiveness. J Virol. 2010;84:1641–7.
Mitchell CJ, Forattini OP, Miller BR. Vector competence experiments with Rocio virus and 3 mosquito species from the epidemic zone in Brazil. Rev Saude Publica. 1986;20:171–7.
Molloy SS, Bresnahan PA, Leppla SH, Klimpel KR, Thomas G. Human furin is a calcium-dependent serine endoprotease that recognizes the sequence Arg-X-X-Arg and efficiently cleaves anthrax toxin protective antigen. J Biol Chem. 1992;267:16396–402.
Posada D. jModelTest: phylogenetic model averaging. Mol Biol Evol. 2008;25:1253–6.
Prias-Landinez E, Bernal-Cubides C, Morales-Alarcon A. Isolation of Ilheus virus from man in Colombia. Am J Trop Med Hyg. 1968;17:112–4.
Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Hohna S, et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61:539–42.
Spence L, Anderson CR, Downs WG. Isolation of Ilheus virus from human beings in Trinidad, West Indies. Trans R Soc Trop Med Hyg. 1962;56:504–9.
Srihongse S, Johnson CM. The isolation of Ilheus virus from man in Panama. Am J Trop Med Hyg. 1967;16:516–8.
Stadler K, Allison SL, Schalich J, Heinz FX. Proteolytic activation of tick-borne encephalitis virus by furin. J Virol. 1997;71:8475–81.
Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–3.
Tang Y, Diao Y, Chen H, Ou Q, Liu X, Gao X, et al. Isolation and genetic characterization of a Tembusu virus strain isolated from mosquitoes in Shandong, China. Transbound Emerg Dis. 2015;62:209–16.
Tiriba AC, Miziara AM, Lorenco R, da Costa RB, Costa CS, Pinto GH. Primary human epidemic encephalitis induced by Arbovirus found at the sea shore south of the state of Sao Paulo. Clinical study in an emergency hospital. AMB Rev Assoc Med Bras. 1976;22:415–20.
Venegas EA, Aguilar PV, Cruz C, Guevara C, Kochel TJ, Vargas J, et al. Ilheus virus infection in human, Bolivia. Emerg Infect Dis. 2012;18:516–8.
Waggoner JJ, Pinsky BA. Zika virus: diagnostics for an emerging pandemic threat. J Clin Microbiol. 2016;54:860–7.
Weaver SC, Costa F, Garcia-Blanco MA, Ko AI, Ribeiro GS, Saade G, et al. Zika virus: history, emergence, biology, and prospects for control. Antivir Res. 2016;130:69–80.
Acknowledgments
This study was supported by the National Institutes of Health awards 5R21AI067281, U19-AI057158 (Northeast Biodefense Center), U19-AI109761 (Center for Research in Diagnostics and Discovery) and AI088647; the United States Department of Defense and an intramural grant from Iowa State University.
Availability of data and materials
The genome sequence of T’Ho virus has been deposited into the Genbank database.
Authors’ contributions
TB and WIL coordinated the high-throughput sequencing experiments, BJB performed the 5′ and 3′ RACE experiments and prepared the first draft of the manuscript, KSD constructed the phylogenetic trees and MAL, JEG, JAF and CMW coordinated the mosquito collections. All authors reviewed and approved the manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not Applicable.
Ethics approval and consent to participate
Not Applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Briese, T., Loroño-Pino, M.A., Garcia-Rejon, J.E. et al. Complete genome sequence of T’Ho virus, a novel putative flavivirus from the Yucatan Peninsula of Mexico. Virol J 14, 110 (2017). https://doi.org/10.1186/s12985-017-0777-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12985-017-0777-6