
Abstract
Background
Progression rates from initial HIV-1 infection to advanced AIDS vary significantly among infected individuals. A distinct subgroup of HIV-1-infected individuals—termed viremic non-progressors (VNP) or controllers—do not seem to progress to AIDS, maintaining high CD4+ T cell counts despite high levels of viremia for many years. Several studies have evaluated multiple host factors, including immune activation, trying to elucidate the atypical HIV-1 disease progression in these patients; however, limited work has been done to characterize viral factors in viremic controllers.
Methods
We analyzed HIV-1 isolates from three VNP individuals and compared the replicative fitness, near full-length HIV-1 genomes and intra-patient HIV-1 genetic diversity with viruses from three typical (TP) and one rapid (RP) progressor individuals.
Results
Viremic non-progressors and typical patients were infected for >10 years (range 10–17 years), with a mean CD4+ T-cell count of 472 cells/mm3 (442–529) and 400 cells/mm3 (126–789), respectively. VNP individuals had a less marked decline in CD4+ cells (mean −0.56, range −0.4 to −0.7 CD4+/month) than TP patients (mean −10.3, −8.2 to −13.1 CD4+/month). Interestingly, VNP individuals carried viruses with impaired replicative fitness, compared to HIV-1 isolates from the TP and RP patients (p < 0.05, 95% CI). Although analyses of the near full-length HIV-1 genomes showed no clear patterns of single-nucleotide polymorphisms (SNP) that could explain the decrease in replicative fitness, both the number of SNPs and HIV-1 population diversity correlated inversely with the replication capacity of the viruses (r = −0.956 and r = −0.878, p < 0.01, respectively).
Conclusion
It is likely that complex multifactorial parameters govern HIV-1 disease progression in each individual, starting with the infecting virus (phenotype, load, and quasispecies diversity) and the intrinsic ability of the host to respond to the infection. Here we analyzed a subset of viremic controller patients and demonstrated that similar to the phenomenon observed in patients with a discordant response to antiretroviral therapy (i.e., high CD4+ cell counts with detectable plasma HIV-1 RNA load), reduced viral replicative fitness seems to be linked to slow disease progression in these antiretroviral-naïve individuals.
Similar content being viewed by others


Background
Infection with human immunodeficiency virus type 1 (HIV-1)—in the absence of antiretroviral therapy—usually follows a well-defined path of virological and immunological changes; however, progression rates to advanced AIDS vary significantly among infected individuals [1, 2]. Based on plasma HIV-1 RNA (viral) load, CD4+ T-cell counts, and symptomatic or asymptomatic HIV-1 infection adult patients have been classified into four groups: (i) rapid progressors (RP), (ii) typical progressors (TP), and two groups of long-term non-progressors (LTNP) namely (iii) elite controllers and (iv) viremic controllers [3, 4]. Among the latter, a rare subgroup of HIV-1-infected individuals has been described who do not seem to progress to AIDS, maintaining high CD4+ T-cell counts despite high levels of viremia (i.e., over 2000 HIV-1 copies RNA/ml of plasma) for many years, also called viremic non-progressors (VNPs) [5–8].
Multiple studies have tried to associate these differences in HIV-1 disease progression with a multitude of host (HIV-1-specific immune response and genetic markers) and viral factors [9–13]. Among the immunological factors, strong HIV-1-specific cytotoxic T cell (CTL) responses have been shown to control HIV-1 infection right after transmission, perhaps determining viral set point in chronic stages of infection, which may help reduce the rate of HIV-1 disease progression [14, 15]. Similarly, a series of host genetic factors that seem to influence HIV-1 progression rates have been well described, including human leukocyte antigen (HLA) class I alleles such as HLA-B*27 and HLA-B*57 [16, 17] and a 32 bp deletion in the CCR5 chemokine receptor gene [18–22]. On the other hand, the impact of restriction cellular factors like APOBEC3, TRIM5α, tetherin, and SAMHD1 on HIV-1 progression is still debated [10, 23]. Altogether, it has been long recognized that immune activation is one of the major contributors to HIV-1 disease and pathogenesis [6, 24–27].
Viral factors—such as impaired HIV-1 replicative fitness—have been associated with slow or limited HIV-1 disease progression [11, 28–31], particularly in patients infected with heavily mutated drug resistant viruses [28, 30, 32–35]. High fitness of transmitted HIV-1 has been related to rapid disease progression [36], while some individuals carrying viruses with decreased replication capacity during acute/early infection have been classified as HIV controllers [37]. Although isolating replication competent HIV-1 strains from elite controllers has been difficult, two studies failed to identify any major defect on the replication ability of these viruses [38, 39]. In contrast, recombinant viruses constructed with HIV-1 gag, pol, and/or env genes from elite controllers exhibited reduced replication capacity compared to recombinant viruses constructed from typical progressors [40–42]. On the other hand, very limited information about the replicative fitness of viruses infecting viremic non-progressor individuals is currently available [6, 43]. If a decrease in HIV-1 replicative fitness is associated with slower/no disease progression, what is causing the virus to have this reduced replication capacity—in the absence of drug resistance mutations—in a certain number of non-progressor patients?
To date, no common pattern or polymorphism(s) shared among HIV-1 variants infecting non-progressor individuals have been clearly defined. Several studies have described changes in the LTR, e.g., in the Sp1 binding site [44] or large deletions [11, 45] that could be associated with slow disease progression. Detrimental mutations in Gag have been associated with viral attenuation [46, 47], which seem to play an important role in HIV-1 disease [48, 49]. Significant replicative fitness loss was observed in recombinant viruses encoding pol genes with mutations affecting HLA-A, but surprisingly not HLA-B, binding [50], although the most attenuated viruses were those constructed from HIV-1 elite controllers expressing HLA-B*57 and HLA-B*51 [40]. Changes in the V2 region of gp120 [51, 52] and a single amino acid deletion in gp41 [53] in the env gene have also been associated with slow disease progression. However, most of the changes in the HIV-1 genome linked to altered pathogenesis have been identified in accessory genes. Single amino acid substitutions [54, 55] and large deletions [56, 57] in Nef have been associated with a decrease in replicative fitness and slow disease progression. Mutations at positions 72 and 77 in Vpr, disrupting the viral protein function, were frequently detected in non-progressors [58, 59]. Finally, single or large insertions in Vpu [60] and amino acid substitutions in Vif [61, 62] have also been associated with non-progression in HIV-1 disease.
In this pilot study we quantified the replicative fitness of HIV-1 isolates obtained from extremely rare viremic non-progressor HIV-infected individuals and compared them to the fitness of those from patients with typical or rapid disease progression. Using deep sequencing we analyzed the full-length HIV-1 genomes and determined intrapatient HIV-1 diversity to investigate if these viral factors could be contributing to the maintenance of stable CD4+ T-cell counts despite persistence viremia for more than 10 years, in the absence of antiretroviral treatment.
Methods
Clinical samples
Blood samples were obtained during routine patient monitoring from a well-characterized cohort of HIV-infected individuals at the Special Immunology Unite (SIU) at Case Western Reserve University/University Hospitals Cleveland Medical Center (CWRU/UHCMC), with the written informed consent. Seven subjects were selected and grouped based on the following definitions: (i) viremic non-progressors (VNP, n = 3) corresponding to patients serologically proven to be HIV-1-infected for at least 10 years, CD4+ T-cell decline of <45 cells/mm3 per year and repeated plasma HIV-1 RNA load >1000 copies/ml in the absence of antiretroviral therapy; (ii) typical progressors (TP, n = 3) corresponding to patients serologically proven to be HIV-1-infected for at least 10 years, CD4+ T-cell decline of >75 cells/mm3 per year and repeated plasma HIV-1 RNA load >1000 copies/ml in the absence of antiretroviral therapy; and (iii) rapid progressor (RP, n = 1) corresponding to a patient serologically proven to be HIV-1-infected for at least 5 years, CD4+ T-cell decline of >77 cells/mm3 per year and repeated plasma HIV-1 RNA load >10,000 copies/ml in the absence of antiretroviral therapy. In the case of the VNP and TP patients, CD4+ T-cell numbers and plasma HIV-1 load levels were monitored for a minimum of 30 months (18 months for the RP patient) with at least 10 determinations over this period. Demographics, clinical and virological characteristics are summarized in Table 1.
Cells and viruses
Peripheral blood mononuclear cells (PBMC), obtained from HIV-seronegative donors, were stimulated with 2.5 µg/ml of phytohemagglutinin (PHA; Gibco BRL) and maintained in RPMI 1640/2 mM l-glutamine media (Cellgro; Mediatech, Herndon, VA) supplemented with 10% fetal bovine serum (Cellgro), 10 mM HEPES buffer (N-2-hydroxyethylpiperazine-N-2-ethanesulfonic acid; Cellgro), 1 ng/ml of interleukin-2 (IL-2) (Gibco, BRL), 100 U of penicillin/m (Cellgro) and 100 μg of streptomycin/ml (Cellgro), for three days before infection with HIV-1 [28]. HIV-1 isolates were obtained from all seven patients by co-cultivating their PBMCs with PBMCs from HIV-seronegative donors as described [28]. Three primary HIV-1 isolates (HIV-1A-92UG029, HIV-1B-92US076, and HIV-1AE-CMU06) were obtained from the AIDS Research and Reference Reagent Program and propagated in PHA-stimulated, IL-2-treated PBMCs. Tissue culture dose for 50% infectivity (TCID50) was determined for each virus in PBMCs in triplicate with serially diluted stocks, based on the reverse transcriptase (RT) activity in culture supernatants on day 7 of culture, using the Reed and Muench method [63]. Viral titers were expressed as infectious units per milliliter (IU/ml).
HIV-1 replicative fitness determination using viral growth kinetics analysis
The ability of the seven patient-derived HIV-1 isolates, plus the primary wild-type HIV-1B-92US076 isolate used as control, to replicate in the absence of drug pressure was determined by measuring viral growth kinetics as described [64, 65]. Briefly, 1 × 106 PHA-stimulated, IL-2-treated PBMCs were infected at a multiplicity of infection (MOI) of 0.001 IU/cell in 1 ml of RPMI 1640 medium and incubated for 2 h at 37 °C in 5% CO2. HIV-infected cells were then washed twice with 1x PBS and split to be cultured in triplicate wells of a 96-well plate (1 × 105 cells/well). Fresh PHA-stimulated, IL-2–treated PBMCs (5 × 104 cells) were added to each well at days 5 and 10 post-infection. Reverse transcriptase activity in the culture supernatant was assayed on days 0, 4, 6, 8, 11, and 14 post-infection as described [28]. Viral replication was quantified using the slope of the growth curves and performing linear regression analysis derived from the equation log(y) = mt + log(h), where y is virus quantity (cpm), t is time in days, and h is the y-intercept (day 0). All slope values for each virus were used to calculate means and standard deviations. Differences in the mean values were evaluated using a one way analysis of variance test and the significance difference from the control HIV-1B-92US076 calculated using the Bonferroni’s Multiple Comparison Test (GraphPad Prism v.6.0b, GraphPad Software).
HIV-1 replicative fitness determination using growth competition experiments
Dual infection/competition experiments were carried out as previously described [28, 35, 66, 67]. Briefly, each query virus (patient-derived subtype B HIV-1 isolates and the HIV-1B-92US076 control) was competed against two different non-subtype B HIV-1 control strains (HIV-1A-92UG029 and HIV-1AE-CMU06) in a 1:1 initial proportion using an MOI of 0.001 IU/cell to infected 1 × 106 PBMCs for 2 h at 37 °C and 5% CO2. Cells were subsequently washed twice with 1× PBS and cultured in a 24-well plate. Cell-free supernatant and cells were harvested at day 10 and stored at −80 °C for subsequent analysis. The final proportions of the two viruses in each competition were quantified using a TaqMan Real-Time PCR assay after normalizing to viral production in the HIV-1 monoinfections as described [28, 35, 67]. Replicative fitness for each patient-derived HIV-1 isolate was calculated and expressed as a percentage of the replicative fitness of the HIV-1B-92US076 control, set as 100%.
Reverse transcription (RT)-PCR amplification of nearly full-length HIV-1 genome
Plasma viral RNA was purified from pelleted virus particles by centrifuging one milliliter of plasma at 20,000g × 60 min at 4 °C, removing 860 µl of cell-free supernatant and resuspending the pellet in the remaining 140 µl, to finally extract viral RNA using QIAamp Viral RNA Mini kit (Qiagen; Valencia, CA). Viral RNA was reverse-transcribed using AccuScript High Fidelity Reverse Transcriptase (Stratagene Agilent; Santa Clara, CA) and five previously described antisense external primers (Pan-HIV-1-1R [68], 1R, 2R, 3R, and 4R [69]) in 20 µl reaction mixtures containing 1 mM dNTPs, 10 mM DTT and 10 units of RNAse inhibitor. Six overlapping fragments, covering almost the entire HIV-1 genome, were amplified using a series of external and nested primers and Phusion High-Fidelity DNA Polymerase (NEB, Ipswich, MA) with defined cycling conditions as previously described [68, 69], i.e., 5′LTR (675 bp; HXB2 coordinates 120 to 794), 5′LTR-gag/p7 (1269 bp; 658 to 1924), gag/p24-pol/RT (2327 bp; 1137 to 3463), pol/RT-vif (2259 bp; 2976 to 5234), pol/int-env/gp120 (2921 bp; 4602 to 7522), and env/gp120-3′LTR (2587 bp; 6858 to 9444).
Population (Sanger) sequence analysis
Nested PCR products were purified with the QIAquick PCR Purification kit (Qiagen) and sequenced by Sanger (population) sequence (GATC Biotech, Constance, Germany). Nucleotide sequences were analyzed using DNASTAR Lasergene Software Suite v.12.3.1.4 (Madison, WI).
Deep sequencing of nearly full-length HIV-1 genome
All six overlapping HIV-1 fragments, from the seven patient-derived plasma samples, were deep sequenced using a variation of the DEEPGEN™HIV assay [70]. Briefly, the six amplicons were purified (Agencourt AMPure XP, Beckman Coulter) and quantified (2100 Bioanalyzer DNA 7500, Agilent Technologies) prior to using the Ion Xpress Fragment Library Kit (Life Technologies, Carlsbad CA) to construct a multiplexed library for shotgun sequencing on the Ion Personal Genome Machine (PGM, Life Technologies). For that, a mixture of all six purified DNA amplicons (16 ng each) was randomly fragmented and blunt-ends repaired using the Ion Shear Plus Reagent (Life Technologies) followed by DNA purification (Agencourt AMPure XP, Beckman Coulter). The P1 adapter and one of seven barcodes were ligated to the repaired fragment ends prior to DNA purification (Agencourt AMPure XP, Beckman Coulter). DNA fragments were then selected by size (i.e., 280–320 bp; Pippin Prep, Life Technologies) and each barcoded library, i.e., a mixture of all six amplicons per sample, was purified (Agencourt AMPure XP, Beckman Coulter) and normalized using the Ion Library Equalizer Kit (Life Technologies). All seven barcoded DNA libraries, corresponding to seven patient-derived amplicons, were pooled in equimolar concentrations and templates prepared and enriched for sequencing on the Ion Sphere Particles (ISPs) using the Ion OneTouch 200 Template Kit v2 (Life Technologies) in the Ion OneTouch 2 System (Life Technologies). Templated ISPs were quantified (Qubit 2.0, Life Technologies) and loaded into an Ion 318 Chip (Life Technologies) to be sequenced on the Ion PGM using the Ion PGM Sequencing 200 Kit v2 (Life Technologies). Sequencing run, signal processing and base calling was performed with Torrent Analysis Suite version 5.0.4. All deep sequencing experiments were performed in the CLIA/CAP-accredited University Hospitals Translational Laboratory under a good laboratory practice framework.
Read mapping, variant calling, phylogenetic and viral diversity analysis
Reads were mapped and aligned (assembled) against the HIV-1HXB2 (GenBank: K03455) reference sequence using SeqMan NGen (DNASTAR Lasergene Software Suite v.12.3.1.4), and the assemblies analyzed using SeqMan Pro (DNASTAR Lasergene Software Suite v.12.3.1.4). Full-length HIV-1 consensus sequences were generated for each patient-derived virus, compared to the corresponding population sequences obtained by Sanger sequencing, aligned using ClustalW [71] and their phylogeny reconstructed using the neighbor-joining statistical method as implemented within MEGA 6.06 [72]. Variant calling (i.e., single nucleotide polymorphisms, including substitutions, deletions and insertions) and their frequencies in the virus population relative to the HIV-1HXB2 reference sequence were quantified using a proprietary pipeline (Alouani and Quiñones-Mateu, unpublished results). Intra-patient HIV-1 quasispecies diversity was determined using near full-length HIV-1 genome (or individual genes and coding region sequences) based on the p-distance model as described for deep sequencing [73].
Statistical analyses
Descriptive results are expressed as mean values, standard deviations, range, and confidence intervals. As described above, differences in the mean of the slope values for the viral growth kinetics curves were determined using a One Way Analysis of Variance test and the difference from the reference HIV-1NL4-3 virus calculated using the Bonferroni’s Multiple Comparison Test. All differences with a P value of <0.05 were considered statistically significant. All statistical analyses were performed using GraphPad Prism v.6.0b (GraphPad Software, La Jolla, CA) unless otherwise specified. Nucleotide sequences of the nearly full-length HIV-1 genomes, both consensus and individual reads, obtained by deep sequencing in this study have been submitted to the Los Alamos National Laboratory HIV-DB Next Generation Sequence Archive (http://www.hiv.lanl.gov/content/sequence/HIV/NextGenArchive/Weber2017).
Results
Clinical, virologic, and immunologic characteristics of viremic non-progressors, typical and rapid progressor patients
As described above, for this study we identified three treatment-naïve HIV-infected individuals classified as VNPs. At the time of the study, these patients were infected with HIV-1 for over 10 years (range 10–17 years), maintaining relatively high CD4+ T-cell counts (mean 472, range 280–814 cells/mm3) with limited decline in CD4+ T-cell count (mean slope −0.56, range −0.4 to −0.7 CD4+ T-cells/month) despite sustained plasma HIV-1 RNA loads above 4000 copies/ml (mean 5.02 log10, range 3.61–5.45 copies/ml) during the time these patients were monitored prior to this study (range 37–123 months) (Table 1; Additional file 1: Figure S1). For comparison, we selected three patients classified as typical progressors (each infected for at least 10 years, range 12–15 years), and one rapid progressor infected for 5 years. Overall, the CD4+ T-cell counts for the TPs were similar to that of the VNPs (mean 400, range 126–789 cells/mm3) albeit with a more marked decline in CD4+ T-cells over time (mean slope −10.3, range −8.2 to −13.1 CD4+ T-cells/month). Plasma HIV-1 RNA levels for the TPs were similarly high (mean 4.45 log10, range 3.89–4.9 copies/ml). As expected, the CD4+ T-cell counts from the RP patient were significantly lower than those for the VNP and TP individuals (mean 57, range 11–124 cells/mm3), declining at a rate of −4.7 CD4+ T-cells/month with high plasma HIV-1 RNA loads (mean 4.49 log10, range 4.09–4.66 copies/ml) (Table 1; Additional file 1: Figure S1). The rest of the clinical baseline characteristics showed no significant differences regarding age, gender distribution, ethnicity, and mode of transmission of HIV-1.
Replicative fitness of HIV-1 isolates from VNP, TP, and RP patients
We used two different but complementary approaches to quantify the ability of all seven patient-derived HIV-1 isolates (i.e., 3 VNPs, 3 TPs, and 1 RP) to replicate in vitro in the absence of any host selective pressure, including antiretroviral drugs. A first glimpse of the replicative fitness of these viruses was obtained using classical viral growth kinetics in PBMCs from HIV-seronegative donors (Fig. 1a). Statistical analysis of the slope of the growth curves showed that the replicative fitness of all three VNP viruses was significantly reduced compared to the replicative fitness of the three TP and RP viruses (p < 0.05, 95% CI, Fig. 1b). Since in vitro dual infection/growth competition experiments are considered the gold standard method to measure viral fitness [30, 74] we competed each one of the seven HIV-1 isolates against two different non-subtype B HIV-1 control strains (HIV-1A-92UG029 and HIV-1AE-CMU06). Interestingly, all three VNP viruses showed a marked decrease in replicative fitness, i.e., 0.1, 7.5, and 15% relative to that of the primary HIV-1B-92US076 isolate used as control (Fig. 1c). With the exception of the TP-1 virus (9%), the replicative fitness of the other two TP viruses was not impaired relative to the fitness of the HIV-1B-92US076 control (167 and 118% for TP-2 and TP-3, respectively). Finally, the RP virus had a 2.6-fold higher replicative fitness than the HIV-1B-92US076 control and, in average, was 35-fold more fit to replicate in PBMCs than the VNP viruses (Fig. 1c).

Replicative fitness of seven patient-derived HIV-1 isolates in the absence of any host selective pressure, including antiretroviral drugs. a Seven HIV-1 isolates from 3 viremic non-progressor (VNP), 3 typical (TP) and one rapid (RP) progressor patients were evaluated for their ability to replicate in peripheral blood mononuclear cells (PBMC). Virus replication was quantified by measuring reverse transcriptase (RT) activity in the cell-free supernatant. b Viral replication slopes were calculated using the slopes between cpm values at days 0 and 4, 0 and 6, 0 and 8, 0 and 11, and 0 and 14. All five slope values for each virus were used to calculate the mean, standard deviation, and 10th and 90th percentiles. Differences in the mean values were calculated using a One Way Analysis of Variance test and the significance difference from the primary wild-type HIV-1B-92US076 control isolate calculated using the Bonferroni’s Multiple Comparison Test. c Each patient-derived HIV-1 isolate was competed against two different non-subtype B HIV-1 control strains (HIV-1A-92UG029 and HIV-1AE-CMU06) in PBMCs and their replicative fitness calculated and expressed as a percentage of the replicative fitness of the reference virus HIV-1B-92US076 (set as 100%) as described [28, 35, 66, 67]. Values represent results obtained from single growth competition experiments. Viral replication kinetics and growth competitions marked with an asterisk were significantly different to the HIV-1B-92US076 control (p < 0.05, 95% CI)
Characterization of HIV-1 isolates from VNP, TP, and RP individuals: subtyping, drug resistance, and coreceptor tropism
To further characterize the HIV-1 isolates, viral RNA was used to amplify and sequence near full-length HIV-1 genomes (HXB2 coordinates 157–99,428) using both Sanger and deep sequencing. For deep sequencing, amplicons corresponding to all 7 viruses were multiplexed into a single 318c chip (78% loading efficiency of Ion Sphere Particles), generating 3,856,115 total quality reads with a mean read length of 131 bp (mean range 130–133 bp). Although the average sequencing coverage at each nucleotide position varied with each sample and HIV-1 genomic region analyzed (p < 0.0001, one-way ANOVA), the mean coverage for each viral sequence ranged from 2079 reads for the RP virus to 8500 for VNP-3 (Fig. 2a). These metrics ensured the minimum coverage of 1000 per nucleotide position sequenced required to guarantee the detection of a minor variant present at least at 1% of the population [75]. As expected, coverage decreases abruptly at the ends of the amplicons (e.g., 5′LTR and 3′LTR in Fig. 2a); however, we observed an inexplicable decrease in coverage around positions 6655–6719 (corresponding to variables regions V1 and V2 in gp120, env gene) that do not correspond with the end of any of the overlapping PCR products covering this genomic region.

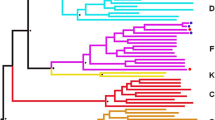
a Coverage, i.e., number of reads per nucleotide position, obtained by deep sequencing the seven patient-derived HIV-1 isolates. Near full-length HIV-1 genomes were RT-PCR amplified and deep sequenced as described in “Methods”. The position relative to the HIV-1 genome of the six overlapping amplicons used to amplify and sequence the near full-length HIV-1 genomes (HXB2 position 157 to 9428, not counting primer sequences) is indicated. b Hierarchical clustering analysis of the single nucleotides polymorphisms (SNPs) was used to group the seven patient-derived HIV-1 isolates by similarity. Dendrograms were calculated using the Euclidean distance and Complete cluster methods with 1000 bootstrap iterations as described (http://www.hiv.lanl.gov/content/sequence/HEATMAP/heatmap.html) Bootstrap values >60% are indicated by an asterisk. Green and grey blocks indicate the presence or absence of SNPs, respectively, in each HIV-1 isolate relative to the HIV-1HXB2 reference. c A Neighbor-joining phylogenetic tree was constructed using the near full-length HIV-1 consensus sequences generated for each patient-derived virus (obtained from deep sequencing reads) and rooted using the HIV-1HXB2 sequence (GenBank accession number AF033819). Bootstrap resampling (1000 data sets) of the multiple alignment tested the statistical robustness of the tree, with percentage values above 75% indicated by an asterisk. s/site substitutions per nucleotide site
The entire HIV-1 genome from all seven viruses was classified as subtype B using the Recombinant Identification Program from the Los Alamos HIV Sequence Database [76] (Table 1; Additional file 2: Figure S2). We used our proprietary DEEPGEN™HIV Software Tool Suite [70] to confirm the absence of mutations (at >1% frequency in the HIV-1 population) associated with drug resistance in the protease (PR), reverse transcriptase (RT), and/or integrase (INT) coding regions. The same tool was used to determine HIV-1 coreceptor tropism based on sequencing reads corresponding to the V3 region of gp120. HIV-1 isolates from VNP and TP patients were all CCR5-tropic viruses, while the RP individual was infected with a dual- or mixed-tropic virus (Table 1).
HIV-1 genetic polymorphisms in VNP, TP, and RP viruses
HIV-1 isolates from viremic non-progressor individuals showed a clear impairment on their ability to replicate in PBMCs in vitro. Since the fitness decrease was not associated with drug resistance mutations, we decided to analyze the entire HIV-1 genomes to try to identify any particular signature(s) that could be responsible for the decrease in replication capacity. First, we scanned all seven near full-length HIV-1 genomes searching for patterns in single-nucleotide polymorphisms (SNPs) among the three groups of patients. As described in Fig. 2b, we observed a higher concentration of SNPs in the variable regions of gp120; however, no clear clustering of the VNP or TP groups was evident when the SNP patterns of the near full-length genomes were analyzed. For example, VNP-1 and VNP-3 clustered with TP-3, while VNP-2 was closer to TP-1 and TP-2. Similar results were observed when SNP patterns were analyzed in each individual HIV-1 genomic region, with the exception of gp41 where significant clustering of VNP-1 with VNP-3 and all three TNP viruses was evident (Additional file 3: Figure S3).
Next, we focused on HIV-1 genetic polymorphisms that have been associated with impaired HIV-1 replicative fitness and/or disease progression [11, 31, 32, 40, 42, 44–62, 77, 78]. We identified a series of SNPs in LTR and amino acid substitutions—at different frequencies in the virus population—in gag, vif, vpr, rev, and nef genes (Table 2). Although no clear pattern of signature mutations was shared among the individual viruses from each group, some of the mutations were present only in VNP and not in TP or RP sequences, e.g., in the LTR (insertion at position 329, G364A, C386T, or G399A), gag (E12Q, A146P, T242 N, or E482D), vif (R132S), vpr (F72S), and nef (T138C). Interestingly, most of these mutations were identified in the VNP-1 isolate, which had the most impaired replicative fitness of the group (Fig. 1). In fact, the number of HIV-1 genetic polymorphisms previously associated with fitness decrease and/or disease progression was significantly higher in the VNP group compared with the TP and RP viruses (mean 39.3, 21.6, and 22, respectively, p = 0.036) (Table 2; Fig. 3a). Moreover, a strong significant inversed correlation was observed between the number of HIV-1 genetic polymorphisms in each viral isolate and the HIV-1 replicative fitness values determined by the slopes of the viral growth curves (r = −0.956, p = 0.0007, Pearson coefficient correlation) (Fig. 3b). Although not significant, most likely perhaps due to the low fitness calculated for the TP-1 virus, a similar trend was observed using the replicative fitness values from the growth competition experiments (r = −0.547, p = 0.203, Pearson coefficient correlation).

a Comparison of the number of HIV-1 genetic polymorphisms in the near full-length HIV-1 genome that have been associated with impaired HIV-1 replicative fitness and/or disease progression [11, 31, 32, 40, 42, 44–62, 77, 78] among the three groups of patients, i.e., VNP, RP, and TP. Unpaired t test was used to assess the statistical significance between VNP and TP patients. A detailed list of the mutations is included in Table 2. b Pearson correlation coefficient was used to determine the strength of association between the number of HIV-1 genetic polymorphisms and the HIV-1 replicative fitness calculated by viral growth kinetics analysis (viral replication slope) described in Fig. 1. r correlation coefficient, p two-tailed p value. Dotted lines represent 95% confidence intervals
Genetic diversity of VNP, TP, and RP viruses
We used the consensus sequences of the near full-length HIV-1 genomes generated by deep sequencing to construct a neighbor-joining phylogenetic tree. As observed in Fig. 2c, the HIV-1 sequences did not cluster together according to their group, i.e., VNP, TP, and RP. Identical results were obtained using the population sequences generated by Sanger sequencing (data not shown). Based on the consensus near full-length HIV-1 sequences, intra-group genetic distances were not significantly different between VNP and TP viruses (0.089 and 0.096 substitutions/site, respectively, p = 0.345; Maximum Composite Likelihood model). Next we used the myriad of reads obtained by deep sequencing to calculate intra-patient HIV-1 population diversity based on the p-distance model [73]. Interestingly, the VNP viruses showed significantly higher genetic diversity than the TP and RP viruses (mean 3.07, 2.52, and 1.93 substitutions/site, p = 0.009) when the near full-length HIV-1 genomes were analyzed (Fig. 4a). Although this trend was consistent across individual HIV-1 genomic regions and genes (data not shown), the higher genetic diversity in VNP viruses was significant in two regions of the gag gene: p2 (4.82, 3.09, and 2.76 substitutions/site, p = 0.008) and p6 (2.9, 1.62, and 1.59 substitutions/site, p = 0.02), and the V4 region of gp120 (13.4, 1.54, and 6.39 substitutions/site, p = 0.018) (Fig. 4a). More importantly, we observed a strongly significant negative correlation between genetic diversity of the near full-length HIV-1 genomes and the replicative fitness values determined by the slopes of the viral growth curves (r = −0.878, p = 0.009, Pearson coefficient correlation) (Fig. 4b). The same significant inverse associations were observed for the two regions of the gag gene: p2 (r = −0.833, p = 0.019, Pearson coefficient correlation) and p6 (r = −0.856, p = 0.013, Pearson coefficient correlation), and the V4 region of gp120 (r = −0.825, p = 0.022, Pearson coefficient correlation) (Fig. 4b) but not for those regions with no significant differences in HIV-1 diversity between the three groups of patients. Similar results, i.e., more heterogeneous virus population (VNPs) having lower viral replicative fitness values, were obtained when replicative fitness was determined using growth competition experiments (regression values ranging from −0.791 to −0.867, p < 0.05, Pearson coefficient correlation; data not shown).

a Comparison of intra-patient HIV-1 population diversity, based on the p-distance model [73], among the three groups of patients, i.e., VNP, RP, and TP. Unpaired t test was used to assess the statistical significance between VNP and TP patients. Only those HIV-1 regions significantly different, i.e., near full-length HIV-1 genome, Gag p2, Gag p6, and Gp120 V4, are included. b Pearson correlation coefficient was used to determine the strength of association between intra-patient HIV-1 population diversity values (p-distance) and the HIV-1 replicative fitness calculated by viral growth kinetics analysis (viral replication slope) described in Fig. 1. r correlation coefficient, p two-tailed p value. Dotted lines represent 95% confidence intervals
Discussion
Since the discovery of HIV-1 as the etiological agent of AIDS [79, 80], multitude of studies have attempted to understand the differences on HIV-1 disease progression, particularly host and/or viral factors that could be associated with HIV-1 pathogenesis [11, 12, 81]. The common (“normal”) inverse correlation between CD4+ T-cell count and plasma HIV RNA load usually governs HIV-1 pathogenesis, i.e., elevated plasma viremia is typically accompanied by a decrease in CD4+ T-cells, leading to disease progression [2]. Some HIV-infected individuals have shown uncommon rates of disease progression (slow, non-progressor or elite controllers) but all of them have reduced or undetected plasma HIV RNA loads [3, 4, 42, 82]. On the other hand, a rare group of patients termed viremic non-progressors (VNP) seem to resemble Simian Immunodeficiency Virus (SIV) infection in sooty mangabeys, i.e., active viral replication without (or very limited) disease progression [26, 82–84]. Here we identified three atypically VNP HIV-infected individuals and proceeded to study their HIV-1 isolates, focusing on replicative fitness and viral genomic analysis to try to understand the maintenance of stable CD4+ T-cell counts despite persistence viremia for more than 10 years, in the absence of antiretroviral treatment.
As described above, viremic non-progressor (controller) patients do not progress to AIDS, maintaining high CD4+ T-cell counts with HIV-1 replication for many years [5–7, 85]. The three VNP individuals described in this study had been infected with HIV-1 for at least 10 years at the time the blood sample was taken, with stable CD4+ T-cell counts and high plasma HIV RNA loads. When compared with three typical (TP) and one rapid progressor (RP) patients, the number of CD4+ T-cells barely declined in the VNPs over this period of time, similar to what have been described for other adult and children VNPs [43, 82–84, 86]. No other significant differences were observed among the VNP and TP patients, including plasma viral load levels and basic virus characteristics such as HIV-1 subtype (all clade B) and coreceptor tropism (all CCR5-tropic viruses).
Most studies involving the characterization of viremic non-progressor (controller) patients have focused in host factors to try to understand their odd HIV-1 disease progression profile. For example, the presence of the HLA-B*27 allele was associated with the VNP phenotype [43], while a higher frequency of the protective single nucleotide polymorphism −35 CC in the HLA-C gene was found in VNP patients [5]. Preferential Gag-specific CD8+ T cell responses have also been observed in viremic controllers [84] and limited infection of CD4+ central memory cells [8], perhaps related to low CCR5 expression on these cells [86], seem to be associated with lack of disease progression in VNPs. Limited deregulation of a series of interferon-stimulated genes correlating with time to disease progression was also identified in VNPs [85]. However, and similar to the absence of immune activation during natural SIV infection in sooty mangabeys [27, 87, 88], healthy CD4+ T cell counts despite detectable viremia seem to be common in most viremic non-progressor patients [86]. Yet, just a limited number of studies have analyzed HIV-1 strains infecting VNPs [6, 43].
Here we showed that the replicative fitness of all three VNP-derived HIV-1 isolates was impaired compared to the fitness of the viruses obtained from the TP and RP patients. We and others have reported that HIV-1 replicative fitness is associated with disease progression [28–30, 42], that is, viruses with decreased replication capacity influence the natural history of HIV-1 infection, correlating with diminished viral burden and HIV-1 pathogenesis. Viruses from long-term non-progressor [28], slow progressor [41, 42] and elite controller [37, 40, 42] patients have been shown to have impaired replication capacity; however, the fitness of viruses from VNP individuals have not been extensively described. Choudhary et al. [6] showed that HIV-1 isolates derived from VNP were as cytopathic as viruses isolated from typical progressor patients. On the other hand, O’Connell et al. [43] observed a slight decrease in the fitness of a virus from a VNP patient, transmitted from a chronic progressor partner, but failed to identify any amino acid substitution(s) potentially responsible for this phenotype.
Reduced HIV-1 replicative fitness is usually the consequence of changes in the viral quasispecies following the evasion of key selective pressures, e.g., host immune response and/or antiretroviral treatment [30, 31]. These changes are accompanied by single or multiple mutations in the targeted HIV-1 genomic region, deviating the virus population from the wild-type quasispecies distribution and impairing their ability to replicate efficiently compared to the original virus [89, 90]. The effect of a multitude of drug resistance mutations (in the pol gene) in viral replicative fitness have been amply described [30, 74]. A number of CTL escape mutations have been identified in HIV and SIV that carry a concomitant replicative fitness cost [46, 91, 92], including HIV-1 mutant variants escaping Gag-specific CD8+ T cell responses—like those identified in viremic controllers [84]—that had reduced replicative fitness [40]. Other studies have shown how multiple mutations and/or deletions in the LTR and all HIV-1 coding regions have a range of effects on the ability of the virus to replicate (virus attenuation) [11, 31, 32, 40, 42, 44–62, 78]. Here we used deep sequencing to analyze near full-length HIV-1 genomes from all VNP-derived viruses, and compared them with the sequences from the TP- and RP-derived viral genomes. Despite identifying a series of SNPs in different genomic regions, some of them previously associated with a reduction in replicative fitness and/or disease progression [11, 31, 32, 40, 42, 44–62, 77, 78], no clear pattern of signature mutations was detected in the viruses from the VNPs that could explain the impairment in replication capacity. Interestingly, it was the number of these HIV-1 genetic polymorphisms that correlated significantly with the replicative fitness of each HIV-1 isolate. In fact, it is reasonable to think that viruses with a higher number of critical SNPs have a more marked decrease in replicative fitness, similar to what has been described for multidrug-resistant HIV-1 variants carrying multiple mutations in the protease, reverse transcriptase and/or integrase coding regions [30, 74]. These results were corroborated by calculating intra-patient HIV-1 population diversity, i.e., the highly replication impaired VNP viruses had a more complex virus quasispecies population, most likely as a consequence of trying to evade the host immune response [93], which was highlighted by the significant correlation between replicative fitness and HIV-1 genetic diversity in highly immunogenic regions such as Gag and Gp120 [31, 42, 92]. We and others have described similar results where drug resistant HIV-1 variants with more heterogeneous virus population had lower viral replicative fitness [94, 95]. It is possible that impaired (less replication competent) HIV-1 strains are capable to ascertain a limited host immune response in VNPs, enough to maintain high levels of viremia, resulting in viral evolution but limiting host immune activation that could exacerbate HIV disease, i.e., decrease CD4+ T-cell counts. It is also possible that the preferential replication of HIV-1 in effector memory T cells, and preservation of central memory T cells, in VNPs [8] could result in greater virus production per HIV-infected cell. All interesting possibilities to be addressed in further studies.
Although prevalence of elite controllers seems to be less than 1% of the HIV-infected population [96, 97], a recent study involving over 13,000 HIV-infected individuals identified 271 viremic controllers for a 2.03% prevalence [82]. Interestingly, viremic pediatric nonprogressor patients with high CD4+ T cell counts seem to be more common than adults VNP individuals [86]. Is it possible that VNP patients are more common than what we originally thought? If so, what can we learn from these individuals that could help monitor and control HIV-1 infection in less fortunate patients? Several studies have pointed to high levels of immune activation as the major cause of HIV-1 pathogenesis [24, 98] and low immune activation seems to be—at least in part- responsible for the lack of disease progression in VNP individuals [86]. Moreover, the VNP-like phenotype during natural SIV infections in sooty mangabeys is definitely not related to a low rate of viral production nor an impairment on replicative fitness of the virus [25, 87]. However, we cannot rule out that original infection with less replication-competent viruses (therefore seeding HIV-1 reservoirs with replication impaired variants) could contribute to the decrease in immune activation, leading to the viremic non-progressor phenotype in these patients. Here we observed that all three VNP patients carried HIV-1 isolates with decreased replicative fitness compared to typical and rapid progressor individuals, and that accumulation of single-nucleotide polymorphisms (mutations) across the HIV-1 genome may be contributing to the overall fitness impairment. Therefore, it is possible that similar to the phenomenon observed in patients with a discordant response to antiretroviral therapy, i.e., high CD4+ cell counts with detectable plasma HIV-1 RNA load [99, 100], reduced viral replicative fitness could be contributing to late disease progression in untreated viremic controller individuals. Additional studies involving host immunology and genetics, together with more detailed analysis of the HIV-1 strains infecting VNP patients, are necessary to better understand HIV-1 pathogenesis, including approaches to prevent HIV-1 disease and potentially eliminate HIV reservoirs.
Abbreviations
- HIV-1:
-
human immunodeficiency virus type 1
- VNP:
-
viremic non-progressors
- TP:
-
typical progressor
- RP:
-
rapid progressor
- LTNP:
-
long-term non-progressors
- SNP:
-
single-nucleotide polymorphism
- HLA:
-
human leukocyte antigen
- CTL:
-
cytotoxic T cell
- PBMC:
-
peripheral blood mononuclear cells
- MOI:
-
multiplicity of infection
References
Poorolajal J, Hooshmand E, Mahjub H, Esmailnasab N, Jenabi E. Survival rate of AIDS disease and mortality in HIV-infected patients: a meta-analysis. Public Health. 2016;139:3–12.
Lackner AA, Lederman MM, Rodriguez B. HIV pathogenesis: the host. Cold Spring Harb Perspect Med. 2012;2(9):a007005.
Casado C, Colombo S, Rauch A, Martinez R, Gunthard HF, Garcia S, Rodriguez C, Del Romero J, Telenti A, Lopez-Galindez C. Host and viral genetic correlates of clinical definitions of HIV-1 disease progression. PLoS ONE. 2010;5(6):e11079.
Okulicz JF, Marconi VC, Landrum ML, Wegner S, Weintrob A, Ganesan A, Hale B, Crum-Cianflone N, Delmar J, Barthel V, et al. Clinical outcomes of elite controllers, viremic controllers, and long-term nonprogressors in the US Department of Defense HIV natural history study. J Infect Dis. 2009;200(11):1714–23.
Ballana E, Ruiz-de Andres A, Mothe B, de Arellano ER, Aguilar F, Badia R, Grau E, Clotet B, del Val M, Brander C, et al. Differential prevalence of the HLA-C −35 CC genotype among viremic long term non-progressor and elite controller HIV+ individuals. Immunobiology. 2012;217(9):889–94.
Choudhary SK, Vrisekoop N, Jansen CA, Otto SA, Schuitemaker H, Miedema F, Camerini D. Low immune activation despite high levels of pathogenic human immunodeficiency virus type 1 results in long-term asymptomatic disease. J Virol. 2007;81(16):8838–42.
Curriu M, Fausther-Bovendo H, Pernas M, Massanella M, Carrillo J, Cabrera C, Lopez-Galindez C, Clotet B, Debre P, Vieillard V, et al. Viremic HIV infected individuals with high CD4 T cells and functional envelope proteins show anti-gp41 antibodies with unique specificity and function. PLoS ONE. 2012;7(2):e30330.
Klatt NR, Bosinger SE, Peck M, Richert-Spuhler LE, Heigele A, Gile JP, Patel N, Taaffe J, Julg B, Camerini D, et al. Limited HIV infection of central memory and stem cell memory CD4+ T cells is associated with lack of progression in viremic individuals. PLoS Pathog. 2014;10(8):e1004345.
Mothe B, Ibarrondo J, Llano A, Brander C. Virological, immune and host genetics markers in the control of HIV infection. Dis Markers. 2009;27(3):105–20.
Santa-Marta M, de Brito PM, Godinho-Santos A, Goncalves J. Host factors and HIV-1 replication: clinical evidence and potential therapeutic approaches. Front Immunol. 2013;4:343.
Wang B. Viral factors in non-progression. Front Immunol. 2013;4:355.
Cohen OJ, Kinter A, Fauci AS. Host factors in the pathogenesis of HIV disease. ImmunolRev. 1997;159:31–48.
O’Brien SJ, Moore JP. The effect of genetic variation in chemokines and their receptors on HIV transmission and progression to AIDS. Immunol Rev. 2000;177:99–111.
Pantaleo G, Demarest JF, Schacker T, Vaccarezza M, Cohen OJ, Daucher M, Graziosi C, Schnittman SS, Quinn TC, Shaw GM, et al. The qualitative nature of the primary immune response to HIV infection is a prognosticator of disease progression independent of the initial level of plasma viremia. Proc Natl Acad Sci USA. 1997;94(1):254–8.
Saez-Cirion A, Lacabaratz C, Lambotte O, Versmisse P, Urrutia A, Boufassa F, Barre-Sinoussi F, Delfraissy JF, Sinet M, Pancino G, et al. HIV controllers exhibit potent CD8 T cell capacity to suppress HIV infection ex vivo and peculiar cytotoxic T lymphocyte activation phenotype. Proc Natl Acad Sci USA. 2007;104(16):6776–81.
Goulder PJ, Bunce M, Krausa P, McIntyre K, Crowley S, Morgan B, Edwards A, Giangrande P, Phillips RE, McMichael AJ. Novel, cross-restricted, conserved, and immunodominant cytotoxic T lymphocyte epitopes in slow progressors in HIV type 1 infection. AIDS Res Hum Retrovir. 1996;12(18):1691–8.
Migueles SA, Sabbaghian MS, Shupert WL, Bettinotti MP, Marincola FM, Martino L, Hallahan CW, Selig SM, Schwartz D, Sullivan J, et al. HLA B*5701 is highly associated with restriction of virus replication in a subgroup of HIV-infected long term nonprogressors. Proc Natl Acad Sci USA. 2000;97(6):2709–14.
Balotta C, Bagnarelli P, Violin M, Ridolfo AL, Zhou D, Berlusconi A, Corvasce S, Corbellino M, Clementi M, Clerici M, et al. Homozygous delta 32 deletion of the CCR-5 chemokine receptor gene in an HIV-1-infected patient. AIDS. 1997;11(10):F67–71.
Dean M, Carrington M, Winkler C, Huttley GA, Smith MW, Allikmets R, Goedert JJ, Buchbinder SP, Vittinghoff E, Gomperts E, et al. Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Hemophilia Growth and Development Study, Multicenter AIDS Cohort Study, Multicenter Hemophilia Cohort Study, San Francisco City Cohort, ALIVE Study. Science. 1996;273(5283):1856–62.
Eugen-Olsen J, Iversen AK, Garred P, Koppelhus U, Pedersen C, Benfield TL, Sorensen AM, Katzenstein T, Dickmeiss E, Gerstoft J, et al. Heterozygosity for a deletion in the CKR-5 gene leads to prolonged AIDS-free survival and slower CD4 T-cell decline in a cohort of HIV-seropositive individuals. AIDS. 1997;11(3):305–10.
Huang Y, Paxton WA, Wolinsky SM, Neumann AU, Zhang L, He T, Kang S, Ceradini D, Jin Z, Yazdanbakhsh K, et al. The role of a mutant CCR5 allele in HIV-1 transmission and disease progression. Nat Med. 1996;2(11):1240–3.
Zimmerman PA, Buckler-White A, Alkhatib G, Spalding T, Kubofcik J, Combadiere C, Weissman D, Cohen O, Rubbert A, Lam G, et al. Inherited resistance to HIV-1 conferred by an inactivating mutation in CC chemokine receptor 5: studies in populations with contrasting clinical phenotypes, defined racial background, and quantified risk. Mol Med. 1997;3(1):23–36.
Merindol N, Berthoux L. Restriction factors in HIV-1 disease progression. Curr HIV Res. 2015;13(6):448–61.
Deeks SG, Kitchen CM, Liu L, Guo H, Gascon R, Narvaez AB, Hunt P, Martin JN, Kahn JO, Levy J, et al. Immune activation set point during early HIV infection predicts subsequent CD4+ T-cell changes independent of viral load. Blood. 2004;104(4):942–7.
Silvestri G, Paiardini M, Pandrea I, Lederman MM, Sodora DL. Understanding the benign nature of SIV infection in natural hosts. J Clin Invest. 2007;117(11):3148–54.
Chahroudi A, Bosinger SE, Vanderford TH, Paiardini M, Silvestri G. Natural SIV hosts: showing AIDS the door. Science. 2012;335(6073):1188–93.
Hirsch VM. What can natural infection of African monkeys with simian immunodeficiency virus tell us about the pathogenesis of AIDS? AIDS Rev. 2004;6(1):40–53.
Quinones-Mateu ME, Ball SC, Marozsan AJ, Torre VS, Albright JL, Vanham G, van der Groen G, Colebunders RL, Arts EJ. A dual infection/competition assay shows a correlation between ex vivo human immunodeficiency virus type 1 fitness and disease progression. J Virol. 2000;74(19):9222–33.
Troyer RM, Collins KR, Abraha A, Fraundorf E, Moore DM, Krizan RW, Toossi Z, Colebunders RL, Jensen MA, Mullins JI, et al. Changes in human immunodeficiency virus type 1 fitness and genetic diversity during disease progression. J Virol. 2005;79(14):9006–18.
Quinones-Mateu ME, Arts EJ. HIV-1 fitness: implications for drug resistance, disease progression, and global epidemic evolution. In: Kuiken C, Foley B, Hahn B, Marx P, McCutchan F, Mellors J, Wolinsky S, Korber B, Los Alamos NM, editors. HIV Sequence Compendium 2001. Los Alamos: Theoretical Biology and Biophysics Group, Los Alamos National Laboratory; 2001. p. 134–70.
Quinones-Mateu ME, Arts EJ. Virus fitness: concept, quantification, and application to HIV population dynamics. Curr Top Microbiol Immunol. 2006;299:83–140.
Armand-Ugon M, Quinones-Mateu ME, Gutierrez A, Barretina J, Blanco J, Schols D, De Clercq E, Clotet B, Este JA. Reduced fitness of HIV-1 resistant to CXCR4 antagonists. Antivir Ther. 2003;8:1–8.
Quinones-Mateu ME, Tadele M, Parera M, Mas A, Weber J, Rangel HR, Chakraborty B, Clotet B, Domingo E, Menendez-Arias L, et al. Insertions in the reverse transcriptase increase both drug resistance and viral fitness in a human immunodeficiency virus type 1 isolate harboring the multi-nucleoside reverse transcriptase inhibitor resistance 69 insertion complex mutation. J Virol. 2002;76(20):10546–52.
Weber J, Chakraborty B, Weberova J, Miller MD, Quinones-Mateu ME. Diminished replicative fitness of primary human immunodeficiency virus type 1 isolates harboring the K65R mutation. J Clin Microbiol. 2005;43(3):1395–400.
Weber J, Rangel HR, Chakraborty B, Tadele M, Martinez MA, Martinez-Picado J, Marotta ML, Mirza M, Ruiz L, Clotet B, et al. A novel TaqMan real-time PCR assay to estimate ex vivo human immunodeficiency virus type 1 fitness in the era of multi-target (pol and env) antiretroviral therapy. J Gen Virol. 2003;84:2217–28.
Claiborne DT, Prince JL, Scully E, Macharia G, Micci L, Lawson B, Kopycinski J, Deymier MJ, Vanderford TH, Nganou-Makamdop K, et al. Replicative fitness of transmitted HIV-1 drives acute immune activation, proviral load in memory CD4+ T cells, and disease progression. Proc Natl Acad Sci USA. 2015;112(12):E1480–9.
Miura T, Brumme ZL, Brockman MA, Rosato P, Sela J, Brumme CJ, Pereyra F, Kaufmann DE, Trocha A, Block BL, et al. Impaired replication capacity of acute/early viruses in persons who become HIV controllers. J Virol. 2010;84(15):7581–91.
Blankson JN, Bailey JR, Thayil S, Yang HC, Lassen K, Lai J, Gandhi SK, Siliciano JD, Williams TM, Siliciano RF. Isolation and characterization of replication-competent human immunodeficiency virus type 1 from a subset of elite suppressors. J Virol. 2007;81(5):2508–18.
Julg B, Pereyra F, Buzon MJ, Piechocka-Trocha A, Clark MJ, Baker BM, Lian J, Miura T, Martinez-Picado J, Addo MM, et al. Infrequent recovery of HIV from but robust exogenous infection of activated CD4(+) T cells in HIV elite controllers. Clin Infect Dis. 2010;51(2):233–8.
Brumme ZL, Li C, Miura T, Sela J, Rosato PC, Brumme CJ, Markle TJ, Martin E, Block BL, Trocha A, et al. Reduced replication capacity of NL4-3 recombinant viruses encoding reverse transcriptase-integrase sequences from HIV-1 elite controllers. J Acquir Immune Defic Syndr. 2011;56(2):100–8.
Lassen KG, Lobritz MA, Bailey JR, Johnston S, Nguyen S, Lee B, Chou T, Siliciano RF, Markowitz M, Arts EJ. Elite suppressor-derived HIV-1 envelope glycoproteins exhibit reduced entry efficiency and kinetics. PLoS Pathog. 2009;5(4):e1000377.
Lobritz MA, Lassen KG, Arts EJ. HIV-1 replicative fitness in elite controllers. Curr Opin HIV AIDS. 2011;6(3):214–20.
O’Connell KA, Pelz RK, Dinoso JB, Dunlop E, Paik-Tesch J, Williams TM, Blankson JN. Prolonged control of an HIV type 1 escape variant following treatment interruption in an HLA-B*27-positive patient. AIDS Res Hum Retroviruses. 2010;26(12):1307–11.
de Arellano RE, Martin C, Soriano V, Alcami J, Holguin A. Genetic analysis of the long terminal repeat (LTR) promoter region in HIV-1-infected individuals with different rates of disease progression. Virus Genes. 2007;34(2):111–6.
Kondo M, Shima T, Nishizawa M, Sudo K, Iwamuro S, Okabe T, Takebe Y, Imai M. Identification of attenuated variants of HIV-1 circulating recombinant form 01_AE that are associated with slow disease progression due to gross genetic alterations in the nef/long terminal repeat sequences. J Infect Dis. 2005;192(1):56–61.
Martinez-Picado J, Prado JG, Fry EE, Pfafferott K, Leslie A, Chetty S, Thobakgale C, Honeyborne I, Crawford H, Matthews P, et al. Fitness cost of escape mutations in p24 Gag in association with control of human immunodeficiency virus type 1. J Virol. 2006;80(7):3617–23.
Miura T, Brockman MA, Schneidewind A, Lobritz M, Pereyra F, Rathod A, Block BL, Brumme ZL, Brumme CJ, Baker B, et al. HLA-B57/B*5801 human immunodeficiency virus type 1 elite controllers select for rare gag variants associated with reduced viral replication capacity and strong cytotoxic T-lymphocyte [corrected] recognition. J Virol. 2009;83(6):2743–55.
Prince JL, Claiborne DT, Carlson JM, Schaefer M, Yu T, Lahki S, Prentice HA, Yue L, Vishwanathan SA, Kilembe W, et al. Role of transmitted Gag CTL polymorphisms in defining replicative capacity and early HIV-1 pathogenesis. PLoS Pathog. 2012;8(11):e1003041.
Wright JK, Novitsky V, Brockman MA, Brumme ZL, Brumme CJ, Carlson JM, Heckerman D, Wang B, Losina E, Leshwedi M, et al. Influence of Gag-protease-mediated replication capacity on disease progression in individuals recently infected with HIV-1 subtype C. J Virol. 2011;85(8):3996–4006.
Mostowy R, Kouyos RD, Hoof I, Hinkley T, Haddad M, Whitcomb JM, Petropoulos CJ, Kesmir C, Bonhoeffer S. Estimating the fitness cost of escape from HLA presentation in HIV-1 protease and reverse transcriptase. PLoS Comput Biol. 2012;8(5):e1002525.
Shioda T, Oka S, Xin X, Liu H, Harukuni R, Kurotani A, Fukushima M, Hasan MK, Shiino T, Takebe Y, et al. In vivo sequence variability of human immunodeficiency virus type 1 envelope gp120: association of V2 extension with slow disease progression. J Virol. 1997;71(7):4871–81.
Wang B, Mikhail M, Dyer WB, Zaunders JJ, Kelleher AD, Saksena NK. First demonstration of a lack of viral sequence evolution in a nonprogressor, defining replication-incompetent HIV-1 infection. Virology. 2003;312(1):135–50.
Alexander L, Weiskopf E, Greenough TC, Gaddis NC, Auerbach MR, Malim MH, O’Brien SJ, Walker BD, Sullivan JL, Desrosiers RC. Unusual polymorphisms in human immunodeficiency virus type 1 associated with nonprogressive infection. J Virol. 2000;74(9):4361–76.
Kirchhoff F, Easterbrook PJ, Douglas N, Troop M, Greenough TC, Weber J, Carl S, Sullivan JL, Daniels RS. Sequence variations in human immunodeficiency virus type 1 Nef are associated with different stages of disease. J Virol. 1999;73(7):5497–508.
Premkumar DRD, Ma XZ, Maitra RK, Chakrabarti BK, Salkowitz J, Yen-Lieberman B, Hirsch MS, Kestler HW. The nef gene from a long-term HIV type 1 nonprogressor. AIDS Res Hum Retrovir. 1996;12:337–45.
Deacon NJ, Tsykin A, Solomon A, Smith K, Ludford-Menting M, Hooker DJ, McPhee DA, Greenway AL, Ellet A, Chatfield C, et al. Genomic structure of an attenuated quasispecies of HIV-1 from a blood transfusion donor and recipients. Science. 1995;270:988–91.
Salvi R, Garbuglia AR, Di Caro A, Pulciani S, Montella F, Benedetto A. Grossly defective nef gene sequences in a human immunodeficiency virus type 1-seropositive long-term nonprogressor. J Virol. 1998;72(5):3646–57.
Caly L, Saksena NK, Piller SC, Jans DA. Impaired nuclear import and viral incorporation of Vpr derived from a HIV long-term non-progressor. Retrovirology. 2008;5:67.
Lum JJ, Cohen OJ, Nie Z, Weaver JG, Gomez TS, Yao XJ, Lynch D, Pilon AA, Hawley N, Kim JE, et al. Vpr R77Q is associated with long-term nonprogressive HIV infection and impaired induction of apoptosis. J Clin Invest. 2003;111(10):1547–54.
Rangel HR, Garzaro D, Rodriguez AK, Ramirez AH, Ameli G, Del Rosario Gutierrez C, Pujol FH. Deletion, insertion and stop codon mutations in vif genes of HIV-1 infecting slow progressor patients. J Infect Dev Ctries. 2009;3(7):531–8.
De Maio FA, Rocco CA, Aulicino PC, Bologna R, Mangano A, Sen L. Unusual substitutions in HIV-1 vif from children infected perinatally without progression to AIDS for more than 8 years without therapy. J Med Virol. 2012;84(12):1844–52.
Hassaine G, Agostini I, Candotti D, Bessou G, Caballero M, Agut H, Autran B, Barthalay Y, Vigne R. Characterization of human immunodeficiency virus type 1 vif gene in long-term asymptomatic individuals. Virology. 2000;276(1):169–80.
Reed LJ, Muench H. A simple method of estimating fifty percent endpoints. Am J Hyg. 1938;27:493–7.
Weber J, Vazquez AC, Winner D, Rose JD, Wylie D, Rhea AM, Henry K, Pappas J, Wright A, Mohamed N, et al. Novel method for simultaneous quantification of phenotypic resistance to maturation, protease, reverse transcriptase, and integrase HIV inhibitors based on 3′Gag(p2/p7/p1/p6)/PR/RT/INT-recombinant viruses: a useful tool in the multitarget era of antiretroviral therapy. Antimicrob Agents Chemother. 2011;55(8):3729–42.
Weber J, Rose JD, Vazquez AC, Winner D, Margot N, McColl DJ, Miller MD, Quinones-Mateu ME. Resistance mutations outside the integrase coding region have an effect on human immunodeficiency virus replicative fitness but do not affect its susceptibility to integrase strand transfer inhibitors. PLoS ONE. 2013;8(6):e65631.
Selhorst P, Vazquez AC, Terrazas-Aranda K, Michiels J, Vereecken K, Heyndrickx L, Weber J, Quinones-Mateu ME, Arien KK, Vanham G. Human immunodeficiency virus type 1 resistance or cross-resistance to nonnucleoside reverse transcriptase inhibitors currently under development as microbicides. Antimicrob Agents Chemother. 2011;55(4):1403–13.
Weber J, Weberova J, Carobene M, Mirza M, Martinez-Picado J, Kazanjian P, Quinones-Mateu ME. Use of a novel assay based on intact recombinant viruses expressing green (EGFP) or red (DsRed2) fluorescent proteins to examine the contribution of pol and env genes to overall HIV-1 replicative fitness. J Virol Methods. 2006;136(1–2):102–17.
Gall A, Ferns B, Morris C, Watson S, Cotten M, Robinson M, Berry N, Pillay D, Kellam P. Universal amplification, next-generation sequencing, and assembly of HIV-1 genomes. J Clin Microbiol. 2012;50(12):3838–44.
Henn MR, Boutwell CL, Charlebois P, Lennon NJ, Power KA, Macalalad AR, Berlin AM, Malboeuf CM, Ryan EM, Gnerre S, et al. Whole genome deep sequencing of HIV-1 reveals the impact of early minor variants upon immune recognition during acute infection. PLoS Pathog. 2012;8(3):e1002529.
Gibson RM, Meyer AM, Winner D, Archer J, Feyertag F, Ruiz-Mateos E, Leal M, Robertson DL, Schmotzer CL, Quinones-Mateu ME. Sensitive deep sequencing-based HIV-1 genotyping assay to simultaneously determine susceptibility to protease, reverse transcriptase, integrase, and maturation inhibitors, as well as HIV-1 coreceptor tropism. Antimicrob Agents Chemother. 2014;58(4):2167–85.
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23(21):2947–8.
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28(10):2731–9.
Shao W, Kearney MF, Boltz VF, Spindler JE, Mellors JW, Maldarelli F, Coffin JM. PAPNC, a novel method to calculate nucleotide diversity from large scale next generation sequencing data. J Virol Methods. 2014;203:73–80.
Quinones-Mateu ME, Arts EJ. Fitness of drug resistant HIV-1: methodology and clinical implications. Drug Res Updates. 2002;5(6):224–33.
Wang C, Mitsuya Y, Gharizadeh B, Ronaghi M, Shafer RW. Characterization of mutation spectra with ultra-deep pyrosequencing: application to HIV-1 drug resistance. Genome Res. 2007;17(8):1195–201.
Siepel AC, Halpern AL, Macken C, Korber BTM. A computer program designed to screen rapidly for HIV type 1 intersubtype recombinant sequences. AIDS Res Hum Retrovir. 1995;11(11):1413–6.
Hiebenthal-Millow K, Greenough TC, Bretttler DB, Schindler M, Wildum S, Sullivan JL, Kirchhoff F. Alterations in HIV-1 LTR promoter activity during AIDS progression. Virology. 2003;317(1):109–18.
Rangel HR, Weber J, Chakraborty B, Gutierrez A, Marotta ML, Mirza M, Kiser P, Martinez MA, Este JA, Quinones-Mateu ME. Role of the human immunodeficiency virus type 1 envelope gene in viral fitness. J Virol. 2003;77(16):9069–73.
Klatzmann D, BarrÇ-Sinoussi F, Nugeyre MT, Dauguet C, Vilmer E, Griscelli C, Brun-Vezinet F, Rouzioux C, Gluckman JC, Cherman JC, et al. Selective tropism of lymphadenopathy associated virus (LAV) for helper induced T lymphocytes. Science. 1984;225:59–63.
Popovic M, Sarngadharan MG, Read E, Gallo RC. Detection, isolation, and continuous production of cytopathic retroviruses (HTLV-III) from patients with AIDS and pre-AIDS. Science. 1984;224:497–500.
Fenyo EM, Fiore J, Karlsson A, Albert J, Scarlatti G. Biological phenotypes of HIV-1 in pathogenesis and transmission. Antibiot Chemother. 1994;46:18–24.
Leon A, Perez I, Ruiz-Mateos E, Benito JM, Leal M, Lopez-Galindez C, Rallon N, Alcami J, Lopez-Aldeguer J, Viciana P, et al. Rate and predictors of progression in elite and viremic HIV-1 controllers. Aids. 2016;30(8):1209–20.
Groves KC, Bibby DF, Clark DA, Isaksen A, Deayton JR, Anderson J, Orkin C, Stagg AJ, McKnight A. Disease progression in HIV-1-infected viremic controllers. J Acquir Immune Defic Syndr. 2012;61(4):407–16.
Tomescu C, Liu Q, Ross BN, Yin X, Lynn K, Mounzer KC, Kostman JR, Montaner LJ. A correlate of HIV-1 control consisting of both innate and adaptive immune parameters best predicts viral load by multivariable analysis in HIV-1 infected viremic controllers and chronically-infected non-controllers. PLoS ONE. 2014;9(7):e103209.
Rotger M, Dalmau J, Rauch A, McLaren P, Bosinger SE, Martinez R, Sandler NG, Roque A, Liebner J, Battegay M, et al. Comparative transcriptomics of extreme phenotypes of human HIV-1 infection and SIV infection in sooty mangabey and rhesus macaque. J Clin Invest. 2011;121(6):2391–400.
Muenchhoff M, Adland E, Karimanzira O, Crowther C, Pace M, Csala A, Leitman E, Moonsamy A, McGregor C, Hurst J, et al. Nonprogressing HIV-infected children share fundamental immunological features of nonpathogenic SIV infection. Sci Transl Med. 2016;8(358):358ra125.
Chakrabarti LA. The paradox of simian immunodeficiency virus infection in sooty mangabeys: active viral replication without disease progression. Front Biosci. 2004;9:521–39.
Klatt NR, Silvestri G, Hirsch V. Nonpathogenic simian immunodeficiency virus infections. Cold Spring Harb Perspect Med. 2012;2(1):a007153.
Najera I, Holguin A, Quinones-Mateu ME, Munoz-Fernandez MA, Najera R, Lopez-Galindez C, Domingo E. Pol gene quasispecies of human immunodeficiency virus: mutations associated with drug resistance in virus from patients undergoing no drug therapy. J Virol. 1995;69(1):23–31.
Domingo E, Escarmis C, Menendez-Arias L, Holland J. Viral quasispecies and fitness variations. In: Domingo E, Webster R, Holland J, editors. Origin and evolution of viruses. San Diego: Academic Press; 1999. p. 141–61.
Peyerl FW, Bazick HS, Newberg MH, Barouch DH, Sodroski J, Letvin NL. Fitness costs limit viral escape from cytotoxic T lymphocytes at a structurally constrained epitope. J Virol. 2004;78(24):13901–10.
Troyer RM, McNevin J, Liu Y, Zhang SC, Krizan RW, Abraha A, Tebit DM, Zhao H, Avila S, Lobritz MA, et al. Variable fitness impact of HIV-1 escape mutations to cytotoxic T lymphocyte (CTL) response. PLoS Pathog. 2009;5(4):e1000365.
Malim MH, Emerman M. HIV-1 sequence variation: drift, shift, and attenuation. Cell. 2001;104(4):469–72.
Gibson RM, Weber J, Winner D, Miller MD, Quinones-Mateu ME. Contribution of human immunodeficiency virus type 1 minority variants to reduced drug susceptibility in patients on an integrase strand transfer inhibitor-based therapy. PLoS ONE. 2014;9(8):e104512.
Kitrinos KM, Nelson JA, Resch W, Swanstrom R. Effect of a protease inhibitor-induced genetic bottleneck on human immunodeficiency virus type 1 env gene populations. J Virol. 2005;79(16):10627–37.
Grabar S, Selinger-Leneman H, Abgrall S, Pialoux G, Weiss L, Costagliola D. Prevalence and comparative characteristics of long-term nonprogressors and HIV controller patients in the French Hospital Database on HIV. Aids. 2009;23(9):1163–9.
Lambotte O, Boufassa F, Madec Y, Nguyen A, Goujard C, Meyer L, Rouzioux C, Venet A, Delfraissy JF, Group S-HS. HIV controllers: a homogeneous group of HIV-1-infected patients with spontaneous control of viral replication. Clin Infect Dis. 2005;41(7):1053–6.
Finkel TH, Tudor-Williams G, Banda NK, Cotton MF, Curiel T, Monks C, Baba TW, Ruprecht RM, Kupfer A. Apoptosis occurs predominantly in bystander cells and not in productively infected cells of HIV- and SIV-infected lymph nodes. Nat Med. 1995;1(2):129–34.
Deeks SG, Barbour JD, Martin JN, Swanson MS, Grant RM. Sustained CD4+ T cell response after virologic failure of protease inhibitor-based regimens in patients with human immunodeficiency virus infection. J Infect Dis. 2000;181(3):946–53.
Mezzaroma I, Carlesimo M, Pinter E, Muratori DS, Di Sora F, Chiarotti F, Cunsolo MG, Sacco G, Aiuti F. Clinical and immunologic response without decrease in virus load in patients with AIDS after 24 months of highly active antiretroviral therapy. Clin Infect Dis. 1999;29(6):1423–30.
Authors’ contributions
JW and MEQM designed the study, collected and assembled the data, wrote and drafted the manuscript. RMG, LS, DS, JH, JW, and MP performed all cell culture, molecular, and sequencing experiments. JW, MML, BR and MEQM contributed to the overall analysis of the data. All authors read and approved the final manuscript.
Acknowledgements
We thank Michelle Gallagher and the Case Western Reserve University/University Hospitals Center for AIDS Research (NIH P30 AI036219) for providing access to critical clinical information.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
All near full-length HIV-1 genome sequences and raw deep sequencing reads generated in this study are available upon request.
Ethics
Blood samples from HIV-infected individuals were obtained with the written informed consent of each participant as described in IRB protocol AIDS 125, University Hospitals Cleveland Medical Center/Case Western Reserve University.
Funding
J.W. was supported by a research grant from the Ministry of Education, Youth and Sports of the Czech Republic (LK11207). M.E.Q-M was partially supported by research Grant NIH-AI-71747, the CWRU/UH Center for AIDS Research (P30 AI036219) and funding from University Hospitals Cleveland Medical Center (UHCMC) for the University Hospitals Translational Laboratory (UHTL).
Author information
Authors and Affiliations
Corresponding authors
Additional files
12981_2017_144_MOESM1_ESM.pdf
Additional file 1: Figure S1. CD4+ T-cell counts and plasma HIV-1 RNA load values available for all seven patients described in this study. Vertical dashed lines indicate the time of sampling. Grey arrows depict the number of years since primary HIV-1 infection. The period of time the patients were under antiretroviral treatment (ART) is indicated by black boxes.
12981_2017_144_MOESM2_ESM.pdf
Additional file 2: Figure S2. Subtyping of the near full-length HIV-1 consensus sequences, obtained by deep sequencing, was performed using the Recombination Identification Program (RIP 3.0) from the Los Alamos HIV Sequence Database as described (1). The similarity plots show distance measurements between the query sequence (patient-derived HIV-1) and the reference HIV-1 sequences from different subtypes.
12981_2017_144_MOESM3_ESM.pdf
Additional file 3: Figure S3. Hierarchical clustering analysis of single nucleotide polymorphisms (SNPs) in all HIV-1 coding regions plus the long-terminal regions (LTRs). Dendograms were calculated using the Euclidian distance and Complete cluster methods with 1000 bootstrap iterations as described in Los Alamos HIV Sequence Database (http://www.hiv.lanl.gov.content/sequence/HEATMAP/heatmap.html). Bootstrap values >60% are indicated by an asterisk. Green and grey blocks indicate the presence and absence of SNPs, respectively.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Weber, J., Gibson, R.M., Sácká, L. et al. Impaired human immunodeficiency virus type 1 replicative fitness in atypical viremic non-progressor individuals. AIDS Res Ther 14, 15 (2017). https://doi.org/10.1186/s12981-017-0144-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12981-017-0144-0