
Abstract
The extreme HIV diversity posts a great challenge on development of an effective anti-HIV vaccine. To solve this problem, it is crucial to discover an appropriate immunogens and strategies that are able to prevent the transmission of the diverse viruses that are circulating in the world. Even though there have been a number of broadly neutralizing anti-HIV antibodies (bNAbs) been discovered in recent years, induction of such antibodies to date has only been observed in HIV-1 infection. Here, in this mini review, we review the progress in development of HIV vaccine in eliciting broad immune response, especially production of bNAbs, discuss possible strategies, such as polyvalent sequential vaccination, that facilitates B cell maturation leading to bNAb response.
Similar content being viewed by others

Background
According to the WHO, there were ~36.7 million people worldwide living with HIV/AIDS by the end of 2015 and 2.1 million new HIV infections in 2015. In Canada, there were estimated 75,500 people living with HIV infection or AIDS at the end of 2014, a 9.7% increase from 2011, with more than 2500 people are newly infected each year. Unfortunately, even after over 30 years of intensive research, there is still no effective anti-HIV vaccine. This mini review will focus on the development of anti-HIV vaccine targeting elicitation of broadly neutralizing antibodies (bNAbs) against extremely diverse HIV strains.
HIV diversity and vaccine development
The extreme genetic diversity of HIV, as a result of high baseline rates of viral mutation and replication, has been a great challenge for HIV vaccine development [1, 2]. There are two types of HIV: HIV-1, predominant throughout the world, and HIV-2, found primarily in West/Central Africa. HIV-1 contains four groups: M (main), O (outlier), N (non-M/non-O), and P (pending). Group M is further subdivided into 9 distinct subtypes [3] and numerous additional circulating recombinant forms (CRF) [4]. Viruses within the same subtype differ by up to 20%, within the highly variable env region by up to 38%. Furthermore, the virus continuously diversifies in infected individuals, resulting in the virus quasispecies varying up to 5% genetic difference in the same patient at different time points. These quasispecies compose of a unique and highly complex mixture of variants in infected individuals, and ultimately give rise to a highly diverse global virus population.
Development of broadly effective anti-HIV vaccine
It is widely thought that an effective strategy to prevent HIV infection will likely come from T cell and B-cell mediated immunity, especially a broadly neutralizing antibody (bNAb) response against the Envelope (Env) protein. The power of bNAbs comes from their ability to recognize epitopes from a variety of viruses, i.e. tackling the extreme viral diversity, and their ability to protect in vivo at low plasma levels [2]. A safe vaccine eliciting bNAbs against HIV could be used to attenuate its spread.
To overcome HIV-1 diversity, one approach is to include different clades to develop a broadly protective polyvalent vaccine. However, early studies on polyvalent vaccine showed inconsistent results regarding elicitation of broad immune responses. Several early studies showed that a polyvalent vaccine, comprising a combination of multiple Env proteins, was better at eliciting broader immune responses than monovalent Env in both rabbits and macaques [5,6,7], while a clinical phase 2b trial of HVTN505 combining three envelope glycoproteins from clade A, B, and C env genes did not reduce either the rate of acquisition or set point viral load of new HIV-1 infections [7].
The HIV-1 T cell vaccine field has recently made some significant advances, such as a novel CMV vector developed by Louis Picker et al. [8], and a polyvalent HIV-1 mosaic antigen strategy which utilizes a genetic algorithm to design small sets of artificial intact viral proteins and collectively optimizes coverage of diverse potential epitopes in a targeted population for a given set size, or valency [9]. Vaccination of a Rhesus Macaque model showed that over 50% of SIVmac infections were effectively cleared in animals that were vaccinated with SIV antigens delivered by the CMV-vectored SIV vaccines [8]. Several recent studies have shown that polyvalent HIV-1 Mosaic antigens result in significantly greater breadth and potency of vaccine-elicited T-cell responses than do natural proteins in NHP studies [10,11,12].
Broadly anti-HIV neutralizing antibodies
Anti-HIV bNAbs were discovered in the early 1990s when researchers found antibodies capable of neutralizing different virus subtypes [13]. Characterization of these responses has shown the bNAbs target sites include the conserved regions near the CD4 binding site (CD4bs) [13], the membrane-proximal external region (MPER) [14], and the base of the V3 and V1/V2 loops [15] of which some bNAbs are glycan-dependent [16,17,18]. Despite the early discovery of broadly neutralizing anti-HIV antibodies (bNAbs), including 447-52D (V3 loop), b12 (CD4 binding site), 17b (co-receptor binding site), 2G12 (viral glycan), 4E10 and 2F5 (gp41 MPER), enthusiasm for an Ab-based vaccine was limited based on the unusual characteristics of these bNAbs: 2G12 has three antigen combining sites, instead of the usual two [19]; 2F5 and 4E10 are self-reactive [20, 21]; and b12 is a phage-derived Ab generated by random pairing of heavy and light chains that may have never existed in nature [22]. However, recent development of single-cell antibody cloning techniques applied to plasma B cells of HIV infected patients uncovered variety of new bNAbs (Table 1), and detailed analyses of these antibodies indicated they are approximately 10- to 100-fold more potent and have an increased breadth compared with the original 4 isolates [23, 24]. To date, there have been a few clinical trials with anti-HIV bNAbs that are successful in reducing viral loads, most notably with 3BNC117 (a CD4bs-specific antibody) currently in phase 2 clinical trials [3,4,5]. Other studies also showed that passive infusion of NAbs could effectively protect macaques from vaginal SHIV challenge [25, 26]. These results suggest a role of Abs in HIV protection and control, but HIV has a tendency to accumulate mutations, making it a difficult target in vaccination strategies. Epitope mapping of the new, potent antibodies has invigorated the vaccine field by providing precise regions to target when designing new protein or subunit vaccine antigens to induce bNAbs [27]. However, even with this new wealth of information at hand, generating bNAbs with improved, redesigned antigens still prove to be problematic, and there are no appropriate immunogens/vaccination strategies that have been discovered to elicit an effectively protective Ab response.
Development of immunogens and vaccination strategies to elicit anti-HIV bNAbs
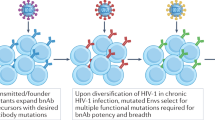
It has been reported that, during chronic infection, potent and cross-reactive bNAbs that are capable of neutralizing heterologous viruses of diverse subtypes develop in a small portion of HIV-1 infected individuals [28,29,30,31]. The effective humoral responses are slow, with NAbs to the initial viral strain appearing after ~12 weeks, and broad NAbs (in 10–30% of individuals) after 2–4 years [30, 32,33,34]. The development of bNAbs was shown to correlate with high plasma viremia and could result from evolving antigen exposure over many years that has allowed sufficient somatic hypermutation in the B-cell receptors (BCRs) and focuses the B-cell response to the conserved neutralization sites on Env [30]. Therefore, delayed bNAb response might be attributed to the slow, antigen-dependent affinity maturation process. Abs typically accumulate mutations in the complementarity determining region (CDR) loops, i.e. the typical antigen contact region [35]. Whereas most human Abs that have undergone affinity maturation carry 15–20 somatic mutations, potent anti-HIV bNAbs carry 40–110 mutations. Reversion of these mutations to the germline sequence drastically reduces their neutralizing potency and breadth [36,37,38,39,40]. These findings suggest that bNAb-producing B cells are the products of clonal evolution which coined the term “B cell maturation”. Thus, selecting the right combination of immunogens and vaccination strategy is crucial for induction of these bNAb-producing B cells via continual increase in affinity-driven selection in the germinal centers (GCs).
A vaccine strategy that aims to mimic the diverse antigenic exposure experienced during natural infection may generate NAbs of greater breadth and potency. Morner et al. achieved greater focusing of the immune response on conserved regions of Env by sequential vaccination of macaques with gp120 core protein followed by trimer boosting [41]. Similarly, the administration of sequential rather than single or mixed patient-derived gp140 Env genes in rabbits showed a marginal improvement in breadth of neutralization [42]. Sellhorn et al. recently described a novel assembly of gp140 heterotrimers which had gp140 subunits from two different genetic sources and improved potency of NAb responses in rabbits as compared to homotrimeric equivalents [43]. Wang et al. also showed through vaccinating mice with three gp120 variants that sequential vaccination is preferred over a cocktail for induction of cross-reactive Abs focused on the shared CD4 binding site epitope [44].
In order to develop an effective and safe polyvalent anti-HIV vaccine, our group has established a SHIV (and HIV) vaccine system which is composed of three DNA plasmids containing complementary subgenomic viral DNAs (Fig. 1), to investigate the optimal polyvalent vaccination strategy. Co-transfection of these three plasmids can produce SHIV (or HIV) virus-like particles (VLPs) containing two distinct subgenomic RNAs that are complementary to each other producing proviral DNA when infecting susceptible cells. The integrated proviral DNA is devoid of most viral coding sequences preventing any viral propagation but retains the env gene for continuous gp120/gp41 expression at the cell surface. The resultant VLPs are morphologically indistinguishable from a wild type virus, and can elicit direct responses by binding surface Ig on B cells, as well as bind target cells through CD4 and CCR5, induce all of the conformational changes, and expose all hidden epitopes. After entering the target cells, expression of HIV-1 Env glycoproteins should also promote proteosomal processing and Env peptide presentation through MHC class I, thus these VLPs can induce both humoral and cellular immune responses. Production of these polyvalent Env-based vaccines may initially appear intuitive, e.g. cloning of multiple HIV-1 env sequences, but until recently this type of cloning represented one of the greatest obstacles to this endeavor. Simply put, unique and conserved restriction sites do not exist for cloning the extremely diverse HIV-1 env gene. We have developed a yeast-based cloning system to clone any HIV-1 env into a common HIV-1 or SHIV backbone through sequence homology recombination/gap repair [45,46,47]. This system was designed for rapid yeast-based cloning and we have introduced >130 HIV-1 env genes with balanced representation from different HIV-1 subtypes (e.g. A, B, C, D) for our polyvalent vaccine construction. Furthermore, the continuous Env protein expression (but not the viral particles) was successfully detected in an in vitro susceptible cell culture.

SHIVenv vaccine system. a The vaccine vector is derived from 293T transfections with three DNA plasmids: 1 pREC_SHIVKB9 _gag/pol vector containing gag and pol coding sequence from SHIVKB9 but lacking the LTRs or RNA packaging elements; 2 pREC_SHIVKB9_env_Δgagpol vector which contains the RNA packaging signal, env and 3′LTR, but lacks 5′LTR, gag and pol sequences; 3 pCMV_SHIVKB9_ cpltRU5 which contains only the 5′LTR, primer binding site, and the RNA packaging signal. b Generation of SHIVenv virus-like particles as HIV vaccine candidate through triple transfection. c Production of defective SHIV-1 proviral DNA and Env glycoproteins by pseudotyped virus containing the two complementary subgenomic viral RNAs
We have tested our polyvalent anti-HIV vaccine in a human CD4 B cell transgenic mouse model which was established to express human CD4 receptor on the surface of B cells, thus can mediate HIV binding, and expose the hidden epitopes on viral gp120. We have tested 25 primary isolates (subtype A, B, C, and D) derived functional Envs and 25 nonfunctional inter-subtype recombinant Env-based VLP vaccines in the human CD4 B cell transgenic mice. The results showed that the sequential vaccination with the 25 functional primary Env-based polyvalent vaccine elicited broader NAb response than any other vaccine combination and vaccination strategy when examining inhibition of a number tier 1 and 2 viruses with different HIV-1 subtype Envs (e.g. A, B, C, and D). More importantly, with each increase in the diversity and number of vaccines (i.e. sequential vaccination), we observed a greater breadth in humoral responses (unpublished data). These results suggest that continuous stimulation with diverse HIV-1 Env immunogens will modulate B cell affinity maturation, educate the immune response to focus on certain regions (most likely on the conserved regions) in Env, and finally have the best success of generating broad NAb response.
Conclusions
A major obstacle for HIV vaccine development is the extreme virus diversity. Although a number of bNAbs have been isolated from HIV patients, the vaccine and procedures capable to elicit such a response remain a mystery. The sequential vaccination with multiple immunogen variants might favor B cell re-circulation within GCs for additional rounds of affinity maturation. This may promote the B cell response to focus on the most conserved regions of immunogens through positive selection, resulting in more potent and broader antibody responses.
References
Gao F, Weaver EA, Lu Z, Li Y, Liao HX, Ma B, et al. Antigenicity and immunogenicity of a synthetic human immunodeficiency virus type 1 group m consensus envelope glycoprotein. J Virol. 2005;79:1154–63.
Gaschen B, Taylor J, Yusim K, Foley B, Gao F, Lang D, et al. Diversity considerations in HIV-1 vaccine selection. Science. 2002;296:2354–60.
Haynes BF, Moody MA, Alam M, Bonsignori M, Verkoczy L, Ferrari G, et al. Progress in HIV-1 vaccine development. J Allergy Clin Immunol. 2014;134:3–10.
Robertson DL, Anderson JP, Bradac JA, Carr JK, Foley B, Funkhouser RK, et al. HIV-1 nomenclature proposal. Science. 2000;288:55–6.
Cho MW, Kim YB, Lee MK, Gupta KC, Ross W, Plishka R, et al. Polyvalent envelope glycoprotein vaccine elicits a broader neutralizing antibody response but is unable to provide sterilizing protection against heterologous simian/human immunodeficiency virus infection in pigtailed macaques. J Virol. 2001;75:2224–34.
Seaman MS, Leblanc DF, Grandpre LE, Bartman MT, Montefiori DC, Letvin NL, et al. Standardized assessment of NAb responses elicited in rhesus monkeys immunized with single- or multi-clade HIV-1 envelope immunogens. Virology. 2007;367:175–86.
Wang S, Pal R, Mascola JR, Chou TH, Mboudjeka I, Shen S, et al. Polyvalent HIV-1 Env vaccine formulations delivered by the DNA priming plus protein boosting approach are effective in generating neutralizing antibodies against primary human immunodeficiency virus type 1 isolates from subtypes A, B, C, D and E. Virology. 2006;350:34–47.
Hansen SG, Sacha JB, Hughes CM, Ford JC, Burwitz BJ, Scholz I, et al. Cytomegalovirus vectors violate CD8+ T cell epitope recognition paradigms. Science. 2013;340:1237874.
Korber BT, Letvin NL, Haynes BF. T-cell vaccine strategies for human immunodeficiency virus, the virus with a thousand faces. J Virol. 2009;83:8300–14.
Barouch DH, O’Brien KL, Simmons NL, King SL, Abbink P, Maxfield LF, et al. Mosaic HIV-1 vaccines expand the breadth and depth of cellular immune responses in rhesus monkeys. Nat Med. 2010;16:319–23.
Hansen SG, Wu HL, Burwitz BJ, Hughes CM, Hammond KB, Ventura AB, et al. Broadly targeted CD8(+) T cell responses restricted by major histocompatibility complex E. Science. 2016;351:714–20.
Santra S, Liao HX, Zhang R, Muldoon M, Watson S, Fischer W, et al. Mosaic vaccines elicit CD8+ T lymphocyte responses that confer enhanced immune coverage of diverse HIV strains in monkeys. Nat Med. 2010;16:324–8.
Corti D, Langedijk JP, Hinz A, Seaman MS, Vanzetta F, Fernandez-Rodriguez BM, et al. Analysis of memory B cell responses and isolation of novel monoclonal antibodies with neutralizing breadth from HIV-1-infected individuals. PLoS ONE. 2010;5:e8805.
Huang J, Ofek G, Laub L, Louder MK, Doria-Rose NA, Longo NS, et al. Broad and potent neutralization of HIV-1 by a gp41-specific human antibody. Nature. 2012;491:406–12.
Walker LM, Huber M, Doores KJ, Falkowska E, Pejchal R, Julien JP, et al. Broad neutralization coverage of HIV by multiple highly potent antibodies. Nature. 2011;477:466–70.
McLellan JS, Pancera M, Carrico C, Gorman J, Julien JP, Khayat R, et al. Structure of HIV-1 gp120 V1/V2 domain with broadly neutralizing antibody PG9. Nature. 2011;480:336–43.
Pejchal R, Doores KJ, Walker LM, Khayat R, Huang PS, Wang SK, et al. A potent and broad neutralizing antibody recognizes and penetrates the HIV glycan shield. Science. 2011;334:1097–103.
Walker LM, Phogat SK, Chan-Hui PY, Wagner D, Phung P, Goss JL, et al. Broad and potent neutralizing antibodies from an African donor reveal a new HIV-1 vaccine target. Science. 2009;326:285–9.
Calarese DA, Scanlan CN, Zwick MB, Deechongkit S, Mimura Y, Kunert R, et al. Antibody domain exchange is an immunological solution to carbohydrate cluster recognition. Science. 2003;300:2065–71.
Haynes BF, Fleming J, St Clair EW, Katinger H, Stiegler G, Kunert R, et al. Cardiolipin polyspecific autoreactivity in two broadly neutralizing HIV-1 antibodies. Science. 2005;308:1906–8.
Yang G, Holl TM, Liu Y, Li Y, Lu X, Nicely NI, et al. Identification of autoantigens recognized by the 2F5 and 4E10 broadly neutralizing HIV-1 antibodies. J Exp Med. 2013;210:241–56.
Burton DR, Pyati J, Koduri R, Sharp SJ, Thornton GB, Parren PW, et al. Efficient neutralization of primary isolates of HIV-1 by a recombinant human monoclonal antibody. Science. 1994;266:1024–7.
Bonsignori M, Liao HX, Gao F, Williams WB, Alam SM, Montefiori DC, et al. Antibody-virus co-evolution in HIV infection: paths for HIV vaccine development. Immunol Rev. 2017;275:145–60.
McCoy LE, Burton DR. Identification and specificity of broadly neutralizing antibodies against HIV. Immunol Rev. 2017;275:11–20.
Klein K, Veazey RS, Warrier R, Hraber P, Doyle-Meyers LA, Buffa V, et al. Neutralizing IgG at the portal of infection mediates protection against vaginal simian/human immunodeficiency virus challenge. J Virol. 2013;87:11604–16.
Mascola JR, Stiegler G, VanCott TC, Katinger H, Carpenter CB, Hanson CE, et al. Protection of macaques against vaginal transmission of a pathogenic HIV-1/SIV chimeric virus by passive infusion of neutralizing antibodies. Nat Med. 2000;6:207–10.
Kwong PD, Mascola JR, Nabel GJ. Broadly neutralizing antibodies and the search for an HIV-1 vaccine: the end of the beginning. Nat Rev Immunol. 2013;13:693–701.
Dhillon AK, Donners H, Pantophlet R, Johnson WE, Decker JM, Shaw GM, et al. Dissecting the neutralizing antibody specificities of broadly neutralizing sera from human immunodeficiency virus type 1-infected donors. J Virol. 2007;81:6548–62.
Doria-Rose NA, Klein RM, Manion MM, O’Dell S, Phogat A, Chakrabarti B, et al. Frequency and phenotype of human immunodeficiency virus envelope-specific B cells from patients with broadly cross-neutralizing antibodies. J Virol. 2009;83:188–99.
Sather DN, Armann J, Ching LK, Mavrantoni A, Sellhorn G, Caldwell Z, et al. Factors associated with the development of cross-reactive neutralizing antibodies during human immunodeficiency virus type 1. Infection. 2009;83:757–69.
Simek MD, Rida W, Priddy FH, Pung P, Carrow E, Laufer DS, et al. Human immunodeficiency virus type 1 elite neutralizers: individuals with broad and potent neutralizing activity identified by using a high-throughput neutralization assay together with an analytical selection algorithm. J Virol. 2009;83:7337–48.
Gray ES, Madiga MC, Hermanus T, Moore PL, Wibmer CK, Tumba NL, et al. The neutralization breadth of HIV-1 develops incrementally over 4 years and is associated with CD4+ T cell decline and high viral load during acute infection. J Virol. 2011;85:4828–40.
Mikell I, Sather DN, Kalams SA, Altfeld M, Alter G, Stamatatos L. Characteristics of the earliest cross-neutralizing antibody response to HIV-1. PLoS Pathog. 2011;7:e1001251.
Stamatatos L, Morris L, Burton DR, Mascola JR. Neutralizing antibodies generated during natural HIV-1 infection: good news for an HIV-1 vaccine? Nat Med. 2009;15:866–70.
Klein F, Diskin R, Scheid JF, Gaebler C, Mouquet H, Georgiev IS, et al. Somatic mutations of the immunoglobulin framework are generally required for broad and potent HIV-1 neutralization. Cell. 2013;153:126–38.
Mouquet H, Scheid JF, Zoller MJ, Krogsgaard M, Ott RG, Shukair S, et al. Polyreactivity increases the apparent affinity of anti-HIV antibodies by heteroligation. Nature. 2010;467:591–5.
Scheid JF, Mouquet H, Ueberheide B, Diskin R, Klein F, Oliveira TY, et al. Sequence and structural convergence of broad and potent HIV antibodies that mimic CD4 binding. Science. 2011;333:1633–7.
Wu X, Zhou T, Zhu J, Zhang B, Georgiev I, Wang C, et al. Focused evolution of HIV-1 neutralizing antibodies revealed by structures and deep sequencing. Science. 2011;333:1593–602.
Xiao X, Chen W, Feng Y, Zhu Z, Prabakaran P, Wang Y, et al. Germline-like predecessors of broadly neutralizing antibodies lack measurable binding to HIV-1 envelope glycoproteins: implications for evasion of immune responses and design of vaccine immunogens. Biochem Biophys Res Commun. 2009;390:404–9.
Zhou T, Georgiev I, Wu X, Yang ZY, Dai K, Finzi A, et al. Structural basis for broad and potent neutralization of HIV-1 by antibody VRC01. Science. 2010;329:811–7.
Morner A, Douagi I, Forsell MN, Sundling C, Dosenovic P, O’Dell S, et al. Human immunodeficiency virus type 1 env trimer immunization of macaques and impact of priming with viral vector or stabilized core protein. J Virol. 2009;83:540–51.
Malherbe DC, Doria-Rose NA, Misher L, Beckett T, Puryear WB, Schuman JT, et al. Sequential immunization with a subtype B HIV-1 envelope quasispecies partially mimics the in vivo development of neutralizing antibodies. J Virol. 2011;85:5262–74.
Sellhorn G, Kraft Z, Caldwell Z, Ellingson K, Mineart C, Seaman MS, et al. Engineering, expression, purification, and characterization of stable clade A/B recombinant soluble heterotrimeric gp140 proteins. J Virol. 2012;86:128–42.
Wang S, Mata-Fink J, Kriegsman B, Hanson M, Irvine DJ, Eisen HN, et al. Manipulating the selection forces during affinity maturation to generate cross-reactive HIV antibodies. Cell. 2015;160:785–97.
Dudley DM, Gao Y, Nelson KN, Henry KR, Nankya I, Gibson RM, et al. A novel yeast-based recombination method to clone and propagate diverse HIV-1 isolates. Biotechniques. 2009;46:458–67.
Marozsan AJ, Arts EJ. Development of a yeast-based recombination cloning/system for the analysis of gene products from diverse human immunodeficiency virus type 1 isolates. J Virol Methods. 2003;111:111–20.
Moore DM, Arts EJ, Gao Y, Marozsan AJ. A yeast recombination-based cloning system to produce chimeric HIV-1 viruses and express HIV-1 genes. Hum Retrovir Protoc Virol Mol Biol. 2005;304:369–85.
Authors’ contributions
All authors contributed to writing and reviewing the manuscript. All authors read and approved the final manuscript.
Acknowledgements
Work in the corresponding author’s laboratory was supported by a Canadian Institute of Health Research Grant (HBF143165), and NIAID, NIH (R0184816).
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
Not applicable.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Funding
CIHR HBF143165, NIAID NIH R0184816.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Ahmed, Y., Tian, M. & Gao, Y. Development of an anti-HIV vaccine eliciting broadly neutralizing antibodies. AIDS Res Ther 14, 50 (2017). https://doi.org/10.1186/s12981-017-0178-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12981-017-0178-3