
Abstract
The sequestering of oxidation-modified low-density lipoprotein by macrophages results in the accumulation of fatty deposits within the walls of arteries. Necrosis of these cells causes a release of intercellular epitopes and the activation of the adaptive immune system, which we predict leads to robust autoantibody production. T cells produce cytokines that act in the plaque environment and further stimulate B cell antibody production. B cells in atherosclerosis meanwhile have a mixed role based on subclass. The current model is that B-1 cells produce protective IgM antibodies in response to oxidation-specific epitopes that work to control plaque formation, while follicular B-2 cells produce class-switched antibodies (IgG, IgA, and IgE) which exacerbate the disease. Over the course of this review, we discuss further the validation of these protective antibodies while evaluating the current dogma regarding class-switched antibodies in atherosclerosis. There are several contradictory findings regarding the involvement of class-switched antibodies in the disease. We hypothesize that this is due to antigen-specificity, and not simply isotype, being important, and that a closer evaluation of these antibodies’ targets should be conducted. We propose that specific antibodies may have therapeutical potential in preventing and controlling plaque development within a clinical setting.
Similar content being viewed by others


Background
Heart disease is the leading cause of death in both the United States and internationally, with cases escalating in recent decades. A vast majority of these deaths are associated with atherosclerosis, the manifestation of fatty plaques within arterial walls, and is estimated to cost Americans over 300 billion annually in healthcare fees [1]. The accumulation of these fatty plaques affect large and medium-sized arteries alike, and leads to many life-threatening clinical complications, including stroke, cardiac infarction, and ischemic damage to the kidneys [2].
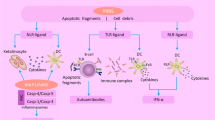
Plaque accumulation is associated with an elevation in total circulating cholesterol and an imbalance between low-density lipoprotein (LDL) and high-density lipoprotein (HDL) levels [3,4,5,6]. Accumulation of LDL within the intimal layer of the major arteries allows these molecules to be targeted by reactive oxygen species, generating oxidation-specific epitopes (OSEs) that are recognized by scavenger receptors present on macrophages [7,8,9]. This binding triggers endocytosis of the lipid-rich substrates by the macrophage, with incessant rounds of LDL engulfment overwhelming the digestive machinery of the cell and converting the macrophage into a foam cell [10, 11]. Continuous infiltration of macrophage-polarized monocytes into the plaque, and their conversion into foam cells, eventually fosters an environment that subjugates and impairs the proper clearance of apoptotic cells within the environment through means such as effectorcytosis. Elevated necroptosis, a non-apoptotic form of cell death, of the macrophages and smooth muscle cells present within these lesions further compounds this issue [12]. This results in necrotic core formation, destabilization of the plaque, and increases release of inflammasome-mediated cytokines that promote sterile inflammation [13, 14]. This pathway causes modified degradation of intracellular components [15], generating end products which upon core rupture spill into the blood alongside cellular debris recognized by the innate immune system as danger-associated molecular patterns (DAMPs) [16]. These two components together have the potential of generating adaptive responses against autoantigens and worsening the arterial inflammation (Fig. 1).

Atherosclerosis begins with the recognition of low-density lipoprotein by scavenger receptors on monocytes and macrophages. These cells infiltrate into the intimal layer of the artery where they uptake these lipid-rich molecules. Inability to process the heavy lipid burden overwhelms the cells and causes conversion to a foam cell. As foam cells accumulate, plaques develop, leading to further occlusion of arteries. In late-stage plaques, the hypoxic environment promotes necrosis of macrophages, smooth muscle cells, and the endothelial lining
Over the course of this review, we will discuss how lymphocytes of the adaptive immune response are involved throughout this process and focus on the role that the antibody responses play in atherosclerosis disease progression and prevention.
T lymphocytes
In addition to macrophage, T cells are also found within arterial plaques. T cells are a major subpopulation of the adaptive immune system and are considered key drivers in the response to foreign and altered-self epitopes. These cells undergo development in the thymus generating a plethora of T cells all containing a unique T cell receptor (TCR) which regulate the antigen specificity of the cell. T cells respond to peptides presented by cells in the context of Major Histocompatibility Complexes (MHC) through engagement with the T cell’s TCR [17,18,19]. The TCR-MHC interaction initiates cellular responses, the production of cytokines, and interaction with other immune cells, including B cells, in order to dictate the nature of the generated response (Fig. 2).

T cells contribute to the development and progression of atherosclerosis in a subset-dependent manner. CD4+ T cells begin as inexperienced naïve cells, upon activation through their TCR via interaction with MHC, polarize to a specific activated population due to signals induced by cytokines present within their surrounding microenvironment. Following activation, these cells secrete additional cytokines and express elevated levels of CD40L on their surface providing T cell help to B cells and other immune cells. CD8+ T cells function through the secretion of cytokines and conduct immunosurveillance, where they can induce cell death in target cells in a TCR-dependent manner using Granzyme B and Fas-FasL signaling
The contribution these cells play in the development of atherosclerosis has been a major focus of research for the past several decades. T cells are seen in the early formation of the plaque environments and have a strong presence throughout many stages of lesion development in humans and mice [20,21,22,23]. Likewise, knocking out these cells by deletion of the Rag Recombinase, which ablates TCR development, results in decreased plaque burden in atherosclerotic ApoE-deficient mice [24, 25]. It should be noted that Rag-deficiency also inhibits B cells development in these animals. T cell expression of CD40L (CD154) provides signals needed for the proper activation of many cytotoxic T cells and B cells through engagement with CD40. This CD40-CD40L interaction is also involved in the effector function of other non-lymphocyte cells, such as macrophages, which has been reviewed elsewhere [26,27,28].
CD8+ cytotoxic T cells
T lymphocytes can be divided into two major functional subgroups: CD8+ cytotoxic T cells, which bind MHC Class I expressed ubiquitously on cells, and CD4+ helper T cells, which bind MHC Class II expressed almost exclusively on antigen-presenting cells (APCs). The role of CD8+ T cells in atherosclerosis is poorly understood. CD8+ T cells have been shown to be present within early plaques, and antibody depletion of CD8+ T cells exhibits decreased plaque burden in atherosclerotic ApoE−/− and LDLR−/− mice. However, similar levels of CD8+ T cell infiltrate into the aorta of ApoE−/− mice regardless of a high-fat diet, suggesting a disconnect between recruitment and disease severity [29,30,31]. In humans, higher CD8+ cells with low CD127 and higher PD-1 expression has been observed in the plaques of patients with cardiovascular disease who have experienced a recent stroke, suggesting that CD8+ effector cells might be involved in the comorbidities of the disease and become exhausted following the cardiovascular event [32].
CD4+ effector T cells
By comparison, CD4+ T cells in atherosclerosis are better characterized, but display a more complex relationship with the disease due to the different possible subtypes of the CD4+ T cell (Fig. 2). CD4+ T cells are divided into several major subclasses based on master transcription factor expression and the cytokines they secrete. Type 1 T Helper cells (TH1) express Tbet and produce high levels of IFNγ upon activation. IFNγ activates macrophages, increasing their inflammatory potential, and during atherosclerosis has a potent proatherogenic effect on plaque formation [33,34,35,36] with deletion of Tbet and IFNγ alleviating this plaque formation in ApoE−/− mice [37, 38]. By comparison, TH2 cells, characterized by their expression of GATA3 and production of IL-4, IL-5, IL-13, and IL-33 are considered to be more anti-atherogenic; although their actual contribution is controversial. IL-4 produced may promote atherosclerosis [39,40,41,42,43], while IL-5 and IL-33 provides protection by promoting atheroprotective antibody production against malondialdehyde-modified LDL [44, 45]. RORγT-expressing TH17 cell involvement is likewise controversial due to the inconsistencies regarding the effect of IL-17A. IL-17A correlates with worsening disease prognosis in humans and mice, and blocking IL-17A with antibodies decreases plaque burden in ApoE−/− mice. However, IL-17A deficiency also causes increased plaque instability, suggesting a differential effect in early compared to late-stage disease [46,47,48,49]. CD25+, Foxp3-expressing T Regulatory (Treg) cells, which secrete TGFβ and IL-10, are widely accepted as atheroprotective by inhibiting atherogenic T Cells and macrophages [50, 51]. It has been proposed that Tregs are important for controlling atherosclerosis in early- and mid-stage plaques, and failure to maintain a high Treg-to-effector ratio results in advanced plaque formation of clinical concern [52,53,54,55,56,57,58,59]. This is further supported by the discovery that responding Tregs from the early stages of plaque formation can undergo transcriptional changes and emerge as TH1 and TH17 effectors cells in late-stage lesions [60].
B lymphocytes
Compared to T cells, the history of research on B cell involvement in atherosclerosis has had a short life, picking up only in the past few decades. Unlike T lymphocytes, B cells produce antibodies that disseminate throughout the body independent of the cell itself. As a result, B cells are not as dependent on localization to the direct site of inflammation with B cells making up only a minute proportion of the cells infiltrating the early plaque environment [31, 61]. Conversely, late-stage plaques show an oligoclonal expansion of B cells within the affected atherosclerotic tissue with reports of artery tertiary lymphoid structures (ATLOs) developing and producing antibodies within the adventitial layer of a diseased aorta [62,63,64,65,66,67,68]. Other B cells likewise can be found within the perivascular adipose tissue (PVAT) surrounding the aorta and are present in greater abundance than on the aorta itself [61, 64, 69]. These cells are elevated in areas surrounding plaques, independent of plaque stability, and produce abundant cytokines and antibodies [61, 69]. Compared to B cells of the adventitia, PVAT B cells may be involved in plaque modulation earlier as B cells do not have a high rate of trafficking to the adventitia following adoptive transfer [63]; however, the nature of this may vary due to differential localization of adoptively transferred B cells around the aorta based on subtype [61].
With regard to development, B cells start out as hematopoietic stem cells in the bone marrow where they progress through a series of checkpoints dependent on expression of transcription factors E2A, PU.1, and Pax5. Each B cell expresses a unique B cell receptor (BCR) which is derived from two independent peptides, the immunoglobulin heavy chain (IgH) and Ig light chain, with the Ig light chain being expressed from either the Ig kappa (Igκ) or Ig lambda (Igλ) locus. To form a unique BCR, a B cell will recombine each loci through the process of V(D)J recombination (Fig. 3A) [70, 71].

Antibody diversity comes from combination of variable and constant domain gene elements. A The B Cell locus is composed of multiple individual Variable, Diversity, and Joining gene segments which are randomly assembled into a functional gene exon through V(D)J recombination. The recombination involves the addition of random nucleotides, increasing the junctional diversity. The sites of the rearrangement form the third complementary determining region of the antibody, which dictates the antigen binding capability of the antibody (as shown in the callout image). B Transcription from intron promoters preceding switch regions allows AID to act and induce double stranded breaks. Repair by Nonhomologous end joining (NHEJ) between two broken switch regions results in class switching
Briefly, V(D)J recombination is the process of bringing a single variable (V), diversity (D), and joining (J) gene segment for IgH, or V and J for Igκ and Igλ, together through genomic recombination. In mice, each loci contains multiple gene segments (3 V and J for Igλ, ~ 80 V and 4 J for Igκ, and ~ 100 V, 13 D, and 4 J for IgH), millions of unique BCR combination can be generated. Through alternative splicing mechanisms, BCRs can be secreted as antibodies, and undergo additional diversity in their V and constant (C) regions after encountering their cognate antigen. Multiple different C regions can be observed across cells sharing the same clonality due to the ability of B cells to undergo the process of class-switching, giving rise to five immunoglobulin classes (IgM, IgD, IgG, IgE, and IgA) with several additional subclasses existing (Fig. 3B). This process is initiated by the activation-induced deaminase (AID) enzyme, which also generates mutations in variable genes to make high affinity antibodies. Following development and further maturation of these cells, self-reactive clones are deleted through the process of negative selection within the bone marrow and the periphery, resulting in immunological tolerance. Failure of this process may occur due to a breakdown of anatomical barriers and the exposure of epitopes that cells would otherwise be ignorant for, or because of improper suppression of clones that become self-reactive as a byproduct of AID-mediated genetic drift. For this reason, the emergence of autoreactive antibodies within atherosclerosis highlights the potential importance of studying these cells and understanding their contribution to the disease.
B-1 cells
B cells throughout the body can be divided into two distinct linages, B-1 and B-2 (Table 1), with both populations suspected to provide distinct roles in the progression of atherosclerosis. B-1 cells initially emerge during the period of fetal and neonatal development [72,73,74,75] and are the major resident B cell populations of the body’s cavities, such as the peritoneal and plural cavity, where they undergo homeostatic proliferation. B-1 responses are often geared towards evolutionarily conserved epitopes, such as bacterial components and apoptotic debris, functioning independent of T cell signaling [76,77,78,79,80,81,82]. The antibodies produced by these cells are primarily IgM, have little junctional nucleotide additions, and have a strong bias for certain V gene segments that compose their BCRs [83, 84]. B-1 cells can be further classified into two additional subpopulations, B-1a and B-1b, which differ based on the expression of CD5. The functional difference between these two cell populations is not fully understood; however, B-1a cells are thought to be the main producers of baseline IgM levels while B-1b cells generate the bulk of neutralizing IgM in response to infection and show class switching to IgA [81, 82, 85,86,87]. Regardless, both B-1a and B-1b cells are largely considered beneficial in atherosclerosis through their secretion of natural, relatively unmutated IgM directed against OSEs, which aid in the attenuation of plaque development in atherosclerotic ApoE−/− mice by blocking foam cell development [61, 88,89,90]. Elevation of these cells, either by knockout of Siglec-G, which negatively regulates B-1 and other myeloid populations and fosters tolerization in these cells [91, 92], or by modulation of TIM-1+ B cells using an anti-TIM-1 antibody [93] has moreover been shown to provide increased atheroprotection. It has also been argued that these cells are critical for preventing inflammation-promoting vascular neutrophilia [94, 95]. Recently it has been reported that the human-equivalent of B-1 cells (CD20+ CD27+ CD38+ CD43+) decline with age and are stunted in their ability to secrete antibodies, providing a potential mechanism for the acceleration of plaques in aged individuals [96]. Likewise, not all B-1 cells are believed to be protective. A subpopulation of B-1a cells, termed Innate Response Activator (IRA) B cells, have been found that aggravate atherosclerosis in mice through the production of IFNγ and GM-CSF. These cytokines act on cells within the plaque and cause an increase in monocyte recruitment and infiltration, drive the polarization of macrophages towards a M1 phenotype, and promote further CD4+ T cell development into inflammatory TH1 cells [97,98,99,100,101].
B-2 cells
While B-1 cells have been a major focus of atherosclerosis research, B-2 lymphocytes make up most of the B cells present within the body. B-2 cells are found preferentially in the spleen and lymph nodes and possess more diverse BCRs compared to their B-1 counterparts. The B-2 compartment is composed of Marginal Zone (MZ) and Follicular (FO) B cells (Table 1), which differ in their naïve form based of their preferential expression of Cμ or Cδ genes, CD21 and CD23 levels, and roles in response to infections. MZ B cells are IgMhi IgDlo CD21hi CD23lo B lymphocytes which have strong similarities to B-1 cells in terms of function and repertoire diversity [78]. By comparison, FO B cells are IgMlo IgDhi CD21lo CD23hi B cells, and show a greater assortment of BCR sequences between clones, allowing the FO cells to bind to a larger array of potential epitopes. Upon activation, FO B cells are dependent on engaging CD40L on CD4+ T cells through CD40 receptor on their cell surface in order to properly activate and produce high titers of antigen-specific antibodies. Upon T cell engagement, FO B cells enter specialized germinal center structures within the spleen and lymph nodes to undergo further diversification. Loss of the CD40/CD40L signaling axis impairs the ability of these cells to generate germinal centers, inhibiting the process of affinity maturation [102]. Furthermore, while evidence proposes that both MZ and FO cells can undergo germinal center formations [103], FO B cells more readily form these complexes upon activation, favoring the production of higher affinity antibodies.
The impact of B-2 cells in atherosclerotic plaque formation is heavily debated, and despite efforts to understand their involvement, the mechanism of their contribution is still unknown. During atherosclerosis, B-2 cells appear to isolate to novel structures within the adventitia of the vessel termed artery tertiary lymphoid organs (ATLOs) [104]. Within the ATLOs, B-2 cells engage T cells and dendritic cells and initiate an adaptive immune response. Reduction of the B cell compartment by splenectomy, reducing B-2 total cell populations, results in increased plaque formation in ApoE−/− mice, indicating that these B cells play a role in the disease [105]. Similar findings were observed in LDLR−/− μMT mice, which lack all B cells [106]. Reintroduction of the B-2 compartment into ApoE−/− μMT mice however gives mixed results as two studies claim atherosclerosis is increased [107, 108], while another states the opposite in a Id3-deficent system [109]. B-2 MZ B cells themselves are thought to provide protection against disease through similar mechanisms as B-1 cells and a reported ability to regulate T Follicular Helper (TFH) cells through PDL1 signaling [110]. Meanwhile, specific antibody depletion of B-2 cells has also been shown to provide protection [108, 111]. Similar results have been observed by knocking out MHC Class II or CD40, which are required for FO B cell activation [112]. While there are non-B cell effects of CD40 disruption on atherosclerosis [113], B cell specific effects support the detrimental role of B-2 activation in plaque formation [114]. This suggests that B-2 FO cells could have a pro-atherogenic role, which is the current understanding of the field. These studies however utilize broad immunomodulatory methods with some inconsistencies existing within the results, indicating that a more intricate evaluation of B-2 cells and their antibodies could aid our understanding of these cells within the disease.
Antibodies in action: IgM
Upon successful recombination of a BCR encoding gene, IgM is the first antibody class able to be produced by the B cell prior to activation. IgM is naturally present within the serum where it exists as a pentamer linked together by the Immunological J chain (Ig J) peptide, which regulates the oligomerization of these complexes [115]. This oligomerization is a critical component of IgM’s ability to bind its cognate antigen due to the binding affinity of a single IgM monomer being a considerably weak interaction. The pentameric formation of IgM increases the valency of the complex from 2 potential binding sites to 10, dramatically increasing the avidity of the molecule to multivalent antigens.
Natural IgM is secreted mostly by B-1 and MZ B Cells, which produce these molecules against evolutionary conserved motifs present on pathogen-associated molecular patterns (PAMPs), such as the cell wall components of bacterium. These natural antibodies are produced without the need for T-cell help and can be considered ‘germline’ due to their low mutation levels and ability to be generated regardless of infection [116]. IgM bound to PAMPs is recognized by C1q, which activates the classical pathway of the complement cascade, resulting in the formation of pore complexes on the plasma membrane of pathogens and cell lysis. IgM-bound C1q can also be recognized by an array of C1q receptors, including C1qRp which mediates C1q-mediated phagocytosis by neutrophils and monocytes [117, 118]. IgM may also directly aid in the recognition and clearance of PAMPs and DAMPs through opsonization and uptake of IgM-antigen complexes by Fcα/μR on phagocytic B cells and monocytes [119], which also negatively regulates humoral responses for T-independent antigens [120]. IgM is likewise recognized by FcμR, expressed on B cells in mice and B, T, and NK lymphocytes in humans, which mediates developing B cell activation and subtype distribution [121, 122], and is responsible for regulating IgM titers [123]. Interestingly, soluble IgM has been reported to be involved in the suppression of autoantibody production [124], with FcμR possibly mediating this function [125], thus making this IgM-FcμR interaction a potential interest for atherosclerosis research.
Atheroprotective nature of IgM
The IgM routinely observed in atherosclerosis is widely accepted to have an atheroprotective role in the prevention and attenuation of the disease in humans [126, 127] and directly in mice [128]. This protection is dependent on IgM secretion as secretion-deficient IgM (sIgM−/−) mice, which lack serum IgM due to the deletion of an exon in the Cμ gene locus, showed exacerbated plaque lesions [129]. While other IgM molecules may also provide protection in the diseases, IgM generated in response to the modification of biomolecules by reactive oxygen species, termed oxidation-specific epitopes (OSE), has been the most studied and these molecules are commonly found within the serum of mice and humans [130]. OSE specific IgM show a strong correlation with the control of atherosclerotic plaque formation across several human studies [130, 131].
In atherosclerosis, antibodies against oxidized forms of low-density lipoprotein (OxLDL) have been of great interest. The term OxLDL is used to encompass several different modifications of the LDL molecule with malondialdehyde (MDA), malondialdehyde-acetaldehyde (MAA), and 4-hydroxynonenal (4HNE) being the most well studied. These modifications are observed on ApoB100 and other proteins of the LDL molecule. Oxidation of phospholipids has also been observed on the LDL molecule, specifically lipid adducts (POVPC, PGPC, PEIPC, etc.) which mimic the structure of phosphorylcholine (PC) [132, 133] and play an important role in early atherogenesis in LDLR−/− mice by mediating monocyte binding [134]. Immunization with oxLDL, namely MDA-OxLDL, has been shown to provide protection in atherosclerotic mice with high titers of IgM against these adducts being observed [135,136,137]. Responses to these LDL OSEs have since been shown to be important in preventing toxicities caused by release of additional reactive oxygen species [138,139,140,141,142,143].
In 1996, the Witztum group generated an IgM-producing hybridoma, E06, which showed ability to bind oxidized phospholipid and plaques within atherosclerotic rabbits [144]. Strikingly, this antibody was found to have an identical peptide sequence to the phosphorylcholine (PC)-binding T15 clone discovered to emerge during fetal development and provide protection from Streptococcus infections [145, 146]. This prompted the discovery that antibodies with the E06/T15 heavy and light variable regions, or idiotype, could neutralize CuOxLDL, and upon passive transfer into mice, reduce plaque formation [147, 148]. Similar results were observed following immunization with PC and S. pneumoniae which generated high titers of these antibodies [149, 150]. Additionally, several studies have found a correlation between anti-PC antibodies and decreased disease in patients [151].
IgM antibodies are mostly thought to function through three mechanisms; 1) receptor-mediated endocytosis, 2) complement-mediated opsonization, and 3) antigen neutralization, each of which may contribute to the protective effect of IgM within atherosclerosis. Additional work with the E06/T15 antibodies showed that these antibodies readily bind POVPC and PEIPC lipids present on apoptotic cells [147, 152], suggesting these antibodies may aid in clearance of apoptotic cells and debris. T15 IgM promotes dendritic cell phagocytosis and clearance of apoptotic cells within plaques as measured by TUNEL staining [90, 153]. Knockout of MERTK, a receptor tyrosine kinase responsible for recognition of apoptotic cells, in ApoE−/− mice results in a dramatic increase in necrosis and TUNEL staining; however, plaques volumes are not significantly increased [154], suggesting that IgM:receptor-mediated endocytosis may play a secondary role in terms of IgM in atherosclerosis. Likewise, loss of IgM-mediated opsonization may only play a minor role in the disease as knockout of C1q, which impairs the clearance of apoptotic cells and debris, shows a less pronounced plaque increase compared to sIgM−/− mice [129]. Based on these findings, we suspect that antigen neutralization may be the major driver of IgM-mediated protection in atherosclerosis as antibodies against OxLDL have been shown to provide significant mediation of atherosclerotic risk by inhibiting macrophage phagocytosis and foam cell formation [11, 130, 155,156,157,158]. This protection is likewise observed to be independent of antibody class as IgG-derived Fabs and single chain variable fragments, which lack the constant domains necessary for Fc receptor binding and thus can only function through neutralization, show a robust ability to bind apoptotic debris and prevent macrophage phagocytosis [142, 157]. Recently, a fourth mechanism of protection has been proposed due to the ability of OSE-specific IgM to block the interaction of Factor Xa with microvesicles and prevent coagulation and thrombosis in C57BL/6 J animals [159]. The effort to understand the impact this novel mechanism is still in an early phase, yet this work provides exciting implications for the role of IgM in the disease; particularly, the control of plaques within late-stage lesions and the prevention of necrotic core rupture.
In a recent study done by our group, we observed that ApoE−/− mice deficient in AID had extensively reduced plaque burden (80%) [160]. The loss of AID results in the inability of B cells to undergo class switch recombination and somatic hypermutation. Consequentially, only low-affinity IgM antibodies can be detected in the serum of these animals and BCR expression is restricted to the Cμ and Cδ genes. This does not change in the distribution of B cell subsets (B-1a, B-1b, MZ, and FO) compared to ApoE−/− mice. Furthermore, there was no difference in splenic germinal center B cell numbers, suggesting that hyperlipidemia alone is sufficient to drive the formation of these reactions. Interestingly, we observed a dramatic decrease in aortic B cells and a massive increase in MDA-OxLDL IgM in the ApoE−/−Aid−/− mice compared to ApoE−/− on a high fat diet. This suggests that AID is somehow impeding proper generation of atheroprotective antibodies. It should be noted that because of the normal mechanisms of V(D)J recombination and negative selection, the ApoE−/− Aid−/− IgM will have a more diverse repertoire than B-1-derived IgM, and MZ-derived IgM (Table 1) which could be why these mice can produce higher amounts of OxLDL specific IgM than an ApoE−/− mouse.
Mechanism of antibody affinity maturation and class switching
Upon activation of the B cell, antibodies are able to undergo further maturation and the process of class switching. Activated B cells form germinal center structures where they undergo continuous cycling between the light and dark zone based on CXCR4 expression. Antigen engagement with the BCR receptor triggers the expression of CXCR4 on the B cell, which migrates into the dark zone against a CXCL12 chemokine gradient generated by follicular dendric cells [161]. Within the dark zone, B cells undergo several rounds of cell division and express the Aicda gene, giving rise to Activation-Induced Deaminase (AID), which introduces mutations into the DNA of the antibody gene and results in changes to the BCR heavy and light chain peptide. These altered B cell clones migrate out of the dark zone into the light zone where they compete for antigen and CD40 ligand signaling from T Follicular Helper (TFH) cells. The binding of these elements are required for proper Myc expression and survival in the B cell, resulting in higher affinity B cell clones being selected [162]. The cycling of this process is referred to as affinity maturation and can result in significant genetic drift from the original BCR peptide sequence.
The switching of antibody classes is accomplished through the activity of AID within switch regions present throughout the constant region of the heavy chain locus. Downstream intronic promoters precede highly repetitive DNA sequences that make up the switch regions prior to each of the different heavy chain constant (C) domains (Fig. 3b). T cell dependent cytokine signaling initiates transcription of these promotors, giving AID access to the DNA sequence. Repair of AID deamination results in double-stranded breaks and allows the recombination between two switch regions, resulting in the complete removal of the intervening DNA by the process of non-homologous end joining. This brings the BCR V region in proximity to a new C gene, altering the antibody isotype. Note that the excised DNA is lost following repair and thus class switching is a unidirectional process.
Class-switched antibodies
Immunoglobulin G (IgG)
IgG is the most abundant class of antibodies found throughout the body and is over half the antibody found in serum. IgG is divided into 4 subclasses in both humans (IgG1, IgG2, IgG3, and IgG4) and mice (IgG1, IgG2a/c, IgG2b, IgG3). IgG antibodies play a large variety of roles through the body, which includes complement activation, neutralization, opsonization of bacteria and cellular debris, and the induction of several immune cell functions through binding to various Fcγ Receptors. The ability of IgG molecules to complete each of these roles is dependent on the specific subclass of the antibody. The overall involvement of IgG antibodies in atherosclerosis is largely debated; however, the current consensus leans towards these molecules being atherogenic. Previously work has shown that treatment of ApoE−/− mice with IgG from an atherosclerotic animal promotes atherosclerosis development [112, 163] and prevention of proper germinal center formation or function, either by blocking AID or CD40 signaling, has an atheroprotective effect [112, 160, 164]. Likewise, it has been shown that deletion of Fcγ receptors reduces atherosclerosis by preventing IgG-induced activation of vascular endothelial cells and the production of pro-inflammatory cytokines [165]. In humans, a meta-analysis of clinical studies suggested that patients with high levels of IgG are at increased risk for myocardial infarctions [127], while another analysis found the opposite [126]. Taken together, this data suggests that IgG function is harmful to plaque control and clearance.
While IgG may be atherogenic, IgG subclasses are not all the same, nor the IgG responses themselves. To get a better understanding of IgGs involvement in atherosclerosis, a more detailed look at the IgG subclasses should be performed. IgG1 is produced in response to IL-4 cytokine signaling and is the major IgG subclass observed following immunization and in ApoE−/− mice [160]. Numerous immunizations have been performed in ApoE−/− and LDLR−/− mice over the years, and the effect of IgG1 on plaques has shown mixed results depending on the antigen. IgG1 responses to β2GP1, HSP60, GRP78, and cardiolipin showed increased plaque development in mice, while IgG1 against MDA-OxLDL, PC, and pCETP showed atheroprotection across mice, humans, and rabbits [135, 150, 166,167,168,169,170,171]. By comparison, mouse IgG2a/c in response to IFNγ signaling has been implicated as a major mechanism for the aggravation of atherosclerosis following IRA B-1 cell activation which activates macrophage through high-affinity binding to CD64 (FcγRI) [98, 172]. Meanwhile, IgG2b produced in response to TGFβ produced primarily by Tregs, seems to have a protective role through binding of CD32b (FcγRIIb). Unlike the other Fcγ receptors which contain c-terminal cell-activating ITAM motifs, CD32b utilizes an inhibitory ITIM motif and has been shown to attenuate mice plaque formation, likely through the repression of inflammation and M2 macrophage polarization [173, 174]. This is further supported by a recently published paper by Ramiro and colleagues that found that IgG2b antibodies mounted against the self-antigen ALDH4A1 provides protection from plaque in LDLR−/− mice on a high-fat diet [175].
Immunoglobulin E (IgE)
Of all the antibodies produced by B cells, IgE is the most unique in how it functions. Unlike IgM and IgG, IgE antibodies are found only at extremely low concentration within the serum and are instead found bound to the Fc epsilon receptor FcεRI expressed primarily on basophils, eosinophils, and mast cells. The binding of IgE to FcεRI is an extremely high-affinity interaction, occurring even in the absence of antigen, and is required for the proper effector function of these cells. Antigen binding to FcεRI-bound IgE induces cellular activation and the mass exocytotic release of many effector molecules, including IL-6, IL-13, granzyme B, histamine, antimicrobial peptides (LL37), and digestive peptidases (trypsin, cathepsin G, etc.) [176].
Mast cells promote inflammation, and while these cells can provide some regulatory function through the production of IL-10 [177], they are correlated with increased adverse clinical effects in human atherosclerosis and worsened plaque formations in mice [178, 179]. Thus, it is of little surprise that a plethora of clinical analyses have found IgE to be positively correlated with increased plaque burden and adverse events in both diabetic and nondiabetic individuals [179,180,181,182,183,184]. Furthermore, while only a few mice studies on IgE in atherosclerosis have been conducted so far, they tend to support this observation. In ApoE−/− IgE−/− mice, IgE was suggested to promote disease through the conversion of M2 macrophages into the M1 phenotype, encouraging foam cell development [185]. IgE was also found to be elevated in LDLR−/−sIgM−/− mice, likely due to dysregulation of the CD23 negative feedback loop on B cells, which could have contributed to the increased plaque burden that was observed [186].
It is unknown what specifically triggers IgE production in atherosclerosis, nor do we know what most of the antigens are these antibodies target within the plaque. The most probable targets are reactive oxygen species-modified self-antigens released by necrotic cells. One study found IgE against the oligosaccharide allergen galactose-α-1,3-galactose (α-Gal) to have an increased correlation with coronary artery disease than total IgE levels in human cohorts. While the mechanism of these antibodies are uncertain, the authors suspect these antibodies may have emerged in response to α-Gal-glycosylated products alongside IgG1 [187]. Like IgG1, IgE is produced in response to IL-4; however, class switching to IgE, but not IgG1, can be inhibited by TGFβ, which may explain why these two antibody classes can show divergent involvement in plaque formation in some studies and similar effects in others depending on the target antigen.
Immunoglobulin A (IgA)
Research on IgA in atherosclerosis is difficult to complete in mice due to concern regarding our ability to translate any findings to humans because of differences in the biology of the two systems [188]. Humans have two IgA constant regions in the heavy chain locus compared to only one in mice. IgA likely also has additional biological functions in humans and other mammals that are not observed in mice due to mice lacking expression of FcαRI (CD89). In humans, IgA is found in high abundance in the serum, just after IgG, and is the most abundant antibody found in the mucosa. IgA is primarily a dimer linked by the Ig J chain and is the major antibody transported through endothelial cell by the FcRn protein. IgA is produced in response to the cytokines IL-5, IL-10, and TGFβ, and plays a critical role in the neutralization and clearance of a large range of environmental antigens.
In terms of atherosclerosis risk, very little is known about IgA. Several studies have discerned a correlation between serum IgA levels and atherosclerosis in terms of patient complications; although it is worth noting that the antigen-targets observed (PC and β2GP1) are strongly correlated with the disease in other Ig classes [171, 189,190,191,192]. IgA may provide protection in an epitope-specific manner as elevated anti-PC IgA seen in individuals from hunter societies in Kitava, as well as hibernating mammals, correlates with decreased incidence of cardiovascular disease, and suggests that immunological memory formed in response to infections may mediate the atherosclerotic risk associated with aging [151, 171, 193,194,195]. We recently reported that ApoE−/− mice have slightly elevated serum IgA compared to C57BL/6 J animals, but this was minor compared to the differences we observed for IgM and IgG1 [160]. Regardless, no detailed study regarding the involvement of IgA in atherosclerosis has been done in mice to our knowledge.
Conclusions
Antibody dynamics: the importance of Isotype
Much of our understanding of antibodies in human atherosclerosis comes from meta-analysis of patient data which are almost entirely correlative in nature [126, 127, 171, 179,180,181,182,183,184, 189,190,191,192]. To investigate antibodies in a more mechanistic manner, hyperlipidemic mice deficient in the APOE or LDLR proteins have been routinely used. The most dramatic results have been seen following the breeding of these mice to mice where entire arms of B cell biology are ablated [106, 112, 160, 164, 196, 197]. This reductionist approach showed that B cells are important for modulating the disease as total loss worsened atherosclerosis while restricting the B cell compartment to B-1 cells aided disease attenuation. In addition, anti-OSE IgM antibodies are suggested to be able to control disease progression [61, 88,89,90, 108, 111, 160]. Interestingly, this neutralization and blocking of recognition by monocytes is maintained even when only the Fab domain of the antibodies were expressed [142, 157].
In ApoE−/− mice, we observed a dramatic 6-fold increase in IgG1 levels, suggesting considerable B cell activation in these mice [160]. A recent study showed a 2.4-fold increase in plaque development by injecting 10 μg of ApoE−/− IgG into Blimp1-deficient mice, which is required for proper plasma cell formation [112]. This suggest that the elevated IgG levels are atherogenic and may be involved in the propagation of the disease. Analysis of antigen specificity suggests that the ApoE−/− IgG binds to self-proteins, with no substantial binding to oxLDL [160]. It was also observed that the antigen specificity has significant overlap between various isotypes, with 72% of antibody targets being shared between the IgM and IgG isotypes in both ApoE−/− [160]. By comparison, C57BL6/J animals, which lack significant atherosclerosis compared to ApoE−/− animals, share only 17% of IgM and 16% of IgG1 targets. Taken together, this may indicate that the generation of atherogenic autoreactive antibodies is dependent on specificity and less so isotype. While technically challenging, it would be interesting to see if the transfer of late-disease ApoE−/− IgM into serum could also propagate disease. Likewise, we previously showed that ApoE−/− Aid−/− animals have high levels of MDA-oxLDL specific IgM [160]. To our knowledge, it has not yet been shown in the literature whether injection of late-stage ApoE−/− antibody can overcome high titers of this protective antibody, or if these antibodies would instead block the detrimental effects as was observed with the ApoE−/− IgG.
Based on these results and the current state of the literature, we propose the following model (Fig. 4). Anti-MDA-OxLDL and other OSEs antibodies produced by B-1 cells maintain normal tissue homeostasis. Decreases in this system due to aging or increases in LDL due to hyperlipidemia overwhelm this system, resulting in early plaque formation and the continuous uptake of OxLDL by macrophages. As the macrophage convert to foam cells, they accumulate in the intima, the resulting hypoxic environment promotes cellular rupture by necrosis and release of their cytosolic content. Intercellular proteins are modified by the plaque environment, permitting them to activate B cells which secrete self-reactive antibodies into the serum. These antibodies then bind Fc receptors expressed by myeloid and vascular smooth muscle cells, triggering the production of inflammatory cytokines and the propagation of disease.

Proposed model for atherosclerosis development. Reactive oxygen species within arteries causes the oxidation of low-density lipoprotein (LDL). These molecules are bound by neutralizing MDA-OxLDL IgM antibodies, preventing the uptake of these molecules by macrophages. Imbalance between these antibodies and OxLDL results in lipid buildup within macrophages and the conversion to foam cells. These foam cells accumulate inside the intimal wall of the artery where they and cells of the surrounding tissue undergo necroptosis. This results in the release of intercellular antigens which may be modified by the plaque environment. These antigens trigger a polyclonal activation of B cells which form tertiary lymphoid organs and produce autoreactive antibodies. These antibodies then bind Fc receptors on macrophages, smooth muscle cells, and other cells of the innate immune system to produce inflammatory cytokines, propagating inflammation and disease progression
Therapeutic potential of antibodies in atherosclerosis
The reason that the protection provided by OSE-specific antibodies is insufficient in hyperlipidemic mice and humans is not understood, but since loss of Aid−/− seems to remediate this issue, we can speculate two possibilities [112, 160]. Inflammation-promoting class-switched B cells might outcompete protective B-1 cell antibodies due to consecutive signals present within the environment of advanced lesions and overwhelm the protective compacity of these cells. Another possibility is that AID activity in OSE-antibody-producing B cells results in genetic drift away from antibodies that can neutralize OSEs in favor of other epitopes present within the plaque environment.
We speculate that injecting mice, and by extension human patients, with MDA-OxLDL IgM antibody may provide attenuation of plaque burden and prevent disease progression in high-risk individuals. Several studies provide further support for this approach, showing that intravenous injection of Ig into ApoE−/− and LDLR−/− mice reduced plaques [128, 148, 198, 199]. Fascinatingly, two of these studies involved only IgG [198, 199]. This in combination with debates previously discussed highlights a major drawback of the reductionist approach used to understand this disease. The current dogma proposes IgM as protective and IgG as harmful, yet findings such as the discovery that anti-ALDH4A1 IgG2b is protective [175] illustrates that this oversimplification, if taken too literally, could be a detriment to our understanding. We suspect that antigen-specificity may play a more important role than isotype and hypothesize that transfer of MDA-OxLDL-specific IgG antibodies should also provide protection. To better translate our knowledge of this disease to the clinic, a closer look at the specific nature of the antibodies themselves and when they emerge needs to be focused. For example, it would be interesting to know how IgG that emerges in early plaques may differ from IgG observed in advanced lesions, and if these molecules could also have clinical benefit. While much ground has been covered, there is still much to be learned about how adaptive immunity impacts atherosclerosis.
Availability of data and materials
No supporting data was generated for this manuscript.
Abbreviations
- HDL:
-
High-Density Lipoprotein
- LDL:
-
Low-Density Lipoprotein
- OxLDL:
-
Oxidized Low-Density Lipoprotein
- OSE:
-
Oxidation-Specific Epitope
- DAMP:
-
Danger-Associated Molecular Patterns
- TCR:
-
T Cell Receptor
- MHC:
-
Major Histocompatibility Complex
- CD:
-
Cluster of Differentiation
- APC:
-
Antigen Presenting Cell
- TH :
-
T Helper Cell (subclasses - TH1, TH2, TH17)
- IFN:
-
Interferon
- IL:
-
Interleukin
- Treg :
-
T Regulatory Cell
- TGFβ:
-
Transforming Growth Factor Beta
- ATLO:
-
Artery Tertiary Lymphoid Organs
- PVAT:
-
Perivascular Adipose Tissue
- BCR:
-
B Cell Receptor
- Ig:
-
Immunoglobulin (Heavy chain subclasses – IgM: Mu; IgD: Delta; IgG: Gamma; IgE: Epsilon; IgA: Alpha; Light chains – Igκ: Kappa; Igλ: Lambda; Joining Chain - IgJ)
- AID:
-
Activation-Induced Deaminase
- IRA:
-
Innate Response Activator B Cells
- GM-CSF:
-
Granulocyte-Macrophage Colony-Stimulating Factor
- MZ:
-
Marginal Zone B Cells
- FO:
-
Follicular Zone B Cells
- APOE:
-
Apolipoprotein E
- LDLR:
-
Low-Density Lipoprotein Receptor
- μMT:
-
Mice unable to make IgM (and thus lack B cells)
- C1q:
-
Component 1 Subcomponent Q
- C1qRp:
-
Component 1 Q Subcomponent Receptor 1 (punitive)
- PAMP:
-
Pathogen-Associated Molecular Patterns
- NK:
-
Natural Killer Cell
- Fc:
-
Antibody Constant Domain (Crystallizable Fragment)
- Fc [x]R:
-
Antibody Receptor for specific Ig constant domains [FcαRI, Fcα/μR, FcμR, FcεRI, Fcγ]
- sIgM−/− :
-
Mice unable to secrete IgM
- MDA:
-
malondialdehyde
- MAA:
-
malondialdehyde-acetaldehyde
- 4HNE:
-
4-hydroxynonenal
- PC:
-
Phosphatidylcholine
- POVPC:
-
1-palmitoyl-2-(5-oxovaleroyl)-sn-glycero-3-phosphocholine
- PGPC:
-
1-palmitoyl-2-glutaroyl-sn-glycero-3-phosphocholine
- PEIPC:
-
1-palmitoyl-2-(5(6)-epoxy-9-oxo-11-hydroxy-7E,14Z-prostadienoyl)-sn-glycero-phosphocholine
- CuOxLDL:
-
Copper-Oxidized Low-Density Lipoprotein
- TUNEL:
-
Terminal deoxynucleotidyl transferase dUTP nick end labeling
- MERTK:
-
MER Receptor Tyrosine Kinase
- Fab:
-
Fragment of Antigen Binding
- C[x]:
-
Antibody Constant Genes (See Ig)
- β2GP1:
-
Beta 2 Glycoprotein 1
- HSP60:
-
Heat-Shock Protein 60
- GRP78:
-
Glucose Regulated Protein 78
- pCETP:
-
Cholesteryl Ester Transfer Protein
- ITAM:
-
Immunoreceptor Tyrosine-based Activation Motif
- ITIM:
-
Immunoreceptor Tyrosine-based Inhibitory Motif
- FcRn:
-
Neonatal Fc Receptor
- ALDH4A1:
-
Aldehyde Dehydrogenase 4 Family Member A1
References
Virani SS, Alonso A, Aparicio HJ, Benjamin EJ, Bittencourt MS, Callaway CW, et al. Heart disease and stroke statistics—2021 update. Circulation. 2021;143(8):e254–743.
Libby P, Lichtman AH, Hansson GK. Immune effector mechanisms implicated in atherosclerosis: from mice to humans. Immunity. 2013;38(6):1092–104.
Gordon T, Castelli WP, Hjortland MC, Kannel WB, Dawber TR. High-density lipoprotein as a protective factor against coronary heart-disease - Framingham study. Am J Med. 1977;62(5):707–14.
Kannel WB, Castelli WP, Gordon T. Cholesterol in the prediction of atherosclerotic disease. New perspectives based on the Framingham study. Ann Intern Med. 1979;90(1):85–91.
Asztalos BF, Schaefer EJ. HDL in atherosclerosis: actor or bystander? Atheroscler Suppl. 2003;4(1):21–9.
Nordestgaard BG, Varbo A. Triglycerides and cardiovascular disease. Lancet. 2014;384(9943):626–35.
Greaves DR, Gordon S. The macrophage scavenger receptor at 30 years of age: current knowledge and future challenges. J Lipid Res. 2009;50 Suppl(Supplement):S282–6.
Binder CJ, Papac-Milicevic N, Witztum JL. Innate sensing of oxidation-specific epitopes in health and disease. Nat Rev Immunol. 2016;16(8):485–97.
Yu H, Ha T, Liu L, Wang X, Gao M, Kelley J, et al. Scavenger receptor a (SR-A) is required for LPS-induced TLR4 mediated NF-kappaB activation in macrophages. Biochim Biophys Acta. 2012;1823(7):1192–8.
Clinton SK, Underwood R, Hayes L, Sherman ML, Kufe DW, Libby P. Macrophage colony-stimulating factor gene expression in vascular cells and in experimental and human atherosclerosis. Am J Pathol. 1992;140(2):301–16.
Kruth HS. Sequestration of aggregated low-density lipoproteins by macrophages. Curr Opin Lipidol. 2002;13(5):483–8.
Martinet W, Schrijvers DM, De Meyer GR. Necrotic cell death in atherosclerosis. Basic Res Cardiol. 2011;106(5):749–60.
Kasikara C, Doran AC, Cai B, Tabas I. The role of non-resolving inflammation in atherosclerosis. J Clin Invest. 2018;128(7):2713–23.
Opoku E, Traughber CA, Zhang D, Iacano AJ, Khan M, Han J, et al. Gasdermin D mediates inflammation-induced defects in reverse cholesterol transport and promotes atherosclerosis. Front Cell Dev Biol. 2021;9:715211.
Wu XW, Molinaro C, Johnson N, Casiano CA. Secondary necrosis is a source of proteolytically modified forms of specific intracellular autoantigens - implications for systemic autoimmunity. Arthritis Rheum. 2001;44(11):2642–52.
Murao A, Aziz M, Wang H, Brenner M, Wang P. Release mechanisms of major DAMPs. Apoptosis. 2021;26(3–4):152–62.
Liston A, Lesage S, Wilson J, Peltonen L, Goodnow CC. Aire regulates negative selection of organ-specific T cells. Nat Immunol. 2003;4(4):350–4.
Gardner JM, Fletcher AL, Anderson MS, Turley SJ. AIRE in the thymus and beyond. Curr Opin Immunol. 2009;21(6):582–9.
Takaba H, Takayanagi H. The mechanisms of T cell selection in the Thymus. Trends Immunol. 2017;38(11):805–16.
Emeson EE, Robertson ALJ. T lymphocytes in aortic and coronary Intimas. Their potential role in Atherogenesis. Am J Pathol. 1988;130(2):369–76.
Shimokama T, Haraoka S, Watanabe T. Immunohistochemical and ultrastructural demonstration of the lymphocyte-macrophage interaction in human aortic intima. Modern Pathol. 1991;4(1):101–7.
Paulsson G, Zhou X, Tornquist E, Hansson GK. Oligoclonal T cell expansions in atherosclerotic lesions of apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2000;20(1):10–7.
Hermansson A, Ketelhuth DF, Strodthoff D, Wurm M, Hansson EM, Nicoletti A, et al. Inhibition of T cell response to native low-density lipoprotein reduces atherosclerosis. J Exp Med. 2010;207(5):1081–93.
Dansky HM, Charlton SA, Harper MM, Smith JD. T and B lymphocytes play a minor role in atherosclerotic plaque formation in the apolipoprotein E-deficient mouse. Proc Natl Acad Sci U S A. 1997;94(9):4642–6.
Daugherty A, Pure E, Delfel-Butteiger D, Chen S, Leferovich J, Roselaar SE, et al. The effects of total lymphocyte deficiency on the extent of atherosclerosis in apolipoprotein E−/− mice. J Clin Invest. 1997;100(6):1575–80.
Diaz A, Gonzalez-Alayon I, Perez-Torrado V, Suarez-Martins M. CD40-CD154: a perspective from type 2 immunity. Semin Immunol. 2021;53:101528.
Daub S, Lutgens E, Munzel T, Daiber A. CD40/CD40L and related signaling pathways in cardiovascular health and disease-the pros and cons for Cardioprotection. Int J Mol Sci. 2020;21(22):8533.
Bosmans LA, Bosch L, Kusters PJH, Lutgens E, Seijkens TTP. The CD40-CD40L dyad as immunotherapeutic target in cardiovascular disease. J Cardiovasc Transl Res. 2021;14(1):13–22.
Kyaw T, Winship A, Tay C, Kanellakis P, Hosseini H, Cao A, et al. Cytotoxic and proinflammatory CD8+ T lymphocytes promote development of vulnerable atherosclerotic plaques in apoE-deficient mice. Circulation. 2013;127(9):1028–39.
Cochain C, Koch M, Chaudhari SM, Busch M, Pelisek J, Boon L, et al. CD8+ T cells regulate Monopoiesis and circulating Ly6C-high monocyte levels in atherosclerosis in mice. Circ Res. 2015;117(3):244–53.
Winkels H, Ehinger E, Vassallo M, Buscher K, Dinh HQ, Kobiyama K, et al. Atlas of the immune cell repertoire in mouse atherosclerosis defined by single-cell RNA-sequencing and mass Cytometry. Circ Res. 2018;122(12):1675–88.
Fernandez DM, Rahman AH, Fernandez NF, Chudnovskiy A, Amir ED, Amadori L, et al. Single-cell immune landscape of human atherosclerotic plaques. Nat Med. 2019;25(10):1576–88.
Liuzzo G, Vallejo AN, Kopecky SL, Frye RL, Holmes DR, Goronzy JJ, et al. Molecular fingerprint of interferon-gamma signaling in unstable angina. Circulation. 2001;103(11):1509–14.
Nathan CF, Murray HW, Wiebe ME, Rubin BY. Identification of interferon-gamma as the lymphokine that activates human macrophage oxidative metabolism and antimicrobial activity. J Exp Med. 1983;158(3):670–89.
Gupta S, Pablo AM, Jiang X, Wang N, Tall AR, Schindler C. IFN-gamma potentiates atherosclerosis in ApoE knock-out mice. J Clin Invest. 1997;99(11):2752–61.
Whitman SC, Ravisankar P, Elam H, Daugherty A. Exogenous interferon-γ enhances atherosclerosis in Apolipoprotein E−/− mice. Am J Pathol. 2000;157(6):1819–24.
Whitman SC, Ravisankar P, Daugherty A. IFN-gamma deficiency exerts gender-specific effects on atherogenesis in apolipoprotein E−/− mice. J Interf Cytokine Res. 2002;22(6):661–70.
Buono C, Binder CJ, Stavrakis G, Witztum JL, Glimcher LH, Lichtman AH. T-bet deficiency reduces atherosclerosis and alters plaque antigen-specific immune responses. Proc Natl Acad Sci U S A. 2005;102(5):1596–601.
Barks JL, McQuillan JJ, Iademarco MF. TNF-alpha and IL-4 synergistically increase vascular cell adhesion molecule-1 expression in cultured vascular smooth muscle cells. J Immunol. 1997;159(9):4532–8.
Sasaguri T, Arima N, Tanimoto A, Shimajiri S, Hamada T, Sasaguri Y. A role for interleukin 4 in production of matrix metalloproteinase 1 by human aortic smooth muscle cells. Atherosclerosis. 1998;138(2):247–53.
Lee YW, Kühn H, Hennig B, Toborek M. IL-4 induces apoptosis of endothelial cells through the caspase-3-dependent pathway. FEBS Lett. 2000;485(2–3):122–6.
Lee YW, Kuhn H, Hennig B, Neish AS, Toborek M. IL-4-induced oxidative stress upregulates VCAM-1 gene expression in human endothelial cells. J Mol Cell Cardiol. 2001;33(1):83–94.
King VL, Szilvassy SJ, Daugherty A. Interleukin-4 deficiency decreases atherosclerotic lesion formation in a site-specific manner in female LDL receptor−/− mice. Arterioscler Thromb Vasc Biol. 2002;22(3):456–61.
Binder CJ, Hartvigsen K, Chang M-K, Miller M, Broide D, Palinski W, et al. IL-5 links adaptive and natural immunity specific for epitopes of oxidized LDL and protects from atherosclerosis. J Clin Invest. 2004;114(3):427–37.
Miller AM, Xu D, Asquith DL, Denby L, Li Y, Sattar N, et al. IL-33 reduces the development of atherosclerosis. J Exp Med. 2008;205(2):339–46.
Erbel C, Chen L, Bea F, Wangler S, Celik S, Lasitschka F, et al. Inhibition of IL-17A attenuates atherosclerotic lesion development in apoE-deficient mice. J Immunol. 2009;183(12):8167–75.
Erbel C, Akhavanpoor M, Okuyucu D, Wangler S, Dietz A, Zhao L, et al. IL-17A influences essential functions of the monocyte/macrophage lineage and is involved in advanced murine and human atherosclerosis. J Immunol. 2014;193(9):4344–55.
Eid RE, Rao DA, Zhou J, Lo SF, Ranjbaran H, Gallo A, et al. Interleukin-17 and interferon-gamma are produced concomitantly by human coronary artery-infiltrating T cells and act synergistically on vascular smooth muscle cells. Circulation. 2009;119(10):1424–32.
Danzaki K, Matsui Y, Ikesue M, Ohta D, Ito K, Kanayama M, et al. Interleukin-17A deficiency accelerates unstable atherosclerotic plaque formation in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2012;32(2):273–80.
Mallat Z, Gojova A, Brun V, Esposito B, Fournier N, Cottrez F, et al. Induction of a regulatory T cell type 1 response reduces the development of atherosclerosis in apolipoprotein E-knockout mice. Circulation. 2003;108(10):1232–7.
Herbin O, Ait-Oufella H, Yu W, Fredrikson GN, Aubier B, Perez N, et al. Regulatory T-cell response to apolipoprotein B100-derived peptides reduces the development and progression of atherosclerosis in mice. Arterioscler Thromb Vasc Biol. 2012;32(3):605–12.
Cheng X, Yu X, Ding YJ, Fu QQ, Xie JJ, Tang TT, et al. The Th17/Treg imbalance in patients with acute coronary syndrome. Clin Immunol. 2008;127(1):89–97.
Zhang J, Hua G, Zhang X, Tong R, Du X, Li Z. Regulatory T cells/T-helper cell 17 functional imbalance in uraemic patients on maintenance haemodialysis: a pivotal link between microinflammation and adverse cardiovascular events. Nephrology (Carlton). 2010;15(1):33–41.
Shen Y, Yuan Z, Yin A, Liu Y, Xiao Y, Wu Y, et al. Antiatherogenic effect of pioglitazone on uremic apolipoprotein E knockout mice by modulation of the balance of regulatory and effector T cells. Atherosclerosis. 2011;218(2):330–8.
Liu M, Xu LJ, Wu JX. Changes of circulating CD4(+)CD25(+)CD127(low) regulatory T cells in patients with acute coronary syndrome and its significance. Genet Mol Res. 2015;14(4):15930–6.
Butcher MJ, Filipowicz AR, Waseem TC, McGary CM, Crow KJ, Magilnick N, et al. Atherosclerosis-driven Treg plasticity results in formation of a dysfunctional subset of plastic IFNgamma+ Th1/Tregs. Circ Res. 2016;119(11):1190–203.
Yang J, Wu J, Zhang R, Yao M, Liu Y, Miao L, et al. Porphyromonas gingivalis oral infection promote T helper 17/Treg imbalance in the development of atherosclerosis. J Dent Sci. 2017;12(1):60–9.
Gao S, Zhang W, Zhao Q, Zhou J, Wu Y, Liu Y, et al. Curcumin ameliorates atherosclerosis in apolipoprotein E deficient asthmatic mice by regulating the balance of Th2/Treg cells. Phytomedicine. 2019;52:129–35.
Fan Q, Liu Y, Rao J, Zhang Z, Xiao W, Zhu T, et al. Anti-atherosclerosis effect of Angong Niuhuang pill via regulating Th17/Treg immune balance and inhibiting chronic inflammatory on ApoE−/− mice model of early and mid-term atherosclerosis. Front Pharmacol. 2020;10:1584.
Wolf D, Gerhardt T, Winkels H, Michel NA, Pramod AB, Ghosheh Y, et al. Pathogenic autoimmunity in atherosclerosis evolves from initially protective Apolipoprotein B100-reactive CD4(+) T-regulatory cells. Circulation. 2020;142(13):1279–93.
Srikakulapu P, Upadhye A, Rosenfeld SM, Marshall MA, McSkimming C, Hickman AW, et al. Perivascular adipose tissue harbors Atheroprotective IgM-producing B cells. Front Physiol. 2017;8:719.
Houtkamp MA, de Boer OJ, van der Loos CM, van der Wal AC, Becker AE. Adventitial infiltrates associated with advanced atherosclerotic plaques: structural organization suggests generation of local humoral immune responses. J Pathol. 2001;193(2):263–9.
Grabner R, Lotzer K, Dopping S, Hildner M, Radke D, Beer M, et al. Lymphotoxin beta receptor signaling promotes tertiary lymphoid organogenesis in the aorta adventitia of aged ApoE−/− mice. J Exp Med. 2009;206(1):233–48.
Srikakulapu P, Hu D, Yin C, Mohanta SK, Bontha SV, Peng L, et al. Artery tertiary lymphoid organs control multilayered territorialized atherosclerosis B-cell responses in aged ApoE−/− mice. Arterioscler Thromb Vasc Biol. 2016;36(6):1174–85.
Watanabe M, Sangawa A, Sasaki Y, Yamashita M, Tanaka-Shintani M, Shintaku M, et al. Distribution of inflammatory cells in adventitia changed with advancing atherosclerosis of human coronary artery. J Atheroscler Thromb. 2007;14(6):325–31.
Burioni R, Canducci F, Saita D, Perotti M, Mancini N, De Marco D, et al. Antigen-driven evolution of B lymphocytes in coronary atherosclerotic plaques. J Immunol. 2009;183(4):2537–44.
Zernecke A, Winkels H, Cochain C, Williams JW, Wolf D, Soehnlein O, et al. Meta-analysis of leukocyte diversity in atherosclerotic mouse aortas. Circ Res. 2020;127(3):402–26.
Graver JC, Boots AMH, Haacke EA, Diepstra A, Brouwer E, Sandovici M. Massive B-cell infiltration and organization into artery tertiary lymphoid organs in the aorta of large vessel Giant cell arteritis. Front Immunol. 2019;10:83.
Farias-Itao DS, Pasqualucci CA, Nishizawa A, Da Silva LFF, Campos FM, Bittencourt MS, et al. B lymphocytes and macrophages in the perivascular adipose tissue are associated with coronary atherosclerosis: an autopsy study. J Am Heart Assoc. 2019;8(24):e013793.
Chi X, Li Y, Qiu X. V(D)J recombination, somatic hypermutation and class switch recombination of immunoglobulins: mechanism and regulation. Immunology. 2020;160(3):233–47.
Chen X, Gellert M, Yang W. Inner workings of RAG recombinase and its specialization for adaptive immunity. Curr Opin Struct Biol. 2021;71:79–86.
Owen JJ, Cooper MD, Raff MC. In vitro generation of B lymphocytes in mouse foetal liver, a mammalian 'bursa equivalent'. Nature. 1974;249(455):361–3.
Owen JJ, Raff MC, Cooper MD. Studies on the generation of B lymphocytes in the mouse embryo. Eur J Immunol. 1976;5(7):468–73.
Montecino-Rodriguez E, Leathers H, Dorshkind K. Identification of a B-1 B cell-specified progenitor. Nat Immunol. 2006;7(3):293–301.
Montecino-Rodriguez E, Dorshkind K. Formation of B-1 B cells from neonatal B-1 transitional cells exhibits NF-kappaB redundancy. J Immunol. 2011;187(11):5712–9.
Choi YS, Dieter JA, Rothaeusler K, Luo Z, Baumgarth N. B-1 cells in the bone marrow are a significant source of natural IgM. Eur J Immunol. 2012;42(1):120–9.
Lalor PA. An evolutionarily-conserved role for murine Ly-1 B cells in protection against bacterial infections. Autoimmunity. 1991;10(1):71–6.
Martin F, Oliver AM, Kearney JF. Marginal zone and B1 B cells unite in the early response against T-independent blood-borne particulate antigens. Immunity. 2001;14(5):617–29.
Vos Q, Lees A, Wu ZQ, Snapper CM, Mond JJ. B-cell activation by T-cell-independent type 2 antigens as an integral part of the humoral immune response to pathogenic microorganisms. Immunol Rev. 2000;176(1):154–70.
Mond JJ, Scher I, Mosier DE, Baese M, Paul WE. T-independent responses in B cell-defective CBA/N mice to Brucella abortus and to trinitrophenyl (TNP) conjugates of Brucella abortus. Eur J Immunol. 1978;8(7):459–63.
Alugupalli KR, Leong JM, Woodland RT, Muramatsu M, Honjo T, Gerstein RM. B1b lymphocytes confer T cell-independent long-lasting immunity. Immunity. 2004;21(3):379–90.
Alugupalli KR, Gerstein RM, Chen J, Szomolanyi-Tsuda E, Woodland RT, Leong JM. The resolution of relapsing fever borreliosis requires IgM and is concurrent with expansion of B1b lymphocytes. J Immunol. 2003;170(7):3819–27.
Forster I, Rajewsky K. Expansion and functional activity of Ly-1+ B cells upon transfer of peritoneal cells into allotype-congenic, newborn mice. Eur J Immunol. 1987;17(4):521–8.
Gu H, Forster I, Rajewsky K. Sequence homologies, N sequence insertion and JH gene utilization in VHDJH joining: implications for the joining mechanism and the ontogenetic timing of Ly1 B cell and B-CLL progenitor generation. EMBO J. 1990;9(7):2133–40.
Haas KM, Poe JC, Steeber DA, Tedder TF. B-1a and B-1b cells exhibit distinct developmental requirements and have unique functional roles in innate and adaptive immunity to S. pneumoniae. Immunity. 2005;23(1):7–18.
Hardy RR, Hayakawa K. Development of B cells producing natural autoantibodies to thymocytes and senescent erythrocytes. Springer Semin Immunopathol. 2005;26(4):363–75.
Cunningham AF, Flores-Langarica A, Bobat S, Dominguez Medina CC, Cook CN, Ross EA, et al. B1b cells recognize protective antigens after natural infection and vaccination. Front Immunol. 2014;5:535.
Tsiantoulas D, Gruber S, Binder CJ. B-1 cell immunoglobulin directed against oxidation-specific epitopes. Front Immunol. 2013;3:415.
Kyaw T, Tay C, Krishnamurthi S, Kanellakis P, Agrotis A, Tipping P, et al. B1a B lymphocytes are atheroprotective by secreting natural IgM that increases IgM deposits and reduces necrotic cores in atherosclerotic lesions. Circ Res. 2011;109(8):830–40.
Rosenfeld SM, Perry HM, Gonen A, Prohaska TA, Srikakulapu P, Grewal S, et al. B-1b cells secrete Atheroprotective IgM and attenuate atherosclerosis. Circ Res. 2015;117(3):e28–39.
Gruber S, Hendrikx T, Tsiantoulas D, Ozsvar-Kozma M, Goderle L, Mallat Z, et al. Sialic acid-binding immunoglobulin-like Lectin G promotes atherosclerosis and liver inflammation by suppressing the protective functions of B-1 cells. Cell Rep. 2016;14(10):2348–61.
Muller J, Lunz B, Schwab I, Acs A, Nimmerjahn F, Daniel C, et al. Siglec-G deficiency leads to autoimmunity in aging C57BL/6 mice. J Immunol. 2015;195(1):51–60.
Hosseini H, Yi L, Kanellakis P, Cao A, Tay C, Peter K, et al. Anti-TIM-1 monoclonal antibody (RMT1-10) attenuates atherosclerosis by expanding IgM-producing B1a cells. J Am Heart Assoc. 2018;7(13):e008447.
Xia N, Hasselwander S, Reifenberg G, Habermeier A, Closs EI, Mimmler M, et al. B lymphocyte-deficiency in mice causes vascular dysfunction by inducing Neutrophilia. Biomedicines. 2021;9(11):1686.
Chistiakov DA, Grechko AV, Myasoedova VA, Melnichenko AA, Orekhov AN. The role of monocytosis and neutrophilia in atherosclerosis. J Cell Mol Med. 2018;22(3):1366–82.
Rodriguez-Zhurbenko N, Quach TD, Hopkins TJ, Rothstein TL, Hernandez AM. Human B-1 cells and B-1 cell antibodies change with advancing age. Front Immunol. 2019;10:483.
Rauch PJ, Chudnovskiy A, Robbins CS, Weber GF, Etzrodt M, Hilgendorf I, et al. Innate response activator B cells protect against microbial sepsis. Science. 2012;335(6068):597–601.
Hilgendorf I, Theurl I, Gerhardt LM, Robbins CS, Weber GF, Gonen A, et al. Innate response activator B cells aggravate atherosclerosis by stimulating T helper-1 adaptive immunity. Circulation. 2014;129(16):1677–87.
Shaposhnik Z, Wang X, Weinstein M, Bennett BJ, Lusis AJ. Granulocyte macrophage colony-stimulating factor regulates dendritic cell content of atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 2007;27(3):621–7.
Haghighat A, Weiss D, Whalin MK, Cowan DP, Taylor WR. Granulocyte colony-stimulating factor and granulocyte macrophage colony-stimulating factor exacerbate atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2007;115(15):2049–54.
Zhu SN, Chen M, Jongstra-Bilen J, Cybulsky MI. GM-CSF regulates intimal cell proliferation in nascent atherosclerotic lesions. J Exp Med. 2009;206(10):2141–9.
Lee BO, Moyron-Quiroz J, Rangel-Moreno J, Kusser KL, Hartson L, Sprague F, et al. CD40, but not CD154, expression on B cells is necessary for optimal primary B cell responses. J Immunol. 2003;171(11):5707–17.
Hendricks J, Bos NA, Kroese FGM. Heterogeneity of memory marginal zone B cells. Crit Rev Immunol. 2018;38(2):145–58.
Mohanta SK, Yin C, Peng L, Srikakulapu P, Bontha V, Hu D, et al. Artery tertiary lymphoid organs contribute to innate and adaptive immune responses in advanced mouse atherosclerosis. Circ Res. 2014;114(11):1772–87.
Caligiuri G, Nicoletti A, Poirier B, Hansson GK. Protective immunity against atherosclerosis carried by B cells of hypercholesterolemic mice. J Clin Invest. 2002;109(6):745–53.
Major AS, Fazio S, Linton MF. B-lymphocyte deficiency increases atherosclerosis in LDL receptor-null mice. Arterioscler Thromb Vasc Biol. 2002;22(11):1892–8.
Tay C, Liu YH, Hosseini H, Kanellakis P, Cao A, Peter K, et al. B-cell-specific depletion of tumour necrosis factor alpha inhibits atherosclerosis development and plaque vulnerability to rupture by reducing cell death and inflammation. Cardiovasc Res. 2016;111(4):385–97.
Kyaw T, Tay C, Khan A, Dumouchel V, Cao A, To K, et al. Conventional B2 B cell depletion ameliorates whereas its adoptive transfer aggravates atherosclerosis. J Immunol. 2010;185(7):4410–9.
Doran AC, Lipinski MJ, Oldham SN, Garmey JC, Campbell KA, Skaflen MD, et al. B-cell aortic homing and atheroprotection depend on Id3. Circ Res. 2012;110(1):e1–12.
Nus M, Sage AP, Lu Y, Masters L, Lam BYH, Newland S, et al. Marginal zone B cells control the response of follicular helper T cells to a high-cholesterol diet. Nat Med. 2017;23(5):601–10.
Ait-Oufella H, Herbin O, Bouaziz JD, Binder CJ, Uyttenhove C, Laurans L, et al. B cell depletion reduces the development of atherosclerosis in mice. J Exp Med. 2010;207(8):1579–87.
Tay C, Liu YH, Kanellakis P, Kallies A, Li Y, Cao A, et al. Follicular B cells promote atherosclerosis via T cell-mediated differentiation into plasma cells and secreting pathogenic immunoglobulin G. Arterioscler Thromb Vasc Biol. 2018;38(5):e71–84.
Bosmans LA, van Tiel CM, Aarts S, Willemsen L, Baardman J, van Os BW, et al. Myeloid CD40 deficiency reduces atherosclerosis by impairing macrophages' transition into a pro-inflammatory state. Cardiovasc Res. 2022.
Tay C, Kanellakis P, Hosseini H, Cao A, Toh BH, Bobik A, et al. B cell and CD4 T cell interactions promote development of atherosclerosis. Front Immunol. 2019;10:1-15.
Brewer JW, Corley RB. Late events in assembly determine the polymeric structure and biological activity of secretory IgM. Mol Immunol. 1997;34(4):323–31.
Haury M, Sundblad A, Grandien A, Barreau C, Coutinho A, Nobrega A. The repertoire of serum IgM in normal mice is largely independent of external antigenic contact. Eur J Immunol. 1997;27(6):1557–63.
Guan E, Robinson SL, Goodman EB, Tenner AJ. Cell-surface protein identified on phagocytic cells modulates the C1q-mediated enhancement of phagocytosis. J Immunol. 1994;152(8):4005–16.
Bobak DA, Gaither TA, Frank MM, Tenner AJ. Modulation of FcR function by complement: subcomponent C1q enhances the phagocytosis of IgG-opsonized targets by human monocytes and culture-derived macrophages. J Immunol. 1987;138(4):1150–6.
Shibuya A, Sakamoto N, Shimizu Y, Shibuya K, Osawa M, Hiroyama T, et al. Fc alpha/mu receptor mediates endocytosis of IgM-coated microbes. Nat Immunol. 2000;1(5):441–6.
Honda S, Kurita N, Miyamoto A, Cho Y, Usui K, Takeshita K, et al. Enhanced humoral immune responses against T-independent antigens in fc alpha/muR-deficient mice. Proc Natl Acad Sci U S A. 2009;106(27):11230–5.
Ouchida R, Mori H, Hase K, Takatsu H, Kurosaki T, Tokuhisa T, et al. Critical role of the IgM fc receptor in IgM homeostasis, B-cell survival, and humoral immune responses. Proc Natl Acad Sci U S A. 2012;109(40):E2699–706.
Ouchida R, Lu Q, Liu J, Li Y, Chu Y, Tsubata T, et al. FcmuR interacts and cooperates with the B cell receptor to promote B cell survival. J Immunol. 2015;194(7):3096–101.
Honjo K, Kubagawa Y, Jones DM, Dizon B, Zhu Z, Ohno H, et al. Altered Ig levels and antibody responses in mice deficient for the fc receptor for IgM (FcmuR). Proc Natl Acad Sci U S A. 2012;109(39):15882–7.
Ehrenstein MR, Cook HT, Neuberger MS. Deficiency in serum immunoglobulin (Ig)M predisposes to development of IgG autoantibodies. J Exp Med. 2000;191(7):1253–8.
Yoshizawa Y, Honda S, Shibuya A. Involvement of Fcalpha/muR (CD351) in autoantibody production. Mol Immunol. 2014;57(2):216–9.
Khamis RY, Hughes AD, Caga-Anan M, Chang CL, Boyle JJ, Kojima C, et al. High serum immunoglobulin G and M levels predict freedom from adverse cardiovascular events in hypertension: a nested case-control substudy of the Anglo-Scandinavian cardiac outcomes trial. EBioMedicine. 2016;9:372–80.
Kovanen PT, Manttari M, Palosuo T, Manninen V, Aho K. Prediction of myocardial infarction in dyslipidemic men by elevated levels of immunoglobulin classes A, E, and G, but not M. Arch Intern Med. 1998;158(13):1434–9.
Cesena FH, Dimayuga PC, Yano J, Zhao X, Kirzner J, Zhou J, et al. Immune-modulation by polyclonal IgM treatment reduces atherosclerosis in hypercholesterolemic apoE−/− mice. Atherosclerosis. 2012;220(1):59–65.
Lewis MJ, Malik TH, Ehrenstein MR, Boyle JJ, Botto M, Haskard DO. Immunoglobulin M is required for protection against atherosclerosis in low-density lipoprotein receptor-deficient mice. Circulation. 2009;120(5):417–26.
Chou MY, Fogelstrand L, Hartvigsen K, Hansen LF, Woelkers D, Shaw PX, et al. Oxidation-specific epitopes are dominant targets of innate natural antibodies in mice and humans. J Clin Invest. 2009;119(5):1335–49.
van den Berg VJ, Vroegindewey MM, Kardys I, Boersma E, Haskard D, Hartley A, et al. Anti-oxidized LDL antibodies and coronary artery disease: a systematic review. Antioxidants (Basel). 2019;8(10):484.
Boullier A, Gillotte KL, Horkko S, Green SR, Friedman P, Dennis EA, et al. The binding of oxidized low density lipoprotein to mouse CD36 is mediated in part by oxidized phospholipids that are associated with both the lipid and protein moieties of the lipoprotein. J Biol Chem. 2000;275(13):9163–9.
Virella G, Thorpe SR, Alderson NL, Derrick MB, Chassereau C, Rhett JM, et al. Definition of the immunogenic forms of modified human LDL recognized by human autoantibodies and by rabbit hyperimmune antibodies. J Lipid Res. 2004;45(10):1859–67.
Subbanagounder G, Leitinger N, Shih PT, Faull KF, Berliner JA. Evidence that phospholipid oxidation products and/or platelet-activating factor play an important role in early atherogenesis : in vitro and in vivo inhibition by WEB 2086. Circ Res. 1999;85(4):311–8.
George J, Afek A, Gilburd B, Levkovitz H, Shaish A, Goldberg I, et al. Hyperimmunization of apo-E-deficient mice with homologous malondialdehyde low-density lipoprotein suppresses early atherogenesis. Atherosclerosis. 1998;138(1):147–52.
Gonen A, Hansen LF, Turner WW, Montano EN, Que X, Rafia A, et al. Atheroprotective immunization with malondialdehyde-modified LDL is hapten specific and dependent on advanced MDA adducts: implications for development of an atheroprotective vaccine. J Lipid Res. 2014;55(10):2137–55.
Zhu L, He Z, Wu F, Ding R, Jiang Q, Zhang J, et al. Immunization with advanced glycation end products modified low density lipoprotein inhibits atherosclerosis progression in diabetic apoE and LDLR null mice. Cardiovasc Diabetol. 2014;13(1):151.
Weismann D, Hartvigsen K, Lauer N, Bennett KL, Scholl HP, Charbel Issa P, et al. Complement factor H binds malondialdehyde epitopes and protects from oxidative stress. Nature. 2011;478(7367):76–81.
Simoes C, Silva AC, Domingues P, Laranjeira P, Paiva A, Domingues MR. Modified phosphatidylethanolamines induce different levels of cytokine expression in monocytes and dendritic cells. Chem Phys Lipids. 2013;175-176:57–64.
Que X, Widhopf GF 2nd, Amir S, Hartvigsen K, Hansen LF, Woelkers D, et al. IGHV1-69-encoded antibodies expressed in chronic lymphocytic leukemia react with malondialdehyde-acetaldehyde adduct, an immunodominant oxidation-specific epitope. PLoS One. 2013;8(6):e65203.
Papac-Milicevic N, Busch CJ, Binder CJ. Malondialdehyde Epitopes as Targets of Immunity and the Implications for Atherosclerosis. Adv Immunol. 2016;131:1-59.
Que X, Hung MY, Yeang C, Gonen A, Prohaska TA, Sun X, et al. Oxidized phospholipids are proinflammatory and proatherogenic in hypercholesterolaemic mice. Nature. 2018;558(7709):301–6.
Kuzan A. Toxicity of advanced glycation end products (review). Biomed Rep. 2021;14(5):46.
Palinski W, Horkko S, Miller E, Steinbrecher UP, Powell HC, Curtiss LK, et al. Cloning of monoclonal autoantibodies to epitopes of oxidized lipoproteins from apolipoprotein E-deficient mice. Demonstration of epitopes of oxidized low density lipoprotein in human plasma. J Clin Invest. 1996;98(3):800–14.
Sigal NH, Pickard AR, Metcalf ES, Gearhart PJ, Klinman NR. Expression of phosphorylcholine-specific B cells during murine development. J Exp Med. 1977;146(4):933–48.
Briles DE, Forman C, Hudak S, Claflin JL. Anti-phosphorylcholine antibodies of the T15 idiotype are optimally protective against Streptococcus pneumoniae. J Exp Med. 1982;156(4):1177–85.
Shaw PX, Horkko S, Chang MK, Curtiss LK, Palinski W, Silverman GJ, et al. Natural antibodies with the T15 idiotype may act in atherosclerosis, apoptotic clearance, and protective immunity. J Clin Invest. 2000;105(12):1731–40.
Faria-Neto JR, Chyu KY, Li X, Dimayuga PC, Ferreira C, Yano J, et al. Passive immunization with monoclonal IgM antibodies against phosphorylcholine reduces accelerated vein graft atherosclerosis in apolipoprotein E-null mice. Atherosclerosis. 2006;189(1):83–90.
Binder CJ, Horkko S, Dewan A, Chang MK, Kieu EP, Goodyear CS, et al. Pneumococcal vaccination decreases atherosclerotic lesion formation: molecular mimicry between Streptococcus pneumoniae and oxidized LDL. Nat Med. 2003;9(6):736–43.
Caligiuri G, Khallou-Laschet J, Vandaele M, Gaston AT, Delignat S, Mandet C, et al. Phosphorylcholine-targeting immunization reduces atherosclerosis. J Am Coll Cardiol. 2007;50(6):540–6.
Frostegard J. Antibodies against phosphorylcholine and protection against atherosclerosis, cardiovascular disease and chronic inflammation. Expert Rev Clin Immunol. 2022;18(5):525–32.
Chang MK, Binder CJ, Miller YI, Subbanagounder G, Silverman GJ, Berliner JA, et al. Apoptotic cells with oxidation-specific epitopes are immunogenic and proinflammatory. J Exp Med. 2004;200(11):1359–70.
Chen Y, Park YB, Patel E, Silverman GJ. IgM antibodies to apoptosis-associated determinants recruit C1q and enhance dendritic cell phagocytosis of apoptotic cells. J Immunol. 2009;182(10):6031–43.
Thorp E, Cui D, Schrijvers DM, Kuriakose G, Tabas I. Mertk receptor mutation reduces efferocytosis efficiency and promotes apoptotic cell accumulation and plaque necrosis in atherosclerotic lesions of apoe−/− mice. Arterioscler Thromb Vasc Biol. 2008;28(8):1421–8.
Chang MK, Bergmark C, Laurila A, Horkko S, Han KH, Friedman P, et al. Monoclonal antibodies against oxidized low-density lipoprotein bind to apoptotic cells and inhibit their phagocytosis by elicited macrophages: evidence that oxidation-specific epitopes mediate macrophage recognition. Proc Natl Acad Sci U S A. 1999;96(11):6353–8.
Horkko S, Bird DA, Miller E, Itabe H, Leitinger N, Subbanagounder G, et al. Monoclonal autoantibodies specific for oxidized phospholipids or oxidized phospholipid-protein adducts inhibit macrophage uptake of oxidized low-density lipoproteins. J Clin Invest. 1999;103(1):117–28.
Shaw PX, Horkko S, Tsimikas S, Chang MK, Palinski W, Silverman GJ, et al. Human-derived anti-oxidized LDL autoantibody blocks uptake of oxidized LDL by macrophages and localizes to atherosclerotic lesions in vivo. Arterioscler Thromb Vasc Biol. 2001;21(8):1333–9.
Friedman P, Horkko S, Steinberg D, Witztum JL, Dennis EA. Correlation of antiphospholipid antibody recognition with the structure of synthetic oxidized phospholipids. Importance of Schiff base formation and aldol condensation. J Biol Chem. 2002;277(9):7010–20.
Obermayer G, Afonyushkin T, Goderle L, Puhm F, Schrottmaier W, Taqi S, et al. Natural IgM antibodies inhibit microvesicle-driven coagulation and thrombosis. Blood. 2021;137(10):1406–15.
Hutchinson MA, Park HS, Zanotti KJ, Alvarez-Gonzalez J, Zhang J, Zhang L, et al. Auto-antibody production during experimental atherosclerosis in ApoE(−/−) mice. Front Immunol. 2021;12:695220.
Becker M, Hobeika E, Jumaa H, Reth M, Maity PC. CXCR4 signaling and function require the expression of the IgD-class B-cell antigen receptor. Proc Natl Acad Sci U S A. 2017;114(20):5231–6.
Luo W, Weisel F, Shlomchik MJ. B cell receptor and CD40 signaling are rewired for synergistic induction of the c-Myc transcription factor in germinal center B cells. Immunity. 2018;48(2):313–26 e5.
Centa M, Jin H, Hofste L, Hellberg S, Busch A, Baumgartner R, et al. Germinal center-derived antibodies promote atherosclerosis plaque size and stability. Circulation. 2019;139(21):2466–82.
Mach F, Schonbeck U, Sukhova GK, Atkinson E, Libby P. Reduction of atherosclerosis in mice by inhibition of CD40 signalling. Nature. 1998;394(6689):200–3.
Sumiyoshi K, Mokuno H, Iesaki T, Shimada K, Miyazaki T, Kume A, et al. Deletion of the fc receptors gamma chain preserves endothelial function affected by hypercholesterolaemia in mice fed on a high-fat diet. Cardiovasc Res. 2008;80(3):463–70.
George J, Afek A, Gilburd B, Levy Y, Blank M, Kopolovic J, et al. Atherosclerosis in LDL-receptor knockout mice is accelerated by immunization with anticardiolipin antibodies. Lupus. 1997;6(9):723–9.
Foteinos G, Afzal AR, Mandal K, Jahangiri M, Xu Q. Anti-heat shock protein 60 autoantibodies induce atherosclerosis in apolipoprotein E-deficient mice via endothelial damage. Circulation. 2005;112(8):1206–13.
Afek A, George J, Shoenfeld Y, Gilburd B, Levy Y, Shaish A, et al. Enhancement of atherosclerosis in beta-2-glycoprotein I-immunized apolipoprotein E-deficient mice. Pathobiology. 1999;67(1):19–25.
Crane ED, Al-Hashimi AA, Chen J, Lynn EG, Won KD, Lhotak S, et al. Anti-GRP78 autoantibodies induce endothelial cell activation and accelerate the development of atherosclerotic lesions. JCI Insight. 2018;3(24):e99363.
Yuan X, Yang X, Cai D, Mao D, Wu J, Zong L, et al. Intranasal immunization with chitosan/pCETP nanoparticles inhibits atherosclerosis in a rabbit model of atherosclerosis. Vaccine. 2008;26(29–30):3727–34.
Samal SK, Qureshi AR, Rahman M, Stenvinkel P, Frostegard J. Different subclasses and isotypes of antibodies against phosphorylcholine in haemodialysis patients: association with mortality. Clin Exp Immunol. 2020;201(1):94–104.
Bruhns P. Properties of mouse and human IgG receptors and their contribution to disease models. Blood. 2012;119(24):5640–9.
Zhao M, Wigren M, Duner P, Kolbus D, Olofsson KE, Bjorkbacka H, et al. FcgammaRIIB inhibits the development of atherosclerosis in low-density lipoprotein receptor-deficient mice. J Immunol. 2010;184(5):2253–60.
Ng HP, Zhu X, Harmon EY, Lennartz MR, Nagarajan S. Reduced atherosclerosis in apoE-inhibitory FcgammaRIIb-deficient mice is associated with increased anti-inflammatory responses by T cells and macrophages. Arterioscler Thromb Vasc Biol. 2015;35(5):1101–12.
Lorenzo C, Delgado P, Busse CE, Sanz-Bravo A, Martos-Folgado I, Bonzon-Kulichenko E, et al. ALDH4A1 is an atherosclerosis auto-antigen targeted by protective antibodies. Nature. 2021;589(7841):287–92.
Kinet JP. The high-affinity IgE receptor (fc epsilon RI): from physiology to pathology. Annu Rev Immunol. 1999;17(1):931–72.
Grimbaldeston MA, Nakae S, Kalesnikoff J, Tsai M, Galli SJ. Mast cell-derived interleukin 10 limits skin pathology in contact dermatitis and chronic irradiation with ultraviolet B. Nat Immunol. 2007;8(10):1095–104.
Bot I, de Jager SC, Zernecke A, Lindstedt KA, van Berkel TJ, Weber C, et al. Perivascular mast cells promote atherogenesis and induce plaque destabilization in apolipoprotein E-deficient mice. Circulation. 2007;115(19):2516–25.
Erdogan O, Gul C, Altun A, Ozbay G. Increased immunoglobulin E response in acute coronary syndromes. Angiology. 2003;54(1):73–9.
Criqui MH, Lee ER, Hamburger RN, Klauber MR, Coughlin SS. IgE and cardiovascular disease. Results from a population-based study. Am J Med. 1987;82(5):964–8.
Korkmaz ME, Oto A, Saraclar Y, Oram E, Oram A, Ugurlu S, et al. Levels of IgE in the serum of patients with coronary arterial disease. Int J Cardiol. 1991;31(2):199–204.
Langer RD, Criqui MH, Feigelson HS, McCann TJ, Hamburger RN. IgE predicts future nonfatal myocardial infarction in men. J Clin Epidemiol. 1996;49(2):203–9.
Kounis NG, Hahalis G. Serum IgE levels in coronary artery disease. Atherosclerosis. 2016;251:498–500.
Guo X, Yuan S, Liu Y, Zeng Y, Xie H, Liu Z, et al. Serum IgE levels are associated with coronary artery disease severity. Atherosclerosis. 2016;251:355–60.
Zhang X, Li J, Luo S, Wang M, Huang Q, Deng Z, et al. IgE contributes to atherosclerosis and obesity by affecting macrophage polarization, macrophage protein network, and foam cell formation. Arterioscler Thromb Vasc Biol. 2020;40(3):597–610.
Tsiantoulas D, Bot I, Ozsvar-Kozma M, Goderle L, Perkmann T, Hartvigsen K, et al. Increased plasma IgE accelerate atherosclerosis in secreted IgM deficiency. Circ Res. 2017;120(1):78–84.
Wilson JM, Nguyen AT, Schuyler AJ, Commins SP, Taylor AM, Platts-Mills TAE, et al. IgE to the mammalian oligosaccharide Galactose-α-1,3-Galactose is associated with increased atheroma volume and plaques with unstable characteristics—brief report. Arterioscler Thromb Vasc Biol. 2018;38(7):1665–9.
Woof JM, Kerr MA. IgA function--variations on a theme. Immunology. 2004;113(2):175–7.
Muscari A, Bozzoli C, Gerratana C, Zaca F, Rovinetti C, Zauli D, et al. Association of serum IgA and C4 with severe atherosclerosis. Atherosclerosis. 1988;74(1–2):179–86.
Rodriguez-Segade S, Camina MF, Carnero A, Lorenzo MJ, Alban A, Quinteiro C, et al. High serum IgA concentrations in patients with diabetes mellitus: agewise distribution and relation to chronic complications. Clin Chem. 1996;42(7):1064–7.
Serrano A, Garcia F, Serrano M, Ramirez E, Alfaro FJ, Lora D, et al. IgA antibodies against beta2 glycoprotein I in hemodialysis patients are an independent risk factor for mortality. Kidney Int. 2012;81(12):1239–44.
Kankaanpaa J, Sampi M, Bloigu R, Wang C, Akhi R, Kesaniemi YA, et al. IgA antibodies to phosphocholine associate with long-term cardiovascular disease risk. Atherosclerosis. 2018;269:294–300.
Frostegård J, Tao W, Georgiades A, Råstam L, Lindblad U, Lindeberg S. Atheroprotective natural anti-phosphorylcholine antibodies of IgM subclass are decreased in Swedish controls as compared to non-westernized individuals from New Guinea. Nutr Metab. 2007;4(1):7.
Frostegård J, Tao W, Råstam L, Lindblad U, Lindeberg S. Antibodies against Phosphorylcholine among new Guineans compared to swedes: an aspect of the hygiene/missing old friends hypothesis. Immunol Investig. 2017;46(1):59–69.
Samal SK, Fröbert O, Kindberg J, Stenvinkel P, Frostegård J. Potential natural immunization against atherosclerosis in hibernating bears. Sci Rep. 2021;11(1):12120.
Kyaw T, Tay C, Hosseini H, Kanellakis P, Gadowski T, MacKay F, et al. Depletion of B2 but not B1a B cells in BAFF receptor-deficient ApoE mice attenuates atherosclerosis by potently ameliorating arterial inflammation. PLoS One. 2012;7(1):e29371.
Sage AP, Tsiantoulas D, Baker L, Harrison J, Masters L, Murphy D, et al. BAFF receptor deficiency reduces the development of atherosclerosis in mice--brief report. Arterioscler Thromb Vasc Biol. 2012;32(7):1573–6.
Nicoletti A, Kaveri S, Caligiuri G, Bariety J, Hansson GK. Immunoglobulin treatment reduces atherosclerosis in apo E knockout mice. J Clin Invest. 1998;102(5):910–8.
Persson L, Boren J, Nicoletti A, Hansson GK, Pekna M. Immunoglobulin treatment reduces atherosclerosis in apolipoprotein E−/− low-density lipoprotein receptor−/− mice via the complement system. Clin Exp Immunol. 2005;142(3):441–5.
Acknowledgements
Figures created with the assistance of Biorender.com.
Funding
Open Access funding provided by the National Institutes of Health (NIH). This work was supported entirely through the Intramural Research Program at the National Institutes of Health (NIH), National Institute on Aging (AG000777).
Author information
Authors and Affiliations
Contributions
The concept was conceived, and the manuscript was written by JAT, MAH, PJG, and RWM. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
No ethical or consent was required for this manuscript.
Consent for publication
All authors have consented to the publication of this manuscript.
Competing interests
The authors have no competing interests to declare.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Taylor, J.A., Hutchinson, M.A., Gearhart, P.J. et al. Antibodies in action: the role of humoral immunity in the fight against atherosclerosis. Immun Ageing 19, 59 (2022). https://doi.org/10.1186/s12979-022-00316-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12979-022-00316-6