
Abstract
Background
Chikungunya virus (CHIKV) is a mosquito-transmitted alphavirus that causes high fever, rash, and recurrent arthritis in humans. It has efficiently adapted to Aedes albopictus, which also inhabits temperate regions and currently causes large outbreaks in the Caribbean and Latin America. Ebola virus (EBOV) is a member of the filovirus family. It causes the Ebola virus disease (EDV), formerly known as Ebola hemorrhagic fever in humans and has a mortality rate of up to 70 %. The last outbreak in Western Africa was the largest in history and has caused approximately 25,000 cases and 10,000 deaths. For both viral infections no specific treatment or licensed vaccine is currently available. The bis-hexasulfonated naphthylurea, suramin, is used as a treatment for trypanosome-caused African river blindness. As a competitive inhibitor of heparin, suramin has been described to have anti-viral activity.
Methods
We tested the activity of suramin during CHIKV or Ebola virus infection, using CHIKV and Ebola envelope glycoprotein pseudotyped lentiviral vectors and wild-type CHIKV and Ebola virus.
Results
Suramin efficiently inhibited CHIKV and Ebola envelope-mediated gene transfer while vesicular stomatitis virus G protein pseudotyped vectors were only marginally affected. In addition, suramin was able to inhibit wild-type CHIKV and Ebola virus replication in vitro. Inhibition occurred at early time points during CHIKV infection.
Conclusion
Suramin, also known as Germanin or Bayer-205, is a market-authorized drug, however shows significant side effects, which probably prevents its use as a CHIKV drug, but due to the high lethality of Ebola virus infections, suramin might be valuable against Ebola infections.
Similar content being viewed by others


Background
The Chikungunya virus (CHIKV) is a mosquito-transmitted alphavirus that causes flu-like symptoms and arthritis. In about 30 % of cases arthritis can last for months or even years, which may cause substantial economic losses [1, 2]. The virus currently spreads from Africa and the Indian Ocean to the Caribbean and Latin America and is now responsible for large, still-ongoing outbreaks with 1.7 million suspected cases as of October 2014 (www.cdc.gov). The mortality rate is very low (0.1 %), but the infection rates are high (about 30 %) and asymptomatic cases are rare (about 15 %) [3]. Due to climate change, globalization, and vector switching, the virus will most likely continue to cause new, worldwide outbreaks also in more temperate regions like Europe or the USA [4, 5].
In contrast, Ebola virus (EBOV) belongs to the filovirus family and is a single (–)-stranded RNA enveloped virus. EBOV infections cause Ebola hemorrhagic fever in humans with mortality rates of up to 70 %. The last outbreak of EBOV in Western Africa was by far the largest outbreak in history. As of May 12th, 2016 an estimated number of 28,616 cases and 11.310 deaths have been reported (www.CDC.gov). For both viral infections no treatment or licensed vaccine exists [2, 6, 7].
CHIKV, like other alphaviruses, enters cells by receptor-mediated endocytosis and a subsequent pH-dependent fusion step. CHIKV has two surface proteins that mediate cell entry: the transmembrane glycoproteins E2 and E1. E2 mediates cell attachment and E1 is a class II viral fusion protein [8]. EBOV cell entry is mediated by a single envelope GP that contains the receptor-binding domain and additionally acts as a fusion protein [9, 10]. Filoviruses utilize a combination of attachment and receptor molecules to enter cells, resulting in a very broad host range. Many factors have been described to interact with EBOV, including proteoglycans. After cell binding, EBOV is taken up by macropinocytosis, followed by low pH-dependent cathepsin B/L-mediated proteolytic cleavage of GP. Finally, binding of the GP to the Niemann-Pick C1 (NPC1) protein as an intracellular receptor is required for the fusion process and virus entry into the cytoplasm [9, 10].
The early steps of viral infections are carried out by the viral glycoproteins which mediate attachment and entry of the virus into the target cell. A tool to investigate the glycoproteins of viruses is the pseudotyping of lentiviral vectors with the desired glycoproteins (here: CHIKV or EBOV) [11]. With this strategy, the lentiviral vectors incorporate a heterologous viral glycoprotein (GP) and thereby acquire the host range of the virus from which the glycoprotein is derived. These pseudotyped vectors enable studies to be carried out without the need for using the native virus, which for Ebola virus reduces the required safety level from the highest level 4 to level 2. We have previously established a chikungunya virus (CHIKV) neutralization assay in a 384-well format [12] and adjusted this multiplex assay for Ebola virus, allowing the analysis of inhibitors of infection in a short period of time, which in cases of virus outbreaks could be of decisive advantage.
Proteoglycans are widely distributed molecules on the cell surface, consisting of a transmembrane protein linked to sulfated glycosaminoglycans (GAGs). Therefore, many pathogens exploit GAGs to cross the cell membrane barrier, using them for initial cell attachment or as entry receptors. These pathogens include several bacteria, parasites, and viruses [13, 14]. It has been shown previously that although CHIKV and Ebola virus enter cells by different pathways, they both use GAGs for their attachment to target cells. Soluble GAGs are able to inhibit pseudotyped vector entry [15–17]. As a competitive inhibitor of GAGs and heparin, suramin has been shown to have anti-viral activity. Suramin, also known as Germanin or Bayer-205, is a market-authorized drug for the treatment of trypanosome-caused river blindness. It has a negative charge and binds to basic side chains of proteins. Several viruses have been described to be inhibited by suramin among them HIV [18, 19] HSV-1 [20], HBV [21], HCV [22], dengue virus [23], EV71 [24], Rift Valley Fever Virus [25] and recently also CHIKV [26, 27]. Here, we extended these observations for CHIKV and EBOV by using pseudotyped-lentiviral vectors carrying envelope glycoproteins and using the wild-type viruses.
Methods
Cell culture
All cells used in this study were cultured at 37 °C under 5 % CO2. HEK 293 T (CRL-1573), MCF7, and Huh7 (CCL-185) cells were grown in Dulbecco’s modified Eagle medium (DMEM; Lonza, Verviers, Belgium). All media were supplemented with 10 % FCS (v/v; PAA, Pasching, Austria), penicillin (50 units/mL), streptomycin (50 μg/mL), and 5 % L-glutamine (200 mM; Lonza, Verviers, Belgium). Suramin was purchased by Sigma-Aldrich (Munich, German) with a purity ≥99 %.
Plasmids and DNA
The gene for the CHIKV E3-E1 envelope polyprotein was expressed from plasmid pIRES2-eGFP-CHIKV E3-E1 [12] and the Zaire Ebola virus envelope polyprotein (pEBOV-GP) gene was obtained from C. Goffinet [28]. Furthermore, the plasmids pMDLg/pRRE, pRSVrev, pRRLsinCMV-GFPpre [29], pCSII-Luc [30] (kind gift of N. Somia) and pHIT-G (encoding VSV-G; [31]) were used for the production of vector particles.
Lentiviral vector particle production
HEK 293 T cells were seeded in 10 cm dishes in 10 ml DMEM. After 16 h, subconfluent cells (~80 % density) were cotransfected with the plasmids pRRLsinCMV-GFPpre or pCSII-Luc (10 μg), pMDLg/pRRE (6.5 μg), pRSVrev (2.5 μg), and pHIT-G, pEBOV-GP (3.5 μg) or pIRES2-eGFP-CHIKV E3-E1 (5.3 μg) using Lipofectamine 2000 (according to the manufacturer’s protocol; Life Technologies). After 24 h of incubation, the medium was replaced with 5 ml fresh DMEM per dish. Another 24 h later, the supernatant containing vector particles was harvested, sterile filtered with 0.45 μm filters (Sartorius, Göttingen, Germany) and frozen at –80 °C.
Transduction of cells with lentiviral vector particles
All transduction experiments were performed using DMEM with 1 % FCS. Transduction of cells with luciferase-encoding lentiviral vectors for luciferase assays was performed by seeding 6000 HEK 293 T cells per well in white CELLSTAR 384-well microtiter plates (Greiner Bio-One, Frickenhausen, Germany) in a volume of 20 μl DMEM using a MultiFlo Microplate Dispenser (BioTek, Bad Friedrichshall, Germany) [12] and incubating for 24 h at 37 °C. Suramin (Sigma, Darmstadt) or rabbit sera (IBT Bioservices, Gaithersburg, MD, USA), were serially diluted with DMEM until the concentrations were twice those required for the experiment, mixed 1:1 with vector particles (either VSV-G, EBOV GP or CHIKV pseudotyped, produced with pCSII-Luc), and incubated in 96-U-well plates (Thermo Scientific, Rockford, IL, USA) at 4 °C for 1 h. The vector particle mixtures were subsequently added to the cells in the 384-well plates using a Matrix Equalizer Multichannel Electronic Pipette (Thermo Scientific). From every dilution/well in the 96-well plates, 20 μl were transferred to three wells each of the 384-well plates (1:2 dilution; triplicate assay). After 16 h incubation, 20 μl BriteLite substrate (PerkinElmer, Rodgau, Germany) were added to each well using the MultiFlo Microplate Dispenser (BioTek). Following an incubation of 5 min at room temperature, the luciferase signal was detected with a PHERAstar FS microplate reader (BMG LABTECH, Ortenberg, Germany).
Cytotoxicity assay
For the cytotoxicity test, 4 × 104 293 T cells per well in a 96-well plate were seeded and incubated with suramin at the same concentrations used in the neutralization assays. Puromycin (2 mg/ml) was used as a positive control for cell killing, while DMEM in the concentration used during the neutralization assay served as a negative control. After incubation for 48 h, the MTT assay was carried out according to the manufacturer’s instructions (Merck Millipore, Darmstadt).
Zaire Ebola virus infections
The Mayinga strain of Zaire Ebola virus (EBOV) (GenBank accession number AF 086833) was used for infections. The virus was propagated in VeroE6 cells and titrated by 50 % tissue culture infectious dose (TCID50) assays. EBOV was preincubated at a multiplicity of infection of 0.1 with different concentrations of suramin (4.6875–300 μg/ml in DMEM containing no FCS) at 37 °C for 30 min. Huh7 cells were then infected with ZEBOV and suramin for 1 h at 37 °C. The inoculum was discarded and the cells were supplied with DMEM (5 % FCS, penicillin, streptomycin) containing different concentration of suramin. Supernatants of cells were collected 48 h post infection and their virus titers were determined by TCID50 analysis. All work with wild-type EBOV was performed in the biosafety level 4 (BSL4) facility of Philipps University, Marburg.
TCID50 analysis of Ebola virus
VeroE6 cells were cultured in 96-well plates to 50 % confluence and infected with 10-fold serial dilutions of supernatants from infected cells (4 replicates). At 7 days post infection (p.i.), when the cytopathic effect had stabilized, cells were analyzed by light microscopy. The TCID50/ml was calculated using the Spearman-Kärber method [32].
CHIKV and VSV infection
The recombinant CHIKV-luci contains the luciferase gene within the CHIKV nsP3 (non-structural protein 3) [33]. The virus was generated by in vitro-transcription of the plasmid pCHIKV-luci after NotI linearization, as described previously [34], followed by transfection of the RNA into BHK-21 cells using Lipofectamine® 2000 (Life Technologies). Supernatants containing virus were harvested 48 h later and the virus was amplified on BHK-21 cells. This replicating luciferase-tagged CHIKV was used to infect 293 T cells at a low MOI of 0.06 and viral replication could be simply detected via luciferase assays. Analogously, VSV infection of target cells was assessed with a VSV encoding luciferase also at a low MOI of 0.2 [35].
Statistical data analysis
Mean values and standard deviation (SD) were analyzed using Microsoft Excel. Half maximal inhibitory concentration (IC50) was calculated using Prism (GraphPad Software).
Results
Analysis of suramin’s toxicity
Although both viruses enter cells by different pathways, CHIKV by receptor-mediated endocytosis and a pH-dependent fusion step and EBOV by a combination of attachment and receptor molecules in the endosomes, both viruses attach to cells via GAGs. Consequently suramin, a competitive inhibitor of GAGs should have anti-viral activity. To exclude that the inhibitory effect of suramin is due to toxicity of the compound, the cytotoxicity of suramin was analyzed by incubating different cell lines with suramin for 16 h and performing a MTT assay (Fig. 1). Suramin displayed only negligible toxicity towards MCF7 however affected Huh7 and 293 T cell viability starting at a concentration of 50 μg/ml (Fig. 1). The CC50 values were 211 μg/ml for 293 T cells and 182 μg/ml for Huh7 cells. CC50 values for MCF7 cells were indeterminable. Therefore effects of suramin at higher concentrations might result from general cell toxicity.

Cytotoxicity of suramin treatment. Cytotoxicity of suramin was analyzed by incubation of MCF7, Huh7 and 293 T cells with suramin for 16 h and conducting a MTT assay by following the manufacturer’s instructions (Merck Millipore, Darmstadt). The graph indicates the amount of viable cells as % of the untreated control and values represent the mean data of two independent experiments done in duplicate
Suramin inhibits CHIKV and EBOV pseudotyped vector transductions
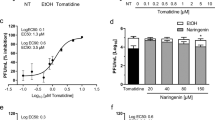
CHIKV and EBOV cell entry are GAG-dependent [15, 16]. The market-authorized drug suramin has been described as a competitive inhibitor of GAGs [36, 37] and thus was tested for its inhibitory activity towards CHIKV and EBOV-pseudotyped vectors. Consequently, transduction of 293 T target cells was performed in the presence of increasing amounts of suramin. VSV-G pseudotyped lentiviral vectors were used as a control to exclude effects on the lentiviral background of the vector system. Figure 2 shows that CHIKV (Fig. 2a) and EBOV GP-mediated transduction (Fig. 2b) was effectively inhibited by suramin with a mean half minimal inhibitory concentration (IC50) of 4,11 ± 0,74 μg/ml for CHIKV and 8,47 ± 3,21 μg/ml for EBOV. VSV-G-mediated gene transfer was less affected by the presence of suramin (IC50 27,37 ± 11,82) and only high suramin concentrations diminished VSV-G mediated transduction (Fig. 2a, b). This implies that suramin is not toxic at the inhibitory concentrations and only partially inhibits VSV-G-mediated entry, but is able to specifically and fully block CHIKV and EBOV GP-mediated entry.

Neutralization assay with suramin. Suramin dissolved in water was serially diluted, incubated with EBOV GP or VSV-G pseudotyped vector particles (a) or CHIKV and VSV-G pseudotyped vector particles (b) and the mixtures were used for transduction of HEK 293 T cells. Neutralizing activity was determined by detection of relative luciferase units (RLUs). The data represent a typical assay with the mean values of triplicates
Suramin blocks CHIKV infection at early time points
The inhibitory effect of suramin on CHIKV-pseudotyped vector particles was confirmed with wild-type CHIKV. HEK 293 T cells were infected with a replicating luciferase-tagged CHIKV (CHIKV-luci) in the presence of suramin. The recombinant CHIKV-luci contains the luciferase gene within the CHIKV nsP3 (non-structural protein 3) gene [33] and viral replication can be detected via luciferase assays. Figure 3a shows a representative assay with the average values of the experiment done in triplicates. Here, 293 T cells were infected for 6 h with CHIKV-luci in the presence of suramin. Increasing doses of suramin drastically inhibited CHIKV infection of cells (IC50 7,38 μg/ml) (Fig. 3). Effects on VSV infection of target cells were assessed with a VSV encoding luciferase [35]. VSV infection of 293 T cells was only inhibited at high suramin concentrations (Fig. 3).

Suramin inhibits CHIKV infection. Suramin was serially diluted, incubated with CHIKV-luci or VSV-luci for 30 min and added to 293 T cells. Its neutralizing activity was detected after 6 h of incubation as relative luciferase activities. The luciferase activity is shown as a percentage, relative to the untreated control. The data show a representative experiment carried out in triplicate
To further analyze the mechanism of viral inhibition by suramin, the drug (10 μg/ml) was added every 30 min for 2.5 h during infection with CHIKV-luci at a low MOI of 0.06 and VSV-luci at a MOI of 0.2 and infection was analyzed after 6 h. Adding the compounds during (0 h) or 30 min after the infection significantly inhibited infection compared to the untreated control. Later addition of the compound had only slight inhibitory effects, however reduced the infectivity to 80 % of the untreated control (Fig. 4a). The same kinetic was also observed when a CHIKV-neutralizing serum was added at the same time points of infection. These data imply that suramin acts on viral entry; however, the influence on later stages of the viral infection cycle might be caused by inhibition of entry of virus released from initially infected cells. In contrast, VSV-luci infection was only inhibited by the neutralizing antibody when added up to 30 min after infection (Fig. 4b). Suramin had only a slight inhibitory effect on VSV when added at all time points of infection.

Suramin acts on early steps of CHIKV infections in vitro. HEK 293 T cells were incubated with CHIKV-luci (a) or VSV-luci (b) and suramin (10 μg/ml). The drug was added during the infection (0 h) and then every 30 min after infection up to 2.5 h after infection. After 6 h, infected cells were detected as relative luciferase activities. CHIKV infection without treatment was set to 100 %
Suramin blocks EBOV replication
To ascertain the relevance of the data obtained with EBOV GP pseudotyped lentiviral vectors, suramin was added in increasing amounts during a wild-type EBOV infection. The virus, at a multiplicity of infection of 0.1, was preincubated with suramin for 30 min. The mixture was then added to Huh7 target cells, washed off after 1 h, and replaced with medium supplemented with suramin at the previous concentration. Incubation was continued for another 48 h. Subsequently, the viral titer was determined as 50 % tissue culture infectious dose (TCID50) values on Vero cells (Fig. 5). Corresponding to the data obtained with EBOV-pseudotyped vectors, there was a distinct inhibition of EBOV replication by suramin with an IC50 of 12,58 ± 5,03 μg/ml: even at a low dose, Ebola virus replication was clearly reduced by up to 1-log. These data confirm that suramin is an inhibitor of EBOV with a selectivity index of 14,46.

Suramin inhibits EBOV replication in vitro. Different concentrations of suramin (4.875–300 μg/ml) were incubated with EBOV (multiplicity of infection of 0.1) prior to infection of Huh7 cells. Supernatants were collected 48 h post infection and virus titers were determined in Vero cells by TCID50 analysis (4 replicates). The data represent the mean values of TCID50 titers from 5 independent experiments. The mock value is indicated as a red square
Discussion
Suramin has been used as an antihelmintic for the treatment of onchocerciasis (African river blindness) since 1920, and is still the only treatment against the adult worms. Suramin is also used with pentamidine to treat early stages of sleeping sickness (African trypanosomiasis) [38]. However, the mode of action against the parasites is still unknown [39]. Suramin is a market-authorized drug and clinical pharmacology data are available. It is metabolically stable, has a long plasma half-life of 30–60 days in humans, is poorly absorbed from the gastrointestinal tract and 80 % of the drug is excreted renally [39, 40].
For the treatment of onchocerciasis, suramin is administered as a single weekly intravenous injection of 1 g suramin for 6 weeks, which might be sufficient to obtain antiviral effects [41]. The most frequent adverse reactions of suramin-treated patients with onchocerciasis are nausea and vomiting. About 90 % of patients develop a reversible urticarial rash. Kidney damage and exfoliative dermatitis occur less commonly. Suramin is also associated with hepatic and bone marrow toxicity, Stephens-Johnson syndrome, and death. There is a greater than 50 % chance of damage to the adrenal cortex [41]. Other toxic effects of suramin in humans have been documented during clinical trials in cancer patients. Reversible liver toxicity, corneal damage, and adrenal insufficiency have been described frequently [42]. The LD50 of suramin in mice is 750 mg/kg [43] after intraperitoneal application and 620 mg/kg following intravenous application [44]. The lowest toxic dose (TDLo) in humans is 46 mg/kg/5 week on an intermittent schedule [45].
Since suramin acts as a competitor of heparin, anticoagulating activity might be expected. However, despite extensive use of suramin, coagulopathy has not been described as a side effect of the treatment. Coagulopathy has only been reported in three female patients receiving suramin as treatment for metastatic adrenocortical carcinoma [46]. For this treatment, a higher suramin dosage of 1.4 g/m2/week was used. The study showed that suramin itself did not prolong the clotting time, but rather a suramin-related anticoagulant, which was heterogeneous in the three patients [46].
Here we have shown that suramin’s toxicity is cell line dependent and highest in HEK 293 T and Huh7 cells, whereas MCF7 cells were only marginally affected. Suramin is able to inhibit CHIKV infections. This has been observed before by others [26, 27]. However the mode of action is still unclear. Inhibition of pseudotyped vector transduction and time of drug-addition experiments indicate an effect on viral entry, as observed by Ho et al. [27]. Nevertheless, except the recurrent arthritis, CHIKV fever is, compared to Ebola virus infections, a rather mild disease and the unwanted side effects of suramin might make it inappropriate for the treatment of CHIKV infections. On the other hand, suramin is able to inhibit Ebola virus pseudotyped lentiviral vectors as well as wild-type Ebola virus in vitro and the severity of the disease makes suramin a therapeutic option as long as more specific drugs are not available. VSV-G pseudotyped vector entry was only slightly inhibited by suramin, but never declined to undetectable levels (Fig. 2). This indicates that the inhibition of CHIKV or EBOV entry by suramin uses a different mechanism to that of VSV. Suramin might bind directly to the cell attachment site of the GPs, similar like soluble GAGs and thereby directly compete with cell binding.
Recently, large screens of Food and Drug Administration (FDA)-approved drugs were performed with EBOV VLPs and identified 53 drugs that are able to inhibit EBOV infections [47]. Microtubule inhibitors were the most potent Ebola virus entry inhibitors, with IC50 values in the low μM range. However, wild-type virus has not yet been used to confirm the drug activity [47]. Others have reported that amiodarone, a multi-ion channel inhibitor and adrenoceptor antagonist, is a potent inhibitor of EBOV cell entry at concentrations reported to be achievable in plasma in humans [48]. All drugs described so far have a broad mode of action and unwanted side effects might be envisioned. Correspondingly, the wide range of toxicities might require improved analogs of suramin. It should, however, be tested in animal models, which can provide indications about the feasibility of using suramin for Ebola treatment. The last EBOV epidemic in Western Africa was the largest in history with over 10,000 confirmed deaths. Earlier outbreaks only caused a few hundred cases. The long time period until the epidemic was under control and the high case fatality rate (about 41 % for the last outbreak) make rapid and effective actions an urgent requirement for future outbreaks. Suramin treatment of EBOV patients might be option for this, as several decades of treatment experience with other pathogens, especially in Africa, are available. Additionally, suramin can be easily produced in high amounts and would be available immediately. The high lethality of Ebola virus infection makes mild side effects of the treatment acceptable as long as there is no better treatment available. Additionally and in parallel, improvements to the drug to reduce unwanted side effects should be attempted.
Conclusion
Suramin inhibits CHIKV and EBOV infections in vitro. Suramin might have too many unwanted side effects for the treatment of CHIKV infections, however might be acceptable for the treatment of Ebola virus infections as long as there is no better treatment available. However, appropriate animal models have to show in vivo applicability first.
References
Suhrbier A, Jaffar-Bandjee M-C, Gasque P. Arthritogenic alphaviruses--an overview. Nat Rev Rheumatol. 2012;8:420–9. doi:10.1038/nrrheum.2012.64.
Weaver SC, Osorio JE, Livengood JA, Chen R, Stinchcomb DT. Chikungunya virus and prospects for a vaccine. Expert Rev Vaccines. 2012;11:1087–101. doi:10.1586/erv.12.84.
Schwartz O, Albert ML. Biology and pathogenesis of chikungunya virus. Nat Rev Microbiol. 2010;8:491–500.
Bajak A. US assesses virus of the Caribbean. Nature. 2014;512:124–5. doi:10.1038/512124a.
Kuehn BM. Chikungunya virus transmission found in the United States: US health authorities brace for wider spread. JAMA. 2014;312:776–7. doi:10.1001/jama.2014.9916.
Feldmann H, Geisbert TW. Ebola haemorrhagic fever. Lancet. 2011;377:849–62. doi:10.1016/S0140-6736(10)60667-8.
Rougeron V, Feldmann H, Grard G, Becker S, Leroy EM. Ebola and Marburg haemorrhagic fever. J Clin Virol. 2015;64:111–9. doi:10.1016/j.jcv.2015.01.014.
Voss JE, Vaney M-C, Duquerroy S, Vonrhein C, Girard-Blanc C, Crublet E, et al. Glycoprotein organization of Chikungunya virus particles revealed by X-ray crystallography. Nature. 2010;468:709–12. doi:10.1038/nature09555.
Takada A. Filovirus tropism: cellular molecules for viral entry. Front Microbiol. 2012;3:34. doi:10.3389/fmicb.2012.00034.
Jae LT, Brummelkamp TR. Emerging intracellular receptors for hemorrhagic fever viruses. Trends Microbiol. 2015;23:392–400. doi:10.1016/j.tim.2015.04.006.
Wool-Lewis RJ, Bates P. Characterization of Ebola virus entry by using pseudotyped viruses: identification of receptor-deficient cell lines. J Virol. 1998;72:3155–60.
Weber C, Konig R, Niedrig M, Emmerich P, Schnierle BS. A neutralization assay for chikungunya virus infections in a multiplex format. J Virol Methods. 2014;201:7–12. doi:10.1016/j.jviromet.2014.02.001.
Kamhi E, Joo EJ, Dordick JS, Linhardt RJ. Glycosaminoglycans in infectious disease. Biol Rev Camb Philos Soc. 2013;88:928–43. doi:10.1111/brv.12034.
Liu J, Thorp SC. Cell surface heparan sulfate and its roles in assisting viral infections. Med Res Rev. 2002;22:1–25.
Salvador B, Sexton NR, Carrion Jr R, Nunneley J, Patterson JL, Steffen I, et al. Filoviruses utilize glycosaminoglycans for their attachment to target cells. J Virol. 2013;87:3295–304. doi:10.1128/JVI.01621-12.
O’Hearn A, Wang M, Cheng H, Lear-Rooney CM, Koning K, Rumschlag-Booms E, et al. Role of EXT1 and glycosaminoglycans in the early stage of filovirus entry. J Virol. 2015. doi:10.1128/JVI.03689-14.
Silva LA, Khomandiak S, Ashbrook AW, Weller R, Heise MT, Morrison TE, Dermody TS. A single-amino-acid polymorphism in Chikungunya virus E2 glycoprotein influences glycosaminoglycan utilization. J Virol. 2014;88:2385–97. doi:10.1128/JVI.03116-13.
Mitsuya H, Popovic M, Yarchoan R, Matsushita S, Gallo RC, Broder S. Suramin protection of T cells in vitro against infectivity and cytopathic effect of HTLV-III. Science. 1984;226:172–4.
Yahi N, Sabatier JM, Nickel P, Mabrouk K, Gonzalez-Scarano F, Fantini J. Suramin inhibits binding of the V3 region of HIV-1 envelope glycoprotein gp120 to galactosylceramide, the receptor for HIV-1 gp120 on human colon epithelial cells. J Biol Chem. 1994;269:24349–53.
Aguilar HC, Anderson WF, Cannon PM. Cytoplasmic tail of Moloney murine leukemia virus envelope protein influences the conformation of the extracellular domain: implications for mechanism of action of the R Peptide. J Virol. 2003;77:1281–91.
Kessler HA, Pottage JC, Trenholme GM, Benson CA, Levin S. Effects of suramin on in vitro HBsAg production by PLC/PRF/5 cells and hepatitis B virus DNA polymerase activity. AIDS Res. 1986;2:63–72.
Garson JA, Lubach D, Passas J, Whitby K, Grant PR. Suramin blocks hepatitis C binding to human hepatoma cells in vitro. J Med Virol. 1999;57:238–42.
Basavannacharya C, Vasudevan SG. Suramin inhibits helicase activity of NS3 protein of dengue virus in a fluorescence-based high throughput assay format. Biochem Biophys Res Commun. 2014;453:539–44. doi:10.1016/j.bbrc.2014.09.113.
Wang Y, Qing J, Sun Y, Rao Z. Suramin inhibits EV71 infection. Antivir Res. 2014;103:1–6. doi:10.1016/j.antiviral.2013.12.008.
Ellenbecker M, Lanchy J-M, Lodmell JS. Inhibition of Rift Valley fever virus replication and perturbation of nucleocapsid-RNA interactions by suramin. Antimicrob Agents Chemother. 2014;58:7405–15. doi:10.1128/AAC.03595-14.
Albulescu IC, van Hoolwerff M, Wolters LA, Bottaro E, Nastruzzi C, Yang SC, et al. Suramin inhibits chikungunya virus replication through multiple mechanisms. Antivir Res. 2015;121:39–46. doi:10.1016/j.antiviral.2015.06.013.
Ho Y-J, Wang Y-M, Lu J-W, Wu T-Y, Lin L-I, Kuo S-C, et al. Suramin Inhibits Chikungunya Virus Entry and Transmission. PLoS One. 2015;10:e0133511. doi:10.1371/journal.pone.0133511.
Goffinet C, Schmidt S, Kern C, Oberbremer L, Keppler OT. Endogenous CD317/Tetherin limits replication of HIV-1 and murine leukemia virus in rodent cells and is resistant to antagonists from primate viruses. J Virol. 2010;84:11374–84. doi:10.1128/JVI.01067-10.
Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, Naldini L. A third-generation lentivirus vector with a conditional packaging system. J Virol. 1998;72:8463–71.
Agarwal S, Nikolai B, Yamaguchi T, Lech P, Somia NV, N1 - Department of Genetics, Cell Biology, and Development, Institute of Human Genetics, University of Minnesota, 420 Delaware Street SE, MMC 206, Minneapolis, MN 55455, USAFAgarwal, Sumit. Construction and use of retroviral vectors encoding the toxic gene barnase. Mol Ther. 2006;14:555–63.
Soneoka Y, Cannon PM, Ramsdale EE, Griffiths JC, Romano G, Kingsman SM, Kingsman AJ. A transient three-plasmid expression system for the production of high titer retroviral vectors. Nucleic Acids Res. 1995;23:628–33.
Hierholzer JC, Killington RA. Virus isolation and quantitation. In: Virology Methods Manual: Elsevier; 1996. p. 25–46. doi:10.1016/B978-012465330-6/50003-8.
von Rhein C, Weidner T, Henß L, Martin J, Weber C, Sliva K, Schnierle BS. Curcumin and Boswellia serrata gum resin extract inhibit chikungunya and vesicular stomatitis virus infections in vitro. Antivir Res. 2016;125:51–7. doi:10.1016/j.antiviral.2015.11.007.
Weber C, Sliva K, von Rhein C, Kümmerer BM, Schnierle BS. The green tea catechin, epigallocatechin gallate inhibits chikungunya virus infection. Antivir Res. 2015;113:1–3. doi:10.1016/j.antiviral.2014.11.001.
Cureton DK, Massol RH, Saffarian S, Kirchhausen TL, Whelan, Sean P J. Vesicular stomatitis virus enters cells through vesicles incompletely coated with clathrin that depend upon actin for internalization. PLoS Pathog. 2009;5:e1000394. doi:10.1371/journal.ppat.1000394
de Clercq E. Suramin in the treatment of AIDS: Mechanism of action. Antiviral Res. 1987;7:1–10. doi:10.1016/0166-3542(87)90034-9.
Schulze A, Gripon P, Urban S. Hepatitis B virus infection initiates with a large surface protein-dependent binding to heparan sulfate proteoglycans. Hepatology. 2007;46:1759–68. doi:10.1002/hep.21896.
Eisenberger MA, Sinibaldi V, Reyno L. Suramin. Cancer Pract. 1995;3:187–9.
McGeary RP, Bennett AJ, Tran QB, Cosgrove KL, Ross BP. Suramin: clinical uses and structure-activity relationships. Mini Rev Med Chem. 2008;8:1384–94.
Cooper MR, Lieberman R, La Rocca RV, Renato V, Gernt PR, Weinberger MS, Headlee DJ, et al. Adaptive control with feedback strategies for suramin dosing. Clin Pharmacol Ther. 1992;52:11–23. doi:10.1038/clpt.1992.97.
Babalola OE. Ocular onchocerciasis: current management and future prospects. Clin Ophthalmol. 2011;5:1479–91. doi:10.2147/OPTH.S8372.
Stein CA, LaRocca RV, Thomas R, McAtee N, Myers CE. Suramin: an anticancer drug with a unique mechanism of action. J Clin Oncol. 1989;7:499–508.
Balzarini J, Mitsuya H, de Clercq E, Broder S. Aurintricarboxylic acid and Evans Blue represent two different classes of anionic compounds which selectively inhibit the cytopathogenicity of human T-cell lymphotropic virus type III/lymphadenopathy-associated virus. Biochem Biophys Res Commun. 1986;136:64–71.
Hawking F. Suramin: with special reference to onchocerciasis. Adv Pharmacol Chemother. 1978;15:289–322.
Weitberg AB, Mayer K, Miller ME, Mikolich DJ. Dysplastic carcinoid tumor and AIDS-related complex. N Engl J Med. 1986;314:1455. doi:10.1056/NEJM198605293142216.
Horne 3rd MK, Stein CA, La Rocca RV, Myers CE. Circulating glycosaminoglycan anticoagulants associated with suramin treatment. Blood. 1988;71:273–9.
Kouznetsova J, Sun W, Martínez-Romero C, Tawa G, Shinn P, Chen CZ, et al. Identification of 53 compounds that block Ebola virus-like particle entry via a repurposing screen of approved drugs. Emerg Microbes Infect. 2014;3:e84. doi:10.1038/emi.2014.88.
Gehring G, Rohrmann K, Atenchong N, Mittler E, Becker S, Dahlmann F, et al. The clinically approved drugs amiodarone, dronedarone and verapamil inhibit filovirus cell entry. J Antimicrob Chemother. 2014;69:2123–31. doi:10.1093/jac/dku091.
Acknowledgements
We thank Markus Eickmann, Gotthard Ludwig and Michael Schmidt from the BSL4 Facility at the Philipps University Marburg and Heike Baumann, Paul-Ehrlich-Institut, for excellent technical assistance.
Funding
This work was supported by a grant of the German Ministry of Health (Bundesministerium für Gesundheit, BMG).
Authors’ contributions
BS designed this study and LH, SiB, TW, NB, KS, CW and StB conceived and designed the experiments. LH, SiB, TW, KS, CW and NB performed the experiments. BS, KS and CW planned and performed the statistical analyses. BS, KS, CW and NB wrote the manuscript. All authors read and approved the manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Henß, L., Beck, S., Weidner, T. et al. Suramin is a potent inhibitor of Chikungunya and Ebola virus cell entry. Virol J 13, 149 (2016). https://doi.org/10.1186/s12985-016-0607-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12985-016-0607-2