
Abstract
Of the members of the primate T cell lymphotropic virus (PTLV) family, only the human T-cell leukemia virus type-1 (HTLV-1) causes disease in humans—as the etiological agent of adult T-cell leukemia/lymphoma (ATLL), HTLV-1-associated myelopathy/tropical spastic paraparesis (HAM/TSP), and other auto-inflammatory disorders. Despite having significant genomic organizational and structural similarities, the closely related human T-cell lymphotropic virus type-2 (HTLV-2) is considered apathogenic and has been linked with benign lymphoproliferation and mild neurological symptoms in certain infected patients. The silencing of proviral gene expression and maintenance of latency are central for the establishment of persistent infections in vivo. The conserved pX sequences of HTLV-1 and HTLV-2 encode several ancillary factors which have been shown to negatively regulate proviral gene expression, while simultaneously activating host cellular proliferative and pro-survival pathways. In particular, the ORF-II proteins, HTLV-1 p30II and HTLV-2 p28II, suppress Tax-dependent transactivation from the viral promoter—whereas p30II also inhibits PU.1-mediated inflammatory-signaling, differentially augments the expression of p53-regulated metabolic/pro-survival genes, and induces lymphoproliferation which could promote mitotic proviral replication. The ubiquitinated form of the HTLV-1 p13II protein localizes to nuclear speckles and interferes with recruitment of the p300 coactivator by the viral transactivator Tax. Further, the antisense-encoded HTLV-1 HBZ and HTLV-2 APH-2 proteins and mRNAs negatively regulate Tax-dependent proviral gene expression and activate inflammatory signaling associated with enhanced T-cell lymphoproliferation. This review will summarize our current understanding of the pX latency-maintenance factors of HTLV-1 and HTLV-2 and discuss how these products may contribute to the differences in pathogenicity between the human PTLVs.
Similar content being viewed by others
Background
The primate T-cell lymphotropic virus (PTLV) family consists of the simian T-cell lymphotropic virus types 1–5 (STLV types 1–5) and human T-cell lymphotropic virus types 1–4 (HTLV types 1–4), which includes the human T-cell leukemia virus type-1 (HTLV-1) and the related human T-cell lymphotropic virus type-2 (HTLV-2, subtypes 2a, 2b, and 2d) [1,2,3,4,5,6,7,8,9,10]. The HTLV-1 is a delta oncoretrovirus that is endemic to tropical equatorial regions, including Southeast Asia (i.e., Japan, China, Taiwan, Malaysia, and the Philippines), Australia and Melanesia, Northern and Central Africa, the Middle East, Central and South America, and certain islands of the Caribbean (in particular, the FWI). Importantly, the HTLV-1 is considered to be an emerging health threat and has been identified in the indigenous populations in Australia and South America. The HTLV-1 infects dendritic cells, monocytes and CD4+ helper T-cells, and oncogenically transforms CD4+ T-cells and causes adult T-cell leukemia/lymphoma (ATLL)—an aggressive and often-fatal hematological malignancy that is poorly responsive to most anticancer treatments, in 3–5% of infected individuals. The HTLV-1 is also etiologically associated with a demyelinating neuro-inflammatory disease, known as HTLV-1-associated myelopathy/tropical spastic paraparesis (HAM/TSP), as well as other autoimmune/inflammatory disorders, including uveitis, rheumatoid arthritis, keratoconjunctivitis, infectious dermatitis, sicca syndrome, and Sjögren’s syndrome. There are currently approximately 10–20 million HTLV-1-infected individuals worldwide; and the virus is transmitted via infected lymphocytes present in blood/blood-products or body fluids through breastfeeding, sexual intercourse, blood transfusions, percutaneous injections, and IV-drug use with contaminated needles. The HTLV-2 was originally isolated from a patient with a rare benign form of hairy T-cell leukemia [9, 10] and, by contrast, this virus is generally considered to be apathogenic. The HTLV-2 infects B-cells and CD4+ and CD8+ T-cells, but preferentially induces oncogenic transformation in CD8+ T-cells in vitro [11,12,13]. Murphy et al. [14] have further suggested that HTLV-2 may be associated with HAM/TSP and other neurological symptoms in certain infected patients. Using a rabbit model of pathogenesis, combined with in vitro T-cell culture/immortalization studies, Kannian et al. [15] demonstrated that the HTLV-1 and HTLV-2 are comparably detected in both the CD4+ and CD8+ T-cell subpopulations as early as 1-week following the initial infection of experimental animals. Their findings further implied that the transformation tropism of these PTLVs (i.e., CD4+ T-cells for HTLV-1 and CD8+ T-cells for HTLV-2) is driven by the clonal expansion and selection of a transformed proviral cellular clone over a latency period of several decades, as occurs in HTLV-1+ ATLL patients [15].
The HTLV-1 and HTLV-2 have complex genomes and encode several regulatory and accessory products within a highly-conserved 3′ nucleotide sequence, known as the pX region (Fig. 1a, b). The HTLV-1 pX sequence encodes the major transactivator protein, Tax-1 (Fig. 2a), the mRNA-splicing regulator, Rex, the open reading frame-I (ORF-I) products: p8I and p12I, and the ORF-II products: p13II and p30II. The HTLV-1 basic leucine zipper (bZIP) protein, HBZ, is encoded by the antisense (i.e., minus) strand of the integrated proviral DNA and the transcriptional initiation of hbz occurs from the 3′ LTR (Fig. 1a). By comparison, the HTLV-2 pX sequence encodes a Tax-2 transactivator (Fig. 2b) and Rex homologue, the accessory products p10 and p11, and the ORF-II p28II protein (a functional synologue of HTLV-1 p30II). The antisense strand of HTLV-2 also encodes a nonconventional bZIP protein, APH-2, which is a synologue of HTLV-1 HBZ. These genes are expressed through alternative mRNA-splicing, and many (i.e., HTLV-1 HBZ, p30II and p13II, and the HTLV-2 APH-2 and p28II proteins; Fig. 1a, b) negatively regulate Tax-dependent transcriptional activity and maintain the latent silencing of proviral gene expression to promote viral persistence in vivo [16,17,18,19,20,21]. Indeed, numerous studies using animal models of pathogenesis have demonstrated that HTLV-1 HBZ, p30II, and p13II and the HTLV-2 p28II proteins are essential for immune-evasion, viral persistence and the maintenance of high proviral titers in vivo [22,23,24,25,26]. This review will discuss what is known about the expression of these pX latency-maintenance genes in HTLV-1 and HTLV-2-infected cell-lines, asymptomatic carriers, and ATLL and HAM/TSP clinical isolates, and how these factors could contribute to retroviral pathogenesis and disease progression.

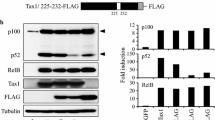
Diagrams of the HTLV-1 and HTLV-2 proviral genomes and their products. a The coding regions of the HTLV-1 genome are represented by filled boxes. The antisense HBZ-SP1 product is shown at the bottom with an arrow indicating its transcriptional initiation site from the 3′ LTR. b The HTLV-2 genome and its products. The coding region for the antisense-encoded APH-2 protein is indicated. The conserved pX nucleotide sequences are indicated in a and b. The pX-encoded latency factors discussed in this review are represented by colored boxes. HBZ-SP1 HTLV-1 basic domain/leucine zipper-spliced-1 isoform, APH-2 antisense protein of HTLV-2, LTR long terminal repeat

Functional domains and predicted structures of the HTLV-1 Tax-1 and HTLV-2B Tax-2 transactivator proteins. The predicted structures were generated using the I-TASSER computer algorithm (https://zhanglab.ccmb.med.umich.edu) and modeled using UCSF-Chimera. a Diagram of the HTLV-1 Tax protein and its functional domains. NLS nuclear localization signal, NES nuclear export sequence, ZF zinc finger motif, LZ leucine zipper region. The sites of the M22 (dimerization), G148 V (NF-κB transactivation), and M47 (activation domain) mutations are indicated. b The HTLV-2 Tax-2B protein and its conserved functional domains are shown. The unique C-terminal aa residues 331–356 are not present within the HTLV-1 Tax protein. NLD nuclear localization determinant
The antisense-encoded proteins, HTLV-1 HBZ and HTLV-2 APH-2
The antisense strand of HTLV-1 encodes unspliced and alternatively-spliced transcripts, hbz, hbz-sp1 and hbz-sp2, which code for different isoforms of a bZIP transcription factor: HBZ, HBZ-SP1 (spliced-1), and HBZ-SP2 (spliced-2), that negatively regulate proviral gene expression and modulate host lymphoproliferative signaling [27,28,29,30,31,32,33]. These transcripts are initiated from the 3′ LTR; and the hbz-sp1 mRNA which codes for the most abundant isoform (HBZ-SP1) present in ATLL cells (Fig. 3a), includes exon 1 (nts 1–367) spliced to an acceptor site at position 1767 on the minus strand. The alternatively-spliced hbz-sp2 mRNA which codes for the HBZ-SP2 protein has its first exon (nts 1–227) spliced to an acceptor site at position 1767 on the antisense RNA strand [28]. Hbz is the only viral gene detectable at every stage of infection in chronically-infected cell-lines and PBMCs derived from HTLV-1+ asymptomatic carriers, HAM/TSP, and ATLL patients. Despite the fact that the 5′ LTR is frequently inactivated in ATLL clinical isolates as a result of DNA hypermethylation or proviral deletions, the 3′ LTR and hbz mRNA and protein expression are usually intact, alluding to their pivotal roles in viral pathogenesis [34]. The HBZ protein is weakly immunogenic and not efficiently translated in ATLL lymphocytes. In 2014, Rowan et al. [35] demonstrated that autologous cytotoxic T-lymphocytes (CTLs), specific for an HBZ26–34 peptide epitope, effectively selected against HTLV-1-infected CD4+ T-cells which expressed a HLA-A*0201 major histocompatibility class I molecule that binds to HBZ-SP1 with high-affinity. These findings suggest a CD8+ cell-mediated immune-response could select for HTLV-1 proviral clones with reduced, steady-state levels of the antisense hbz products and intermittent tax expression in vivo.

Functional domains and predicted structures of the antisense-encoded HTLV-1 HBZ and HTLV-2 APH-2 proteins. The predicted structures were generated using the I-TASSER algorithm and modeled using UCSF-Chimera. a Diagram and structure of the HTLV-1 HBZ-SP1 (spliced-1) isoform. The amphipathic helical motifs (1 and 2) which bind the KIX domains of p300/CBP are indicated. AD, activation domain; NES, nuclear export signal; NLS, nuclear localization signal. b The core IXXL and terminal LXXL modulatory sequences of the HTLV-2 APH-2 protein are indicated in the diagram. nc bZIP nonconventional basic domain/leucine zipper region, NFAR NF90/NF110-binding Nuclear factors associated with double-stranded RNA proteins NF90/NF110-binding region, PLMnb-localization PML nuclear body-localization/APH-2 stabilization domain. The basic regions in HBZ and APH-2 are represented by blue boxes with +++
HBZ nuclear interactions and repression of Tax-dependent transcription
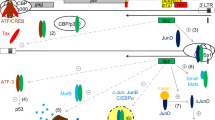
The antisense bZIP protein of HTLV-1, HBZ, antagonizes Tax-dependent viral gene expression through its nuclear interactions with the transcriptional coactivators p300/CBP and chromatin-remodeling components. The HBZ protein localizes in nuclear speckles and contains three nuclear localization signals, with NLS1 (aa 87–92) and NLS2 (aa 116–120) located within the two upstream basic domains and NLS3 (aa 137–163) in the DNA-binding region (Fig. 3a) [36, 37]. Mukai and Ohshima [38] have also shown that HBZ contains a nuclear export signal (NES), spanning amino acid (aa) residues 21–33 (Fig. 3a), and is shuttled to the cytoplasm in a CRM1-dependent manner where it binds and suppresses the growth-arrest and DNA-damage gene 34 (GADD34) to activate the mammalian target of rapamycin (mTOR) signaling pathway which could promote the growth and proliferation of HTLV-1-infected cells. Using LC–MS/MS analyses, Dissinger et al. [39] demonstrated that HBZ is posttranslationally modified by phosphorylation on serine residue S49, acetylation on lysines K66 and K155, and methylation on residues K35, K37, K181, and K186; however, these modifications did not appear to influence the stability or biological functions of the protein. HBZ negatively regulates Tax-dependent LTR transactivation and proviral gene expression by interacting with the bZIP domains of CREB/ATF-family transcription factors and inhibits their binding to the 21-bp-repeat Tax-responsive elements (TREs) of the HTLV-1 promoter [27, 30]. The N-terminal region of HBZ binds to the KIX domain of the transcriptional coactivators, p300/CREB-binding protein (p300/CBP), and interferes with the recruitment of p300/CBP by the viral transactivator to Tax/CREB/21-bp-repeat complexes on the 5′ LTR [40]. HBZ also inhibits the catalytic acetyltransferase activity of p300/CBP and prevented the acetylation of histone and non-histone targets, including the p53 tumor suppressor and NF-κB p65RelA subunit [41, 42]. Moreover, Alasiri et al. [43] have demonstrated that HBZ interacts with the brahma-related gene 1 (BRG1) and BRG/hBRM-associated factor 200 (BAF200) components of the SWI/SNF (PBAF) chromatin-remodeling complexes and modulates their recruitment to Tax-containing 5′ LTR complexes to repress proviral gene expression. It is possible feedback interactions may coordinately regulate the expression of the proviral sense (plus-strand) and antisense (minus-strand) products, as Tax has been shown to transactivate the 3′LTR to drive hbz expression. The HBZ protein suppresses AP-1-dependent transcription through interactions with the bZIP factors, c-Jun and Jun B [44]. By contrast, HBZ activates JunD and stimulates the human telomerase reverse transcriptase (hTERT) gene promoter [45]. HBZ also preferentially induces the expression of the ∆JunD isoform through the repression of a ribosomal small subunit protein, RPS25, which promotes translational leaky-scanning past an upstream ORF and initiation on a downstream ORF [46]. This study further demonstrated that HBZ cooperates with the pro-proliferative ∆JunD isoform and enhanced its oncogenic colony-forming potential in vitro [46]. HBZ could also promote proviral latency through its interactions with the NF-κB p65RelA subunit which inhibits p65RelA DNA-binding and NF-κB transactivation [47]. Moreover, in 2011, Zhi et al. [48] reported that HBZ counters the cellular senescence and cytotoxicity associated with Tax-induced NF-κB hyper-activation in stable HeLa-G/FLAG-HBZ cell-lines that were transduced with adenoviral Tax expression vectors.
Dynamic regulation of HTLV-1 gene expression by HBZ and Tax
The regulation of HTLV-1 proviral gene expression occurs through dynamic coordinate interactions between the sense and antisense pX-encoded products. In 2017, Billman et al. [49] used RNA-fluorescence in situ hybridization (RNA-FISH) to quantify the single-cell expression of tax and hbz transcripts within individual T-cell clones established from HTLV-1-infected patients. These findings demonstrated that hbz is not constantly expressed in every cell, but, rather, the tax and hbz mRNAs are produced in intermittent bursts—with hbz exhibiting a predominantly intranuclear localization and corresponding to cells in either the S or G2/M-phases of the cell-cycle [49]. Mahgoub et al. [50] further demonstrated that the viral transactivator Tax is persistently expressed at low levels in the HTLV-1-infected cell-line MT-1 and switches between ‘on’ and ‘off’ states within individual cells. Tax expression was important to protect these cells against apoptosis and delayed their transition into the G2/M-phase; and shRNA-knockdown of tax resulted in significant cellular cytotoxicity, suggesting that low levels of Tax are needed to promote the continuous survival of HTLV-1-infected leukemic cells in vivo. The hbz mRNA has been shown to indirectly increase levels of the Tax protein by inhibiting the expression of pX-orfII-p30II transcripts [51]. Interestingly, Rushing et al. [52] reported that HBZ causes genotoxic stress, resulting in the accumulation of double-stranded DNA breaks, through its interactions with the Ku70/Ku80 subunits of DNA-PK and inhibition of the nonhomologous end-joining (NHEJ) repair pathway. It is likely a balance exists between HTLV-1 pX regulatory factors which may have ancillary roles in viral pathogenesis. Indeed, Hutchison et al. [53] have shown that the ORF-II p30II protein cooperates with Tax and HBZ and countered their cytotoxicity due to oxidative stress, and enhanced the oncogenic potential of these viral proteins in vitro.
Induction of T-cell lymphoproliferation by HBZ
The HBZ protein induces T-cell lymphoproliferation and enhances cellular survival through several different mechanisms. HBZ inhibits the classical Wnt signaling pathway through binding to lymphoid enhancer-binding factor 1 (LEF1), while also activating the noncanonical Wnt5a signaling pathway which could promote ATLL cell proliferation [54]. In 2013, Zhao et al. [55] demonstrated that HBZ interacts with the bZIP factor CCAAT/enhancer binding protein-alpha (C/EBPα) and inhibits its negative growth-suppressive functions in transfected Jurkat and 293T cells, in a Smad3-dependent manner. The HBZ protein also activates E2F-1-dependent transcription, associated with G1/S cell-cycle progression and apoptosis, through interactions with retinoblastoma protein (Rb)/E2F-1 complexes and displacement of the histone deacetylase, HDAC3 [56]. HBZ induces enhanced lymphoproliferation mediated by T-cell receptor (TCR) signaling, as a result of interfering with the recruitment of SHP-1/2 tyrosine phosphatases to inhibitory coreceptors, PD-1 and TIGIT, on the surfaces of HTLV-1-infected CD4+ T-cells [57]. Moreover, Forlani et al. [58] have shown that the HBZ protein exclusively localizes in the cytoplasm of cells isolated from HTLV-1+ asymptomatic carriers and HAM/TSP patients. The expression of hbz correlates with disease severity in HAM/TSP patients and could also potentially serve as a surrogate marker for therapy-responsiveness [59].
Latency-maintenance and in vivo functions of HBZ
Although hbz is dispensable for the infection and immortalization of primary T-cells by HTLV-1 in vitro, it is required for viral persistence and the maintenance of a high proviral titer in vivo [22]. By inoculating rabbits with irradiated 729 B-cell-lines that contained HTLV-1 ACH proviral clones, expressing either wild-type HBZ or deletion mutants of HBZ (i.e., HTLV-1HBZ∆LZ or HTLV-1∆HBZ), Arnold et al. [22] demonstrated the antibody response against HTLV-1 antigens (p19Gag) and viral persistence in vivo as measured by quantitative RT-PCR is dependent upon hbz gene expression. A study of the kinetics of viral gene expression demonstrated the levels of tax/rex, gag/pol, and env mRNAs decreased and inversely correlated with higher levels of the hbz transcripts in infected rabbits [60]. Rende et al. [61] have reported that 90% of the hbz mRNAs are compartmentalized and sequestered in the nuclei of cultured HTLV-1-infected T-cell clones established from ATLL and HAM/TSP patients. Valeri et al. [23] have further shown that hbz is required for viral persistence in rabbits and Rhesus macaques inoculated with lethally-irradiated 729 B-cell-lines expressing the wild-type HTLV-1 provirus or mutants ablated for hbz expression (or other ORF-I and ORF-II products), with genetic reversion to the wild-type sequence observed in 3 out of 4 seropositive macaques inoculated with the HBZ-knockout mutant. The hbz mRNA and HBZ protein differentially promote T-cell activation, lymphoproliferation and cell survival [33, 34]. The HBZ protein induces cellular apoptosis, whereas the hbz mRNA protects against programmed cell-death and induces the expression of cell-cycle regulatory and anti-apoptotic genes (e.g., survivin) in transduced primary murine T-cells. Both the HBZ protein and mRNA were able to induce T-cell lymphoproliferation and aberrant S-phase entry [33]. Small-interfering or short hairpin RNAs that inhibit hbz expression blocked the in vitro proliferation of HTLV-1-transformed T-cell-lines and ATLL cells [31, 34]. Arnold et al. [31] also demonstrated that HTLV-1-transformed SLB1 lymphoma cells, transduced with shRNA lentiviral vectors targeted against hbz, exhibit reduced cell proliferation, tumorigenesis, and secondary tissue infiltration in engrafted NOD/scidγchain−/− animals. The hbz mRNA posttranscriptionally increased the expression of oncogenic microRNAs, miR17 and miR21, in CD4+ T-cell clones established from HTLV-1-infected HAM/TSP patients [62]. Importantly, the expression of HBZ in the CD4+ T-cells of hbz-transgenic mice resulted in the formation of skin and lung lesions associated with systemic inflammation and lymphocyte infiltration [32, 63]. Many of these animals also developed T-cell lymphomas following a prolonged latency. The HBZ protein interacts with FoxP3/NFAT transcription complexes and inhibited FoxP3-dependent immunosuppressive-signaling in CD4+ Treg cells which resulted in increased inflammation [32]. In 2011, Zhao et al. [64] demonstrated that HBZ forms ternary complexes with Smad3 and the p300 transcriptional coactivator and enhances transforming growth factor-beta (TGF-β) signaling, associated with the increased expression of FoxP3 and conversion of HTLV-1-infected CD4+ cells into Treg cells. Esser et al. [65] have further shown that granzyme B promoter-hbz transgenic mice developed CD45+ mixed-cellular tumors, with enlargement of the spleen, elevated white blood cell counts, and osteolytic bone metastases associated with the increased expression of inflammatory cytokines and factors involved in hypercalcemia, including RANKL, PTHrP and DKK1.
The HTLV-2 counterpart of HBZ, APH-2
The antisense protein of HTLV-2, or APH-2, is a functional synologue of the HTLV-1 HBZ factor and is generated through alternative mRNA-splicing which uses a donor site at position 8544 and a splice acceptor site at position 7173 on the antisense strand of the pH6neo molecular clone of HTLV-2 [66]. APH-2 is a nuclear protein comprised of 183 aa residues and contains two core modulatory aliphatic sequences: IXXLL (aa 64–68) and LXXLL (aa 179–183), and a basic region located upstream from a noncanonical bZIP motif (Fig. 3b) [66]. In 2009, Halin et al. [66] demonstrated that APH-2 interacts with the CREB transcription factor and inhibits Tax-2-mediated transactivation from the HTLV-2 LTR in luciferase reporter assays and represses proviral gene expression (p19Gag) by the pH6neo HTLV-2 clone, suggesting APH-2 antagonizes Tax-2 functions and promotes viral latency in vivo. The repression of Tax-2-dependent transactivation and binding to CREB were dependent upon the C-terminal modulatory sequence, LXXLL, of APH-2 [67]. Unlike HBZ, however, the APH-2 protein does not interact with the p300/CBP transcriptional coactivators [66]. The aph-2 mRNA is constitutively expressed in chronically-infected cell-lines and PBMCs derived from HTLV-2-infected carriers [66]. Although the aph-2 mRNA levels coincided with the proviral loads in HTLV-2-infected patients, neither the aph-2 mRNA nor APH-2 protein were able to induce lymphoproliferation in vitro [68]. Bender et al. [69] have reported that the majority of aph-2 transcripts are sequestered in the nuclei of HTLV-2-infected cells, similar to the subcellular compartmentalization of hbz mRNAs. In 2012, Yin et al. [67] demonstrated that APH-2 is dispensable for viral infectivity and the immortalization of primary T-cells in vitro; and rabbits inoculated with an irradiated 729 B-cell-line that contained an HTLV-2 proviral deletion mutant of APH-2 (∆Aph-2) exhibited increased antibody titers and proviral loads, as compared to animals inoculated with 729/wild-type HTLV-2 clones. These findings suggest HBZ and APH-2 are functionally divergent for the maintenance of viral persistence in vivo. Both HBZ and APH-2 inhibit NF-κB p65RelA-dependent transcriptional activation; however, by contrast, APH-2 does not augment TGF-β signaling [70]. The HBZ protein was discovered to be significantly more stable than APH-2 in half-life assays using cycloheximide-treated cells [70]. While both HBZ and APH-2 were shown to interact with the E3 ubiquitin ligase, UBR5, only HBZ was stabilized by knocking down UBR5 expression [71]. Dubuisson et al. [72] have further demonstrated that the APH-2 protein is shuttled to PML nuclear bodies, in a manner dependent upon APH-2-SUMOylation, where it is degraded by the proteasome. The noncanonical bZIP domain of APH-2 interacts with c-Jun and JunB; and APH-2 activates AP-1-dependent transcription [73]. Marban et al. [73] have also demonstrated that the C-terminal region of APH-2 binds to Tax-2 and inhibits Tax-dependent Ap-1 transactivation in cotransfected 293T cells. The central domain of HBZ and aa residues 1–93 of APH-2 interact with the nuclear factors associated with double-stranded RNA (NFAR) proteins, NF90 and NF110 (Fig. 3a, b), which are involved in innate immunity as targets of PKR-activation and, in addition, enhance Tax-dependent transactivation from the viral LTR and transcriptionally activate the survivin gene associated with the cellular anti-apoptotic response [74]. The siRNA-knockdown of NFAR did not significantly affect the ability of APH-2 to repress Tax-2-dependent LTR transactivation; and APH-2 inhibited survivin promoter transactivation by the NFAR NF110a [74]. These results suggest that HBZ and APH-2 interact with the NFARs to modulate viral gene expression and latency, as well as host innate-immunity and anti-apoptotic signals.
The ORF-II latency-maintenance factors, HTLV-1 p30II and HTLV-2 p28II
The ORF-II proteins which suppress the expression of proviral antigens represent an understudied area in the HTLV field, yet—in the light of recent evidence, there is reason to believe these factors may have key ancillary functions that could provide clues into the different pathogenic properties of the human PTLVs. The conserved pX regions of HTLV-1 and HTLV-2 encode the ORF-II products: p30II and p28II, respectively, through alternative mRNA-splicing, which negatively regulate the Tax-dependent expression of viral antigens [16,17,18,19,20, 75] and are required for the maintenance of proviral latency and persistence in vivo [23, 24, 26]. In this regard, they are considered functional synologues [17]. However, mounting evidence indicates that p30II and p28II differ significantly in their capacity to modulate host signaling pathways and cooperate with other viral and cellular oncoproteins and, therefore, these factors are likely to have divergent roles in pathogenesis [18, 19, 53, 76,77,78,79,80,81,82,83,84,85,86]. This section will highlight the major similarities and differences between these ORF-II products, and discuss how they may contribute to mitotic proviral replication, T-cell immortalization, and the establishment and progression of neoplastic disease.
Expression of the ORF-II products in HTLV-infected cell-lines, asymptomatic carriers, and ATLL and HAM/TSP patients
The HTLV-1 p30II protein (also known as Tax-ORFII, or Tof-II) is comprised of 241 aa residues, contains arginine and serine/threonine-rich regions, and shares aa sequence similarities with the Oct-1/Pit-1/POU-family of homeodomain transcription factors (Fig. 4a) [87,88,89]. The C-terminus of p30II (aa residues 155–241) also corresponds with the reading frame for the p13II protein sequence (Fig. 4b) [88, 90,91,92]. The related HTLV-2 synologue, p28II, is comprised of 216 aa residues; and a peptide sequence (aa residues 1–49) within its N-terminus shares 78% sequence homology with residues 193–241 of HTLV-1 p30II [17, 93]. However, there are no other sequence or structural similarities between these proteins outside of this region (Fig. 4c). Although p30II has been reported to contain two intrinsically disordered sequences, spanning aa residues 75–155 and 197–241 [94], the p30II protein is predicted to contain at least five alpha-helices which may contribute to its unique biological functions and molecular interactions (Fig. 4a). The HTLV-2 p28II protein is predicted to be largely unstructured and exists as random coils, and little is known about the specific regions of p28II that mediate its interactions with cellular factors (Fig. 4c). An NCBI-BLAST analysis identified a region (aa residues 82–105) that shares 58% similarity with aa residues 741–764 of the human chondroitin sulfate proteoglycan core protein 1, although the functional relevance of this sequence, if any, remains to be determined. The HTLV-1 pX-orfII mRNA which codes for p30II is generated through alternative-splicing that includes exon 1 (nts 1–119) with exon 2 (nts 4641–4831) spliced to an acceptor site at position 6478 of the downstream pX sequence that is also used for the bicistronic pX-tax/rex mRNA [90, 95]. The alternatively-spliced pX mRNAs, including pX-orfII-p30II, have been detected by RT-PCR in cultured HTLV-1-infected T-cell-lines and primary uncultured ATLL clinical isolates, as well as in cells from asymptomatic HTLV-1-infected carriers [87, 90, 96]. In 2003, Princler et al. [95] demonstrated that the pX-orfII mRNA is expressed in chronically-infected T-cell-lines; and Cereseto et al. [97] have detected the pX-orfII mRNA in HTLV-1-transformed T-cell-lines, PBMCs from HTLV-1-infected carriers, and cells isolated from HAM/TSP patients using a non-PCR-based ribonuclease-protection assay. The HTLV-2 alternatively-spliced pX-orfII-p28II mRNA, which is generated by splicing exon 1 (nts 316–449) to an acceptor site at position 6944 of the pX sequence, has been detected in the chronically-infected MoT cell-line by RT-PCR analysis [93]. Further, Pique et al. [91] have isolated CD8+ cytotoxic T-lymphocytes (CTLs) that specifically target the ORF-II p30II and p13II peptides from HTLV-1-infected carriers, HAM/TSP and ATLL patients, suggesting these proteins are chronically expressed and could contribute to the establishment of persistent infections in vivo as well as viral pathogenesis.

Functional domains and predicted structures of the HTLV-1 p30II, p13II, and HTLV-2 p28II proteins. The predicted structures were generated using the I-TASSER algorithm and modeled using UCSF-Chimera. a Diagram and structure of HTLV-1 p30II. TAD, transcriptional activation domain; NLS, nuclear localization signal; NoRS, nucleolar retention sequence. b The p13II aa sequence overlaps with the C-terminal region of HTLV-1 p30II (aa 155–241) and is colored green in the p30II structure (lower panel, a). The SH3-binding domain (aa 75–87) of HTLV-1 p13II which contains a PXXP motif is colored orange in the diagram and in the modeled structure (b). MTS, mitochondrial targeting sequence; Hel, helical region; TM, transmembrane domain; H, flexible hinge region; Beta, predicted beta-sheet secondary structure. c The amino-terminus of the HTLV-2 p28II protein (aa 1–49) shares 78% sequence homology with aa residues 193–241 of HTLV-1 p30II. A region of 58% similarity with a peptide sequence of the Chondroitin sulfate proteoglycan core protein 1 (aa 741–764) is also indicated
In vivo requirement for the ORF-II latency-maintenance factors of HTLV-1 and HTLV-2
The functional roles of the ORF-II p30II and p28II proteins can be divided into: (a) the establishment of early-stage latency and viral persistence in vivo, and (b) their interactions with host proliferative-signaling pathways and cooperation with other viral (i.e., Tax and HBZ) and cellular factors to drive mitotic proviral replication. The HTLV-1 p30II protein contains three putative nuclear localization signals (NLS1/2/3) as well as a nucleolar retention sequence (NoRS; Fig. 4a) and is primarily nuclear and/or nucleolar in its localization, although it is also frequently detectable in the cytoplasm [16, 20, 53, 84, 86, 88, 98]. The HTLV-2 p28II protein is predominantly nuclear in its subcellular distribution [16, 98]. Using an established rabbit model of pathogenesis, Bartoe et al. [24] demonstrated that pX-orfII products are essential for the maintenance of a high proviral titer in experimental animals inoculated with PBMCs that contained either a wild-type molecular clone of HTLV-1 ACH.1, or an ACH.30II/13II.1 mutant defective for the expression of ORF-II proteins. While the pX-orfII products are widely thought to be dispensable for viral infectivity and the immortalization of primary T-cells in vitro [99, 100], the data presented in Table 1 of Robek et al. [100] indicate that the ACH.p30II mutant exhibited a 50% reduced capacity to immortalize T-cells in in vitro co-culture assays, suggesting p30II may contribute to HTLV-1-induced leukemogenesis. Indeed, Romeo et al. [86] have demonstrated that lentiviral p30II induced the long-term proliferation beyond crisis (> 4 months) of transduced human PBMCs selected on blasticidin and cultured in the presence of recombinant interleukin-2 (IL-2), although these transiently-amplified clones were observed to undergo a second crisis at around 7 months and it is presumed that other viral and/or cellular factors are necessary for T-cell immortalization [86]. By contrast, a p28II-defective HTLV-2 mutant provirus, derived from the pH6neo molecular clone, exhibited viral infectivity and immortalized T-cells in vitro similar to the wild-type virus, but failed to promote proviral replication and T-cell survival in vivo in a rabbit model of HTLV-2 pathogenesis [26]. A study of the kinetics of HTLV-1 gene expression in cultured PBMCs isolated from ATLL and HAM/TSP patients, using splice-site specific quantitative RT-PCR analysis, revealed two-phase kinetics in the ATLL cells where the levels of pX-tax/rex mRNA were inversely correlated with the expression of the other pX-orfII, pX-orfI, and hbz-sp1 transcripts [61]. A similar study which used 293T cells transiently transfected with the HTLV-1 ACHneo proviral clone failed to detect significant expression of the pX-orfII mRNA in vitro [60]. By contrast, in 2012, Bender et al. [69] investigated the kinetics of HTLV-2 gene expression and reported the pX-tax/rex and pX-orfII-p28II transcripts were detected at comparable levels in the chronically-infected cell lines, MoT and BJAB-Gu, and in PBMCs isolated from 2 out of 3 HTLV-2-infected patients. The HTLV-1 p30II protein has been shown to promote aberrant S-phase entry and lymphoproliferation, and induces the expression of T-cell activation and pro-survival genes [53, 77, 80, 84,85,86, 101]. In 2011, Anupam et al. [80] demonstrated that lentiviral p30II enhanced the survival of transduced 293T and Jurkat T-cells, associated with p30II-interactions with the ataxia telangiectasia mutated (ATM) and REGγ proteins. A follow-up study by Doueiri et al. [82] demonstrated that an S-tagged p30II protein interacts with the nuclear 20S proteasome activator REGγ. This study combined biochemical affinity purification with mass spectrometry analysis and identified several unique binding partners and three common interacting factors (i.e., protein arginine methyltransferase 5, hnRNP K, and large ribosomal subunit protein L8) that associate with the HTLV-1 p30II and HTLV-2 p28II proteins [82].
Latency and the repression of Tax-dependent proviral gene expression by p30II and p28II
The HTLV-1 p30II and HTLV-2 p28II proteins negatively regulate proviral gene expression and function as latency-maintenance factors which could help HTLV-infected cells evade host immune-surveillance pathways for the establishment of persistent infections in vivo. In 2000, Zhang et al. [19] used Gal4-p30II fusion constructs and luciferase reporter assays and demonstrated that p30II contains a functional transcriptional activation domain (Fig. 4a), and that p30II differentially induces CREB-dependent transcription from the 21-bp repeat TREs in the HTLV-1 promoter, yet represses CREB-dependent transactivation from cellular CREB-responsive elements (CREs). The p30II protein binds to the KIX domain of the transcriptional coactivators, p300/CBP, and competes against the viral transactivator Tax for the recruitment of p300/CBP to CREB/21-bp-repeat TRE complexes on the HTLV-1 promoter and represses the expression of viral antigens [18]. The transcriptional repression of the HTLV-1 5′ LTR by p30II was dependent upon a single lysine residue at position K106 within the p30II protein (Fig. 4a), and required p300-binding and the catalytic acetyltransferase domain of the p300 coactivator [20]. Interestingly, Datta et al. [76] have shown that p30II interacts with the Ets domain of the PU.1 transcription factor, inhibits its DNA-binding, and represses the PU.1-dependent expression and activation of Toll-like receptor-4 (TLR4) in transfected cells. The inhibitory effect of p30II upon PU.1-dependent transcriptional activation was countered through overexpression of the p300 coactivator. p30II also inhibited the pro-inflammatory cytokines, MCP-1, TNF-α, and IL-8, and increased the release of the anti-inflammatory factor IL-10 following the stimulation of TLR4 in THP-1 monocytic cells with lipopolysaccharide, suggesting p30II might interfere with adaptive immunological signaling in the early stages of viral pathogenesis [76]. These findings were supported in a study by Fenizia et al. [83] which demonstrated that p30II inhibits the expression of interferon-responsive genes by interfering with the PU.1-dependent expression of TLR4 in THP-1 monocytes and dendritic cells. The inhibition of interferon-responsive signaling and innate immunity by p30II could contribute to the early-stage establishment of infection and viral persistence in vivo. Valeri et al. [23] have also shown that p30II is required for the productive infection of human dendritic cells by HTLV-1, and further demonstrated that Rhesus macaques inoculated with an irradiated 729 B-cell-line, containing an ACH.p30-knockout mutant ablated for p30II expression, either failed to seroconvert or exhibited genetic reversion to the wild-type ACH sequence.
In 2004, Nicot et al. [16] reported that p30II posttranscriptionally inhibits the nuclear export of the doubly-spliced bicistronic pX-tax/rex mRNA and negatively regulates HTLV-1 gene expression. For these studies, 293T cells were cotransfected with an HTLV-1 molecular clone, p-BST, and an expression construct for p30II and the inhibition of viral gene expression was demonstrated by Anti-p19Gag ELISAs and the nuclear accumulation of pX-tax/rex transcripts was detected with RT-PCR. This study further demonstrated that p30II was associated with the splice-junction of the pX-tax/rex mRNA using biotinylated RNA-precipitation experiments, and that lentiviral p30II negatively regulated proviral gene expression in transduced HTLV-1-transformed T-cell-lines (i.e., MT-2, C91PL, and HUT-102) [16]. Ghorbel et al. [98] have shown the nuclear/nucleolar retention of p30II is dependent upon its interactions with large ribosomal subunit L18a within cell nucleoli. Younis et al. [17] subsequently demonstrated that both p30II and the synologue p28II posttranscriptionally inhibit proviral gene expression from molecular clones of HTLV-1 or HTLV-2. The HTLV-2 p28II (AU1-tagged) protein was immunoprecipitated bound to pX-tax/rex mRNA complexes in vivo; and p28II resulted in increased nuclear sequestration of the pX-tax/rex mRNA as detected by RT-PCR [17]. Interestingly, both p30II and p28II are recruited to their respective RNA targets through co-transcriptional interactions with the retroviral transactivator protein Tax on the promoters of the HTLV-1 and HTLV-2 proviruses [102]. In 2006, using chromatin-immunoprecipitation analyses, Younis et al. [102] demonstrated that p30II and p28II interact with Tax-1 or Tax-2 and are recruited and travel with RNA Polymerase II-containing transcriptional elongation complexes until they reach their nascent RNA targets. Using confocal microscopy and biochemical glutathione-S-transferase (GST)-pull-downs, Baydoun et al. [103] have shown that p30II interacts with the retroviral splicing regulator, Rex, and sequesters Rex/CRM1 complexes in nucleoli in cotransfected COS and 293T cells. However, this somewhat contradicts findings by Sinha-Datta et al. [104] which demonstrated that aa residues 131–164 of p30II interact with Rex complexed with the Rex-responsive RNA element (RxRE) of tax/rex mRNA transcripts, but this interaction did not interfere with the shuttling of Rex/CRM1/mRNA complexes out of the nucleus. The regulation of HTLV-1 gene expression and latency by Tax, HBZ, and p30II (or HTLV-2 gene expression by Tax-2, Aph-2, and p28II) is orchestrated by coordinate and dynamic molecular interactions at the transcriptional and posttranscriptional levels. Choudhary and Ratner [51] have also shown that the hbz-sp1 anti-sense mRNA indirectly increases expression of the Tax transactivator from a molecular clone of HTLV-1 by reducing the expression of the pX-orfII-p30II mRNA, which lends additional complexity to the control of proviral gene expression by pX products.
Activation of cellular pro-survival and proliferative signals by HTLV-1 p30II, but not p28II
Viruses that induce latent infections replicate their genomes by inducing mitotic host-cell division and proliferation. The p30II protein of HTLV-1 helps to accomplish this by activating cellular oncogenic and pro-survival pathways through its molecular interactions with the TIP60 acetyltransferase—a transcriptional cofactor for both c-Myc and p53. In 2005, Awasthi et al. [84] demonstrated that p30II cooperates with the c-Myc oncoprotein, induces aberrant lymphoproliferation in Molt-4 T-cells, and enhances the oncogenic potential of c-Myc in cellular transformation/foci-formation assays using human fibroblasts. Mechanistically, aa residues 99–154 of p30II were shown to interact with the MYST-family acetyltransferase TIP60, using biochemical GST-pull-downs and co-immunoprecipitation experiments (Fig. 4a); and the p30II protein was present in c-Myc/TIP60-containing NuA4 transcriptional activation complexes (together with the scaffold subunit TRRAP/p434, hGCN5, and the ATP-dependent DNA-helicases TIP48/TIP49 [105]) recruited to E-box enhancer elements within the cyclin D2 gene promoter [84]. p30II transcriptionally activated the cyclin D2 promoter as well as a minimal M4-tk-luciferase reporter construct that contains four tandem E-box elements [84, 85]. Using dominant-negative mutants of TIP60 and TRRAP, it was found that the cooperation and oncogenic transformation by p30II/c-Myc were dependent upon the TRRAP transcriptional cofactor and the catalytic acetyltransferase domain of TIP60 [84]. At least three studies have examined the global gene expression profiles of cells containing HTLV-1 p30II [78, 84, 101]. In 2005, Awasthi et al. [84] performed Affymetrix U133-Plus microarray gene expression analyses which included a dominant-negative acetyltransferase-defective mutant of TIP60, and identified 250 target genes that were transcriptionally activated or repressed by p30II in a TIP60-dependent or -independent manner. In a follow-up study, Romeo et al. [85] used the HO15.19 myc-null rat fibroblast cell-line, together with various acetylation-defective Lys → Arg substitution mutants of c-Myc, and demonstrated that oncogenic cellular transformation by p30II/c-Myc required acetylation of the c-Myc oncoprotein and that p30II co-immunoprecipitated with acetylated c-Myc [85]. The oncogenic foci-formation by p30II/c-Myc was dependent upon the acetyltransferase domains of TIP60, p300, and PCAF; however, this study did not determine which acetyltransferase(s) was primarily responsible for acetylation of the c-Myc oncoprotein [85]. The p53 tumor suppressor is a downstream target of c-Myc; and acute and lymphoma-stage ATLL clinical isolates frequently contain c-Myc overexpression and elevated levels of wild-type p53 [106,107,108]. Intriguingly, the p53 gene is rarely mutated in HTLV-1-infected ATLL patient samples [109,110,111,112], which leads to speculation that p53-dependent gene expression may somehow contribute to retroviral pathogenesis. Several studies have demonstrated that TIP60-mediated acetylation of the p53 protein on lysine residue K120 differentially regulates the induction of p53-dependent pro-apoptotic versus pro-survival/growth-regulatory genes [113,114,115]. In 2018, Romeo et al. [86] and Hutchison et al. [53] demonstrated that the HTLV-1 p30II protein induces p53 and inhibits TIP60-mediated K120-acetylation of p53, and transcriptionally activates the expression of p53-regulated pro-survival genes. Most notably, lentiviral p30II induced the expression and mitochondrial targeting of the TP53-induced glycolysis and apoptosis regulator (TIGAR [53, 86])—a fructose-2,6-bisphosphatase which prevents the intracellular accumulation of reactive oxygen species (ROS) by increasing the levels of free NADPH and the antioxidant effector, reduced glutathione (GSH), in transduced cells [116,117,118]. The p30II protein suppressed ROS-production by either c-Myc or the viral oncoproteins Tax and HBZ, dependent upon the induction of TIGAR, and inhibited genomic and mitochondrial DNA-damage and cytotoxicity/apoptosis as a result of the aberrant expression of cellular or viral oncoproteins [53, 86]. These studies further demonstrated that p30II cooperates with c-Myc, Tax and HBZ in cellular transformation/foci-formation assays through the induction of TIGAR, and enhanced the colony-forming potential of these oncoproteins in vitro. As Baydoun et al. [81] have reported that p30II inhibits homologous recombination-directed DNA-damage repair and favors the error-prone NHEJ pathway, it is possible p30II could destabilize the genome and promote the accumulation of somatic mutations that may contribute to viral carcinogenesis. Moreover, HT1080 fibrosarcoma clones expressing the infectious HTLV-1 ACH provirus exhibited higher levels of TIGAR expression associated with reduced oxidative DNA-damage, mitophagy, and apoptosis, as compared to clones that contained a mutant ACH.p30II provirus defective for p30II production [24, 53, 86, 119]. Although the viral transactivator Tax has been reported to inhibit p53 functions [120,121,122], Zane et al. [106] have demonstrated that Tax does not completely inhibit p53, rather, the p53 protein was shown to contribute to Tax-induced tumorigenesis in Tax+ p53+/+ transgenic mice. Wright et al. [42] have also reported that HBZ inhibits p53 functions by inhibiting the p300-dependent acetylation of p53 and recruitment of the p53-cofactor HBO1 to the p21/CDKN1A promoter in transfected cells. However, the HBZ protein induces genotoxic stress and is not highly expressed in vivo [58]; and Billman et al. [49] using RNA-FISH have shown that tax and hbz are alternately expressed in intermittent bursts in HTLV-1-infected patient cells and observed that many cells do not express hbz. It is possible that HBZ could interfere with the p300-dependent acetylation of p53 on lysine residue K372 which is also targeted for methylation by the SET7/SET9 methyltransferases and creates a docking site for the TIP60 chromo-domain for the induction of K120-acetylation and p53-dependent pro-apoptotic signaling [114, 115]. It is therefore likely that p30II-interactions with TIP60 may counter the cytotoxicity and oxidative stress caused by viral and/or cellular oncogenes—consistent with the demonstration that p30II cooperated with and enhanced the transforming potential of Tax and HBZ in vitro [53]. p30II could further enhance the lymphoproliferative activity of Tax and/or HBZ by preventing the accumulation of damaging mitochondrial ROS and inhibiting cellular apoptosis induced by these oncoproteins [53, 86]. Recently, Malu et al. [123] demonstrated that p30II prevents Tax-induced genomic instability and mitotic catastrophe induced by NF-κB hyperactivation in the HTLV-1 ACH proviral clone, through the activation of p53 and the p53-dependent repression of Stathmin/oncoprotein-18—a p65RelA-binding cofactor and tubulin-destabilizing protein. These findings allude to a possible key ancillary role for p30II and the induction of p53-regulated pro-survival signals in HTLV-1 pathogenesis. In 2018, Romeo et al. [86] demonstrated that HTLV-1-transformed T-cell-lines (MJG11, SLB1, ATL-1, and ATL-7) and primary uncultured HTLV-1-infected ATLL clinical samples contain elevated levels of TIGAR that correlated with oncogenic c-Myc expression as compared to activated hu-PBMCs. Using a highly-penetrant NOD/scid xenograft model of HTLV-1-induced T-cell lymphoma, Hutchison et al. [53] demonstrated that TIGAR is expressed at high levels in engrafted HTLV-1-infected SLB1 or Met-1 tumor lymphocytes, associated with c-Myc dysregulation in the primary tumor masses and infiltrated secondary tissues. The elevated levels of TIGAR in HTLV-1+ tumor cells also correlated with increased angiogenesis and infiltration of the tumor stroma and secondary tissues by murine endothelial progenitors (CD31/Flk1-positive cells); and 2 animals developed splenic hemangiomas associated with HTLV-1-induced T-cell lymphomas [53]. By contrast, there is no evidence that the HTLV-2 p28II synologue possesses transcriptional activity and this functional disparity, taken together with its inability to promote cell-survival, could, in part, account for the different pathogenic properties of HTLV-1 and HTLV-2.
HTLV-1 p13II—a ubiquitinated antagonist of Tax-transactivation
The HTLV-1 p13II protein is a mitochondrial targeting factor, comprised of 87 aa residues, and corresponds to the C-terminus of the p30II sequence beginning with a methionine start codon at position 155 (Fig. 4a, b) [25, 124, 125]. p13II is produced from a singly-spliced pX-orfII-p13II mRNA which splices a donor nucleotide at position 119 to an acceptor site at position 6875 [87, 90]. Interestingly, the pX region of HTLV-2 does not encode a functional synologue of p13II and this represents a significant point of divergence between these PTLV family members. In 1997, however, Mahieux et al. [126] identified a phylogenetically distinct isolate of STLV-1 (STLV-1marc1) from an Asian monkey species, Macaca arctoides, which lacked the methionine initiation codons for both p12I and p13II and serologically more closely resembled HTLV-2. The mitochondrial targeting signal (MTS) of p13II spans the amino-proximal residues 20–35 (Fig. 4b) which are predicted to form an amphipathic alpha-helix [124]. In 1999, Ciminale et al. [124], using nested deletions and site-directed mutagenesis, demonstrated that mitochondrial targeting of the p13II protein is atypical and does not require the basic residues within its MTS. p13II is required for viral infectivity and the maintenance of a high proviral titer in vivo, which was demonstrated by experimentally inoculating rabbits with a human B-cell line that contained a mutant infectious clone of HTLV-1 defective for p13II production (729.ACH.p13) [25]. Although the in vivo functions of p13II remain to be completely defined, Andresen et al. [21] have shown that the p13II protein is mono-ubiquitinated on a non-lysine residue and localizes in nuclear speckles in the presence of the viral transactivator Tax and, consequently, interferes with recruitment of the p300 coactivator to Tax-containing complexes and represses transcriptional activation from the HTLV-1 5′ LTR. These findings suggest that ubiquitinated-p13II may help to promote viral latency for the establishment of persistent infections in vivo. The unmodified p13II protein targets the inner membrane of mitochondria, induces membrane depolarization and mitochondrial swelling, opens the apoptogenic permeability transition pore, and results in an increased flux of K+ and Ca2+ ions and the production of ROS [124, 125, 127,128,129]. Interestingly, Tibaldi et al. [130] have shown that the proline-rich Src-homology 3 (SH3) domain of p13II (Fig. 4b) interacts with and recruits Src-family tyrosine kinases to the intermembrane space of mitochondria, which resulted in increased mitochondrial tyrosine-phosphorylation and abrogated the physiological effects of p13II upon mitochondrial membranes. In 2004, Silic-Benussi et al. [125] demonstrated that p13II inhibited tumorigenesis and the growth of c-Myc/Ha-Ras-transformed rat embryo fibroblasts, as well as p13II-expressing HeLaTet-On cell-lines, in engrafted nude mice. p13II also inhibited cellular proliferation in vitro and caused delayed cell-cycle progression and growth-arrest in nocodozole-treated cells. The p13II protein resulted in an increased sensitivity to C2 ceramide-induced apoptosis as detected by poly(ADP-ribose) polymerase (PARP)-cleavage, and also enhanced the levels of nuclear phospho-CREB in response to Ca2+-stimulation in histamine-treated cells [125]. By comparison, the related G4 protein of the bovine leukemia virus (BLV) cooperates with the Ha-Ras oncoprotein and induced tumors in engrafted nude mice [131]; and Lefèbvre et al. [132] have demonstrated that both the BLV G4 and HTLV-1 p13II proteins localize to mitochondria and interact with farnesyl pyrophosphate synthetase (FPPS)—a cofactor involved in targeting oncogenic Ras to the plasma membrane, suggesting there may be some functional overlap between these factors. Further, a mutant BLV proviral clone, defective for G4 production, was impaired in its pathogenic potential and failed to induce leukemia or lymphosarcomas in infected sheep [131]. In 2005, Hiraragi et al. [133] demonstrated that p13II inhibited the growth of Jurkat T-cells at high culture densities, and sensitized these cells to apoptosis induced by either Fas Ligand or ceramide-treatment. The ability of p13II to promote cellular apoptosis was countered by treating p13II-expressing Jurkat cells with a farnesyl transferase-inhibitor which prevents the posttranslational modification of the Ras protein and interferes with its membrane localization [133]. Silic-Benussi et al. [127] have further demonstrated that increased ROS production by p13II was associated with the activation of resting primary T-cells which was countered by ROS scavengers, whereas p13II sensitized transformed Jurkat T-cells to apoptosis under conditions of glucose-deprivation. It is thus intriguing to speculate that the ORF-II products, p13II and p30II, could act coordinately in HTLV-1-infected cells to promote carcinogenesis—with p30II suppressing ROS-dependent apoptosis by p13II through the p53-regulated induction of the antioxidant effector, TIGAR [53, 86]. The absence of a p13II synologue and dissimilar functions of HTLV-1 p30II and HTLV-2 p28II, in terms of their ability to activate cellular pro-survival signals, may, at least in part, account for the different pathogenic properties of these related PTLVs.
Conclusions
It remains an enigmatic mystery why the HTLV-1 is the only member of the PTLV family that is pathogenic in humans. Both the HTLV-1 and HTLV-2 can infect and immortalize primary T-cells cultured in vitro. Despite its similar genomic organization and structural relatedness, the HTLV-2 is not causally linked with any specific disease, although it has been associated with non-malignant lymphoproliferation and mild neurological symptoms in some infected patients [9, 10, 14]. The major viral transactivator proteins, Tax-1 and Tax-2, exhibit > 77% aa sequence homology and activate CREB/ATF and NF-κB-dependent transcriptional signaling in a nearly identical manner (Fig. 2a, b) [134, 135]. However, the HTLV-2 Tax-2 oncoprotein was observed to be less efficient at transforming rat fibroblasts in vitro [135]; and Semmes et al. [134] have demonstrated that Tax-2 does not induce significant genomic DNA-damage resulting in the formation of micronuclei/microsatellites, as compared to Tax-1 in transfected COS cells.
Similar to other transforming viruses that encode latency-maintenance factors, such as Epstein–Barr virus and the Kaposi’s sarcoma-associated herpesvirus, the HTLVs have developed several strategies to suppress the expression of viral antigens, while simultaneously driving mitotic proviral replication through the activation of cellular proliferative pathways. Indeed, the divergent and dissimilar functions of the pX-encoded latency-maintenance factors of HTLV-1 and HTLV-2 may provide clues into the differences in pathogenicity of these PTLVs. The antisense bZIP proteins, HBZ and APH-2, both repress Tax-dependent transactivation and gene expression from the viral 5′ LTR and inhibit NF-κB-signaling through interactions with the p65RelA subunit which prevents its binding to κB-responsive enhancer elements [27, 30, 47, 66, 67, 70]. However, whereas HBZ inhibits AP-1-dependent transcription and modulates FoxP3 and TGF-β-mediated inflammatory signaling, the APH-2 protein activates AP-1 and doesn’t affect TGF-β immunomodulatory signaling. Interestingly, although hbz is required for HTLV-1 proviral persistence in vivo, Yin et al. [67] demonstrated that rabbits experimentally inoculated with a 729 B-cell/HTLV-2 proviral clone deleted for aph-2 (∆Aph-2) had higher antibody titers and proviral loads than animals infected with wild-type HTLV-2. These results are somewhat surprising and suggest that HBZ and APH-2 have different roles for the maintenance of viral persistence in vivo, and allude to the potential importance of the other pX latency factors: p30II, p28II, and p13II. While the HTLV-1 p30II and HTLV-2 p28II proteins are functionally similar in their ability to negatively regulate Tax-dependent transactivation from the proviral LTR [16,17,18,19,20], unlike p30II, there is no evidence that p28II contains transcriptional activity. p30II interacts with the cellular acetyltransferases p300/CBP and TIP60 [18, 20, 84,85,86], interferes with the recruitment of p300/CBP to Tax/CREB/21-bp-repeat TRE complexes on the HTLV-1 promoter [18], and modulates host cellular gene expression through transcriptional and posttranscriptional mechanisms [19, 53, 78, 84, 86, 101]. In vivo evidence has demonstrated that p30II is required for viral persistence and the maintenance of a high proviral titer—presumably, through the suppression of viral antigens which could help HTLV-1-infected cells evade host immune-surveillance pathways [23, 24]. Alternatively, p30II could enhance the survival of infected T-cells by activating cellular pro-survival genes and antioxidant effectors, such as TIGAR, to prevent the accumulation of cytotoxic metabolic byproducts (e.g., ROS) and counter the oxidative-stress caused by the aberrant expression of viral (Tax and HBZ) and/or cellular oncoproteins [53, 86]. The mitochondrial targeting of the unmodified HTLV-1 p13II protein has been shown to promote the activation of primary T-cells through the induction of low levels of ROS; however, higher levels of ROS, as can be present in oncogenically-transformed cells, induce apoptosis [127]. It is thus plausible that p30II, p13II, and HBZ may act coordinately to promote enhanced lymphoproliferation and mitotic proviral replication, while preventing the buildup of excessive levels of damaging ROS. The absence of a p13II synologue, together with the disparate functions of APH-2 and p28II relative to their HTLV-1 pX counterparts, could provide a molecular explanation for the weakened pathogenic nature of HTLV-2.
Availability of data and materials
Not applicable.
Abbreviations
- APH-2:
-
antisense protein of HTLV-2
- ATLL:
-
adult T-cell leukemia/lymphoma
- CREB:
-
cyclic AMP-responsive element binding protein
- p300/CBP:
-
p300/CREB-binding protein
- HAM/TSP:
-
HTLV-1-associated myelopathy/tropical spastic paraparesis
- HBZ:
-
HTLV-1 basic leucine zipper factor
- HDAC3:
-
histone deacetylase-3
- HTLV-1:
-
human T-cell leukemia virus type-1
- HTLV-2:
-
human T-cell lymphotropic virus type-2
- LTR:
-
long terminal repeat
- NFAR:
-
nuclear factors associated with double-stranded RNA
- NF-κB:
-
nuclear factor kappa light-chain enhancer of activated B cells
- NHEJ:
-
non-homologous end-joining
- ORF-I/II:
-
open reading frame-I/II
- PTLV:
-
primate T-cell lymphotropic virus
- ROS:
-
reactive oxygen species
- STLVs:
-
simian T-cell lymphotropic viruses
- TGF-β:
-
transforming growth factor-beta
- TIGAR:
-
TP53-induced glycolysis and apoptosis regulator
- TREs:
-
Tax-responsive elements
References
Wolfe ND, Heneine W, Carr JK, Garcia AD, Shanmugam V, Tamoufe U, et al. Emergence of unique primate T-lymphotropic viruses among central African bushmeat hunters. Proc Natl Acad Sci USA. 2005;102(22):7994–9.
Liégeois F, Lafay B, Switzer WM, Locatelli S, Mpoudi-Ngolé E, Loul S, et al. Identification and molecular characterization of new STLV-1 and STLV-3 strains in wild-caught nonhuman primates in Cameroon. Virology. 2008;371(2):405–17.
Calattini S, Chevalier SA, Duprez R, Bassot S, Froment A, Mahieux E, et al. Discovery of a new human T-cell lymphotropic virus (HTLV-3) in Central Africa. Retrovirology. 2005;2:30.
LeBreton M, Switzer WM, Djoko CF, Gillis A, Jia H, Sturgeon MM, et al. A gorilla reservoir for human T-lymphotropic virus type 4. Emerg Microbes Infect. 2014;3(1):e7.
Kornfeld H, Riedel N, Viglianti GA, Hirsch V, Mullins JI. Cloning of HTLV-4 and its relation to simian and human immunodeficiency viruses. Nature. 1987;326(6113):610–3.
Poiesz BJ, Ruscetti FW, Reitz MS, Kalyanaraman VS, Gallo RC. Isolation of a new type C retrovirus (HTLV) in primary uncultured cells of a patient with Sezary T-cell leukaemia. Nature. 1981;294(5838):268–71.
Poiesz BJ, Ruscetti FW, Gazdar AF, Bunn PA, Minna JD, Gallo RC. Detection and isolation of type C retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T-cell lymphoma. Proc Natl Acad Sci USA. 1980;77(12):7415–9.
Kalyanaraman VS, Sarngadharan MG, Nakao Y, Ito Y, Aoki T, Gallo RC. Natural antibodies to the structural core protein (p24) of the human T-cell leukemia (lymphoma) retrovirus found in sera of leukemia patients in Japan. Proc Natl Acad Sci USA. 1982;79(5):1653–7.
Kalyanaraman VS, Samgadharan MG, Robert-Guroff M, Miyoshi I, Golde D, Gallo RC. A new subtype of human T-cell leukemia virus (HTLV-II) associated with a T-cell variant of hairy cell leukemia. Science. 1982;218(4572):571–3.
Chen IS, McLaughlin J, Gasson JC, Clark SC, Golde DW. Molecular characterization of genome of a novel human T-cell leukaemia virus. Nature. 1983;305(5934):502–5.
Chen IS, Quan SG, Golde DW. Human T-cell leukemia virus type II transforms normal human lymphocytes. Proc Natl Acad Sci USA. 1983;80(22):7006–9.
Casoli C, Cimarelli A, Bertazzoni U. Cellular tropism of human T-cell leukemia virus type II is enlarged to B lymphocytes in patients with high proviral load. Virology. 1995;206(2):1126–8.
Ijichi S, Ramundo MB, Takahashi H, Hall WW. In vivo cellular tropism of human T cell leukemia virus type II (HTLV-II). J Exp Med. 1992;176(1):293–6.
Murphy EL, Fridey J, Smith JW, Engstrom J, Sacher RA, Miller K, et al. HTLV-associated myelopathy in a cohort of HTLV-I and HTLV-II-infected blood donors. The REDS investigators. Neurology. 1997;48(2):315–20.
Kannian P, Yin H, Doueiri R, Lairmore MD, Fernandez S, Green PL. Distinct transformation tropism exhibited by human T lymphotropic virus type 1 (HTLV-1) and HTLV-2 is the result of postinfection T cell clonal expansion. J Virol. 2012;86(7):3757–66.
Nicot C, Dundr M, Johnson JM, Fullen JR, Alonzo N, Fukumoto R, et al. HTLV-1-encoded p30II is a post-transcriptional negative regulator of viral replication. Nat Med. 2004;10(2):197–201.
Younis I, Khair L, Dundr M, Lairmore MD, Franchini G, Green PL. Repression of human T-cell leukemia virus type 1 and 2 replication by a viral mRNA-encoded posttranscriptional regulator. J Virol. 2004;78(20):11077–83.
Zhang W, Nisbet JW, Albrecht B, Ding W, Kashanchi F, Bartoe JT, et al. Human T-lymphotropic virus type 1 p30(II) regulates gene transcription by binding CREB binding protein/p300. J Virol. 2001;75(20):9885–95.
Zhang W, Nisbet JW, Bartoe JT, Ding W, Lairmore MD. Human T-lymphotropic virus type 1 p30(II) functions as a transcription factor and differentially modulates CREB-responsive promoters. J Virol. 2000;74(23):11270–7.
Michael B, Nair AM, Datta A, Hiraragi H, Ratner L, Lairmore MD. Histone acetyltransferase (HAT) activity of p300 modulates human T lymphotropic virus type 1 p30II-mediated repression of LTR transcriptional activity. Virology. 2006;354(2):225–39.
Andresen V, Pise-Masison CA, Sinha-Datta U, Bellon M, Valeri V, Washington Parks R, et al. Suppression of HTLV-1 replication by Tax-mediated rerouting of the p13 viral protein to nuclear speckles. Blood. 2011;118(6):1549–59.
Arnold J, Yamamoto B, Li M, Phipps AJ, Younis I, Lairmore MD, et al. Enhancement of infectivity and persistence in vivo by HBZ, a natural antisense coded protein of HTLV-1. Blood. 2006;107(10):3976–82.
Valeri VW, Hryniewicz A, Andresen V, Jones K, Fenizia C, Bialuk I, et al. Requirement of the human T-cell leukemia virus p12 and p30 products for infectivity of human dendritic cells and macaques but not rabbits. Blood. 2010;116(19):3809–17.
Bartoe JT, Albrecht B, Collins ND, Robek MD, Ratner L, Green PL, et al. Functional role of pX open reading frame II of human T-lymphotropic virus type 1 in maintenance of viral loads in vivo. J Virol. 2000;74(3):1094–100.
Hiraragi H, Kim SJ, Phipps AJ, Silic-Benussi M, Ciminale V, Ratner L, et al. Human T-lymphotropic virus type 1 mitochondrion-localizing protein p13(II) is required for viral infectivity in vivo. J Virol. 2006;80(7):3469–76.
Yamamoto B, Li M, Kesic M, Younis I, Lairmore MD, Green PL. Human T-cell leukemia virus type 2 post-transcriptional control protein p28 is required for viral infectivity and persistence in vivo. Retrovirology. 2008;5:38.
Gaudray G, Gachon F, Basbous J, Biard-Piechaczyk M, Devaux C, Mesnard JM. The complementary strand of the human T-cell leukemia virus type 1 RNA genome encodes a bZIP transcription factor that down-regulates viral transcription. J Virol. 2002;76(24):12813–22.
Cavanaugh MH, Landry S, Audet B, Arpin-André C, Hivin P, Paré ME, et al. HTLV-I antisense transcripts initiating in the 3′LTR are alternatively spliced and polyadenylated. Retrovirology. 2006;3:15.
Murata K, Hayashibara T, Sugahara K, Uemura A, Yamaguchi T, Harasawa H, et al. A novel alternative splicing isoform of human T-cell leukemia virus type 1 bZIP factor (HBZ-SI) targets distinct subnuclear localization. J Virol. 2006;89(5):2495–505.
Lemasson I, Lewis MR, Polakowski N, Hivin P, Cavanaugh MH, Thébault S, et al. Human T-cell leukemia virus type 1 (HTLV-1) bZIP protein interacts with the cellular transcription factor CREB to inhibit HTLV-1 transcription. J Virol. 2007;81(4):1543–53.
Arnold J, Zimmerman B, Li M, Lairmore MD, Green PL. Human T-cell leukemia virus type-1 antisense-encoded gene, Hbz, promotes T-lymphocyte proliferation. Blood. 2008;112(9):3788–97.
Satou Y, Yasunaga J, Zhao T, Yoshida M, Miyazato P, Takai K, et al. HTLV-1 bZIP factor induces T-cell lymphoma and systemic inflammation in vivo. PLoS Pathog. 2011;7(2):e1001274.
Mitobe Y, Yasunaga J, Furuta R, Matsuoka M. HTLV-1 bZIP factor RNA and protein impart distinct functions on T-cell proliferation and survival. Cancer Res. 2015;75(19):4143–52.
Satou Y, Yasunaga J, Yoshida M, Matsuoka M. HTLV-I basic leucine zipper factor gene mRNA supports proliferation of adult T cell leukemia cells. Proc Natl Acad Sci USA. 2006;103(3):720–5.
Rowan AG, Suemori K, Fujiwara H, Yasukawa M, Tanaka Y, Taylor GP, et al. Cytotoxic T lymphocyte lysis of HTLV-1 infected cells is limited by weak HBZ protein expression, but non-specifically enhanced on induction of Tax expression. Retrovirology. 2014;11:116.
Raval GU, Bidoia C, Forlani G, Tosi G, Gessain A, Accolla RS. Localization, quantification and interaction with host factors of endogenous HTLV-1 HBZ protein in infected cells and ATL. Retrovirology. 2015;12:59.
Hivin P, Frederic M, Arpin-André C, Basbous J, Gay B, Thébault S, et al. Nuclear localization of HTLV-I bZIP (HBZ) is mediated by three distinct motifs. J Cell Sci. 2005;118:1355–62.
Mukai R, Ohshima T. HTLV-1 HBZ positively regulates the mTOR signaling pathway via inhibition of GADD34 activity in the cytoplasm. Oncogene. 2014;33(18):2317–28.
Dissinger N, Shkriabai N, Hess S, Al-Saleem J, Kvaratskhelia M, Green PL. Identification and characterization of HTLV-1 HBZ post-translational modifications. PLoS ONE. 2014;9(11):e112762.
Clerc I, Polakowski N, André-Arpin C, Cook P, Barbeau B, Mesnard JM, et al. An interaction between the human T cell leukemia virus type 1 basic leucine zipper factor (HBZ) and the KIX domain of p300/CBP contributes to the down-regulation of tax-dependent viral transcription by HBZ. J Biol Chem. 2008;283(35):23903–13.
Wurm T, Wright DG, Polakowski N, Mesnard JM, Lemasson I. The HTLV-1-encoded protein HBZ directly inhibits the acetyl transferase activity of p300/CBP. Nucleic Acids Res. 2012;40(13):5910–25.
Wright DG, Marchal C, Hoang K, Ankney JA, Nguyen ST, Rushing AW, et al. Human T-cell leukemia virus type-1-encoded protein HBZ represses p53 function by inhibiting the acetyltransferase activity of p300/CBP and HBO1. Oncotarget. 2016;7(2):1687–706.
Alasiri A, Abboud Guerr J, Hall WW, Sheehy N. Novel interactions between the HTLV-1 antisense protein HBZ and the SWI/SNF chromatin remodeling family: implications for viral life cycle. J Virol. 2019. https://doi.org/10.1128/jvi.00412-19 (epub ahead of print).
Basbous J, Arpin C, Gaudray G, Piechaczyk M, Devaux C, Mesnard JM. The HBZ factor of human T-cell leukemia virus type I dimerizes with transcription factors JunB and c-Jun and modulates their transcriptional activity. J Biol Chem. 2003;278(44):43620–7.
Kuhlman AS, Villaudy J, Gazzolo L, Castellazzi M, Mesnard JM, Duc Dodon M. HTLV-1 HBZ cooperates with JunD to enhance transcription of the human telomerase reverse transcriptase gene (hTERT). Retrovirology. 2007;4:92.
Terol M, Gazon H, Lemasson I, Duc-Dodon M, Barbeau B, Césaire R, et al. HBZ-mediated shift of JunD from growth suppressor to tumor promoter in leukemic cells by inhibition of ribosomal protein S25 expression. Leukemia. 2017;31(10):2235–43.
Zhao T, Yasunaga J, Satou Y, Nakao M, Takahashi M, Fujii M, et al. Human T-cell leukemia virus type 1 bZIP factor selectively suppresses the classical pathway of NF-kappaB. Blood. 2009;113(12):2755–64.
Zhi H, Yang L, Kuo YL, Ho YK, Shih HM, Giam CZ. NF-kB hyper-activation by HTLV-1 tax induces cellular senescence, but can be alleviated by the viral anti-sense protein HBZ. PLoS Pathog. 2011;7(4):e1002025.
Billman MR, Rueda D, Bangham CRM. Single-cell heterogeneity and cell-cycle related viral gene bursts in the human leukaemia virus HTLV-1. Wellcome Open Res. 2017;2:87.
Mahgoub M, Yasunaga JI, Iwami S, Nakaoka S, Koizumi Y, Shimura K, et al. Sporadic on/off switching of HTLV-1 Tax expression is crucial to maintain the whole population of virus-induced leukemic cells. Proc Natl Acad Sci USA. 2018;115(6):e1269–78.
Choudhary G, Ratner L. The HTLV-1 hbz antisense gene indirectly promotes tax expression via down-regulation of p30(II) mRNA. Virology. 2011;410(2):307–15.
Rushing AW, Hoang K, Polakowski N, Lemasson I. The human T-cell leukemia virus type 1 basic leucine zipper factor attenuates repair of double-stranded DNA breaks via nonhomologous end joining. J Virol. 2018;92(15):e00672-18.
Hutchison T, Malu A, Yapindi L, Bergeson R, Peck K, Romeo M, et al. The TP53-Induced Glycolysis and Apoptosis Regulator mediates cooperation between HTLV-1 p30II and the retroviral oncoproteins Tax and HBZ and is highly expressed in an in vivo xenograft model of HTLV-1-induced lymphoma. Virology. 2018;520:39–58.
Ma G, Yasunaga J, Fan J, Yanagawa S, Matsuoka M. HTLV-1 bZIP factor dysregulates the Wnt pathways to support proliferation and migration of adult T-cell leukemia cells. Oncogene. 2013;32(36):4222–30.
Zhao T, Coutts A, Xu L, Yu J, Ohshima K, Matsuoka M. HTLV-1 bZIP factor supports proliferation of adult T cell leukemia cells through suppression of C/EBPα signaling. Retrovirology. 2013;10:159.
Kawatsuki A, Yasunaga JI, Mitobe Y, Green PL, Matsuoka M. HTLV-1 bZIP factor protein targets the Rb/E2F-1 pathway to promote proliferation and apoptosis of primary CD4(+) T cells. Oncogene. 2016;35(34):4509–17.
Kinosada H, Yasunaga JI, Shimura K, Miyazato P, Onishi C, Iyoda T, et al. HTLV-1 bZIP factor enhances T-cell proliferation by impeding the suppressive effects of co-inhibitory signals. PLoS Pathog. 2017;13(1):e1006120.
Forlani G, Baratella M, Tedeschi A, Pique C, Jacobson S, Accolla RS. HTLV-1 HBZ protein resides exclusively in the cytoplasm of infected cells in asymptomatic carriers and HAM/TSP patients. Front Microbiol. 2019;10:819.
Saito M, Matsuzaki T, Satou Y, Yasunaga J, Saito K, Arimura K, et al. In vivo expression of HBZ gene of HTLV-1 correlates with proviral load, inflammatory markers and disease severity in HTLV-1 associated myelopathy/tropical spastic paraparesis (HAM/TSP). Retrovirology. 2009;6:19.
Li M, Kesic M, Yin H, Yu L, Green PL. Kinetic analysis of human T-cell leukemia virus type 1 gene expression in cell culture and infected animals. J Virol. 2009;83(8):3788–97.
Rende F, Cavallari I, Corradin A, Silic-Benussi M, Toulza F, Toffolo GM, et al. Kinetics and intracellular compartmentalization of HTLV-1 gene expression: nuclear retention of HBZ mRNAs. Blood. 2011;117(18):4855–9.
Vernin C, Thernoz M, Pinatel C, Gessain A, Gout O, Delfau-Larue MH, et al. HTLV-1 bZIP factor HBZ promotes cell proliferation and genetic instability by activating OncomiRs. Cancer Res. 2014;74(21):6082–93.
Yamamoto-Taguchi N, Satou Y, Miyazato P, Ohshima K, Nakagawa M, Katagiri K, et al. HTLV-1 bZIP factor induces inflammation through labile Foxp3 expression. PLoS Pathog. 2013;9(9):e1003630.
Zhao T, Satou Y, Sugata K, Miyazato P, Green PL, Imamura T, et al. HTLV-1 bZIP factor enhances TGF-β signaling through p300 coactivator. Blood. 2011;118(7):1865–76.
Esser AK, Rauch DA, Xiang J, Harding JC, Kohart NA, Ross MH, et al. HTLV-1 viral oncogene HBZ induces osteolytic bone disease in transgenic mice. Oncotarget. 2017;8(41):69250–63.
Halin M, Douceron E, Clerc I, Journo C, Ko NL, Landry S, et al. Human T-cell leukemia virus type 2 produces a spliced antisense transcript encoding a protein that lacks a classic bZIP domain but still inhibits Tax2-mediated transcription. Blood. 2009;114(12):2427–38.
Yin H, Kannian P, Dissinger N, Haines R, Niewiesk S, Green PL. Human T cell leukemia virus type 2 antisense viral protein 2 is dispensable for in vitro immortalization but functions to repress early virus replication in vivo. J Virol. 2012;86(16):8412–21.
Douceron E, Kaidarova Z, Miyazato P, Matsuoka M, Murphy EL, Mahieux R. HTLV-2 APH-2 expression is correlated with proviral load but APH-2 does not promote lymphocytosis. J Infect Dis. 2012;205(1):82–6.
Bender C, Rende F, Cotena A, Righi P, Ronzi P, Cavallari I, et al. Temporal regulation of HTLV-2 expression in infected cell lines and patients: evidence for distinct expression kinetics with nuclear accumulation of APH-2 mRNA. Retrovirology. 2012;9:74.
Panfil AR, Dissinger NJ, Howard CM, Murphy BM, Landes K, Fernandez SA, et al. Functional comparison of HBZ and the related APH-2 protein provides insight into human T-cell leukemia virus type 1 pathogenesis. J Virol. 2016;90(7):3760–72.
Panfil AR, Al-Saleem J, Howard CM, Shkriabai N, Kvaratskhelia M, Green PL. Stability of the HTLV-1 antisense-derived protein, HBZ, is regulated by the E3 ubiquitin-protein ligase, UBR5. Front Microbiol. 2018;9:80.
Dubuisson L, Lormières F, Fochi S, Turpin J, Pasquier A, Douceron E, et al. Stability of HTLV-2 antisense protein is controlled by PML nuclear bodies in a SUMO-dependent manner. Oncogene. 2018;37(21):2806–16.
Marban C, McCabe A, Bukong TN, Hall WW, Sheehy N. Interplay between the HTLV-2 Tax and APH-2 proteins in the regulation of the AP-1 pathway. Retrovirology. 2012;9:98.
Murphy J, Hall WW, Ratner L, Sheehy N. Novel interactions between the HTLV antisense proteins HBZ and APH-2 and the NFAR protein family: implications for the HTLV lifecycles. Virology. 2016;494:129–42.
Lairmore MD, Albrecht B, D’Souza C, Nisbet JW, Ding W, Bartoe JT, et al. In vitro and in vivo functional analysis of human T cell lymphotropic virus type 1 pX open reading frames I and II. AIDS Res Hum Retroviruses. 2000;16(16):1757–64.
Datta A, Sinha-Datta U, Dhillon NK, Buch S, Nicot C. The HTLV-I p30 interferes with TLR4 signaling and modulates the release of pro- and anti-inflammatory cytokines from human macrophages. J Biol Chem. 2006;281(33):23414–24.
Datta A, Silverman L, Phipps AJ, Hiraragi H, Ratner L, Lairmore MD. Human T-lymphotropic virus type-1 p30 alters cell cycle G2 regulation of T lymphocytes to enhance cell survival. Retrovirology. 2007;4:49.
Taylor JM, Ghorbel S, Nicot C. Genome wide analysis of human genes transcriptionally and post-transcriptionally regulated by the HTLV-I protein p30. BMC Genomics. 2009;10:311.
Baydoun HH, Pancewicz J, Bai X, Nicot C. HTLV-I p30 inhibits multiple S phase entry checkpoints, decreases cyclin E-CDK2 interactions and delays cell cycle progression. Mol Cancer. 2010;9:302.
Anupam R, Datta A, Kesic M, Green-Church K, Shkriabai N, Kvaratskhelia M, et al. Human T-lymphotropic virus type 1 p30 interacts with REGgamma and modulates ATM (ataxia telangiectasia mutated) to promote cell survival. J Biol Chem. 2011;286(9):7661–8.
Baydoun HH, Pancewicz J, Nicot C. Human T-lymphotropic type 1 virus p30 inhibits homologous recombination and favors unfaithful DNA repair. Blood. 2011;117(22):5897–906.
Doueiri R, Anupam R, Kvaratskhelia M, Green KB, Lairmore MD, Green PL. Comparative host protein interactions with HTLV-1 p30 and HTLV-2 p28: insights into difference in pathobiology of human retroviruses. Retrovirology. 2012;9:64.
Fenizia C, Fiocchi M, Jones K, Parks RW, Ceribelli M, Chevalier SA, et al. Human T-cell leukemia/lymphoma virus type 1 p30, but not p12/8, counteracts toll-like receptor 3 (TLR3) and TLR4 signaling in human monocytes and dendritic cells. J Virol. 2014;88(1):393–402.
Awasthi S, Sharma A, Wong K, Zhang J, Matlock EF, Rogers L, et al. A human T-cell lymphotropic virus type 1 enhancer of Myc transforming potential stabilizes Myc-TIP60 transcriptional interactions. Mol Cell Biol. 2005;25(14):6178–98.
Romeo MM, Ko B, Kim J, Brady R, Heatley HC, He J, et al. Acetylation of the c-MYC oncoprotein is required for cooperation with the HTLV-1 p30II accessory protein and the induction of oncogenic cellular transformation by p30II/c-MYC. Virology. 2015;476:271–88.
Romeo M, Hutchison T, Malu A, White A, Kim J, Gardner R, et al. The human T-cell leukemia virus type-1 p30II protein activates p53 and induces the TIGAR and suppresses oncogene-induced oxidative stress during viral carcinogenesis. Virology. 2018;518:103–15.
Ciminale V, Pavlakis GN, Derse D, Cunningham CP, Felber BK. Complex splicing in the human T-cell leukemia virus (HTLV) family of retroviruses: novel mRNAs and proteins produced by HTLV type I. J Virol. 1992;66(3):1737–45.
Koralnik IJ, Fullen J, Franchini G. The p12I, p13II, and p30II proteins encoded by human T-cell leukemia/lymphotropic virus type I open reading frames I and II are localized in three different cellular compartments. J Virol. 1993;67(4):2360–6.
Nicot C, Harrod RL, Ciminale V, Franchini G. Human T-cell leukemia/lymphoma virus type 1 nonstructural genes and their functions. Oncogene. 2005;24(39):6026–34.
Koralnik IJ, Gessain A, Klotman ME, Lo Monico A, Berneman ZN, Franchini G. Protein isoforms encoded by the pX region of human T-cell leukemia/lymphotropic virus type I. Proc Natl Acad Sci USA. 1992;89(18):8813–7.
Pique C, Ureta-Vidal A, Gessain A, Chancerel B, Gout O, Tamouza R, et al. Evidence for the chronic in vivo production of human T cell leukemia virus type I Rof and Tof proteins from cytotoxic T lymphocytes directed against viral peptides. J Exp Med. 2000;191(3):567–72.
Johnson JM, Harrod R, Franchini G. Molecular biology and pathogenesis of the human T-cell leukaemia/lymphotropic virus type-1 (HTLV-1). Int J Exp Pathol. 2001;82(3):135–47.
Ciminale V, D’Agostino DM, Zotti L, Franchini G, Felber BK, Chieco-Bianchi L. Expression and characterization of proteins produced by mRNAs spliced into the X region of the human T-cell leukemia/lymphotropic virus type II. Virology. 1995;209:445–56.
Namdev P, Lyngdoh DL, Dar HY, Chaurasiya SK, Srivastava R, Tripathi T, et al. Intrinsically disordered human T lymphotropic virus type 1 p30 protein: experimental and computational evidence. AIDS Res Hum Retroviruses. 2019;35(5):477–87.
Princler GL, Julias JG, Hughes SH, Derse D. Roles of viral and cellular proteins in the expression of alternatively spliced HTLV-1 pX mRNAs. Virology. 2003;317:136–45.
Berneman ZN, Gartenhaus RB, Reitz MS Jr, Blattner WA, Manns A, Hanchard B, et al. Expression of alternatively spliced human T-lymphotropic virus type I pX mRNA in infected cell lines and in primary uncultured cells from patients with adult T-cell leukemia/lymphoma and healthy carriers. Proc Natl Acad Sci USA. 1992;89:3005–9.
Cereseto A, Berneman Z, Koralnik I, Vaughn J, Franchini G, Klotman ME. Differential expression of alternatively spliced pX mRNAs in HTLV-I-infected cell lines. Leukemia. 1997;11(6):866–70.
Ghorbel S, Sinha-Datta U, Dundr M, Brown M, Franchini G, Nicot C. Human T-cell leukemia virus type I p30 nuclear/nucleolar retention is mediated through interactions with RNA and a constituent of the 60S ribosomal subunit. J Biol Chem. 2006;281(48):37150–8.
Derse D, Mikovits J, Ruscetti F. X-I and X-II open reading frames of HTLV-I are not required for virus replication or for immortalization of primary T-cells in vitro. Virology. 1997;237(1):123–8.
Robek MD, Wong FH, Ratner L. Human T-cell leukemia virus type 1 pX-I and pX-II open reading frames are dispensable for the immortalization of primary lymphocytes. J Virol. 1998;72(5):4458–62.
Michael B, Nair AM, Hiraragi H, Shen L, Feuer G, Boris-Lawrie K, et al. Human T lymphotropic virus type-1 p30II alters cellular gene expression to selectively enhance signaling pathways that activate T lymphocytes. Retrovirology. 2004;1:39.
Younis I, Boris-Lawrie K, Green PL. Human T-cell leukemia virus open reading frame II encodes a posttranscriptional repressor that is recruited at the level of transcription. J Virol. 2006;80(1):181–91.
Baydoun H, Duc-Dodon M, Lebrun S, Gazzolo L, Bex F. Regulation of the human T-cell leukemia virus gene expression depends on the localization of regulatory proteins Tax, Rex and p30II in specific nuclear subdomains. Gene. 2007;386:191–201.
Sinha-Datta U, Datta A, Ghorbel S, Dodon MD, Nicot C. Human T-cell lymphotropic virus type I rex and p30 interactions govern the switch between virus latency and replication. J Biol Chem. 2007;282(19):14608–15.
McMahon SB, Van Buskirk HA, Dugan KA, Copeland TD, Cole MD. The novel ATM-related protein TRRAP is an essential cofactor for the c-Myc and E2F oncoproteins. Cell. 1998;94(3):363–74.
Zane L, Yasunaga J, Mitagami Y, Yedavalli V, Tang SW, Chen CY, et al. Wip1 and p53 contribute to HTLV-1 Tax-induced tumorigenesis. Retrovirology. 2012;9:114.
Mengle-Gaw L, Rabbitts TH. A human chromosome 8 region with abnormalities in B cell, HTLV-I+ T cell and c-myc amplified tumours. EMBO J. 1987;6(7):1959–65.
Peller S, Rotter V. TP53 in hematological cancer: low incidence of mutations with significant clinical relevance. Hum Mutat. 2003;21(3):277–84.
Reid RL, Lindholm PF, Mireskandari A, Dittmer J, Brady JN. Stabilization of wild-type p53 in human T-lymphocytes transformed by HTLV-I. Oncogene. 1993;8:3029–36.
Yamato K, Oka T, Hiroi M, Iwahara Y, Sugito S, Tsuchida N, et al. Aberrant expression of the p53 tumor suppressor gene in adult T-cell leukemia and HTLV-I-infected cells. Jpn J Cancer Res. 1993;84(1):4–8.
Takemoto S, Trovato R, Cereseto A, Nicot C, Kislyakova T, Casareto L, et al. p53 stabilization and functional impairment in the absence of genetic mutation or the alteration of the p14(ARF)-MDM2 loop in ex vivo cultured adult T-cell leukemia/lymphoma cells. Blood. 2000;95:3939–44.
Yoshida N, Imaizumi Y, Utsunomiya A, Miyoshi H, Arakawa F, Tsukasaki K, et al. Mutation analysis for TP53 in chronic-type adult T-cell leukemia/lymphoma. J Clin Exp Hematop. 2015;55:13–6.
Tang Y, Luo J, Zhang W, Gu W. Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol Cell. 2006;24(6):827–39.
Sykes SM, Mellert HS, Holbert MA, Li K, Marmorstein R, Lane WS, et al. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol Cell. 2006;24(6):841–51.
Kurash JK, Lei H, Shen Q, Marston WL, Granda BW, Fan H, et al. Methylation of p53 by Set7/9 mediates p53 acetylation and activity in vivo. Mol Cell. 2008;29(3):392–400.
Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, et al. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126(1):107–20.
Bensaad K, Cheung EC, Vousden KH. Modulation of intracellular ROS levels by TIGAR controls autophagy. EMBO J. 2009;28(19):3015–26.
Cheung EC, Ludwig RL, Vousden KH. Mitochondrial localization of TIGAR under hypoxia stimulates HK2 and lowers ROS and cell death. Proc Natl Acad Sci USA. 2012;109(50):20491–6.
Kimata JT, Wong FH, Wang JJ, Ratner L. Construction and characterization of infectious human T-cell leukemia virus type 1 molecular clones. Virology. 1994;204:656–64.
Akagi T, Ono H, Tsuchida N, Shimotohno K. Aberrant expression and function of p53 in T-cells immortalized by HTLV-I Tax1. FEBS Lett. 1997;406:263–6.
Pise-Masison CA, Choi KS, Radonovich M, Dittmer J, Kim SJ, Brady JN. Inhibition of p53 transactivation function by the human T-cell lymphotropic virus type 1 Tax protein. J Virol. 1998;72(2):1165–70.
Mulloy JC, Kislyakova T, Cereseto A, Casareto L, LoMonico A, Fullen J, et al. Human T-cell lymphotropic/leukemia virus type 1 Tax abrogates p53-induced cell cycle arrest and apoptosis through its CREB/ATF functional domain. J Virol. 1998;72(11):8852–60.
Malu A, Hutchison T, Yapindi L, Smith K, Nelson K, Bergeson R, et al. The human T-cell leukemia virus type-1 tax oncoprotein dissociates NF-κB p65RelA-Stathmin complexes and causes catastrophic mitotic spindle damage and genomic instability. Virology. 2019;535:83–101.
Ciminale V, Zotti L, D’Agostino DM, Ferro T, Casareto L, Franchini G, et al. Mitochondrial targeting of the p13II protein coded by the x-II ORF of human T-cell leukemia/lymphotropic virus type I (HTLV-I). Oncogene. 1999;18(31):4505–14.
Silic-Benussi M, Cavallari I, Zorzan T, Rossi E, Hiraragi H, Rosato A, et al. Suppression of tumor growth and cell proliferation by p13II, a mitochondrial protein of human T cell leukemia virus type 1. Proc Natl Acad Sci USA. 2004;101(17):6629–34.
Mahieux R, Pecon-Slattery J, Gessain A. Molecular characterization and phylogenetic analyses of a new, highly divergent simian T-cell lymphotropic virus type 1 (STLV-1marc1) in Macaca arctoides. J Virol. 1997;71(8):6253–8.
Silic-Benussi M, Cavallari I, Vajente N, Vidali S, Chieco-Bianchi L, Di Lisa F, et al. Redox regulation of T-cell turnover by the p13 protein of human T-cell leukemia virus type 1: distinct effects in primary versus transformed cells. Blood. 2010;116(1):54–62.
Biasiotto R, Aguiari P, Rizzuto R, Pinton P, D’Agostino DM, Ciminale V. The p13 protein of human T cell leukemia virus type 1 (HTLV-1) modulates mitochondrial membrane potential and calcium uptake. Biochim Biophys Acta. 2010;1797(6–7):945–51.
Silic-Benussi M, Cannizzaro E, Venerando A, Cavallari I, Petronilli V, La Rocca N, et al. Modulation of mitochondrial K(+) permeability and reactive oxygen species production by the p13 protein of human T-cell leukemia virus type 1. Biochim Biophys Acta. 2009;1787(7):947–54.
Tibaldi E, Venerando A, Zonta F, Bidoia C, Magrin E, Marin O, et al. Interaction between the SH3 domain of Src family kinases and the proline-rich motif of HTLV-1 p13: a novel mechanism underlying delivery of Src family kinases to mitochondria. Biochem J. 2011;439(3):505–16.
Kerkhofs P, Heremans H, Burny A, Kettmann R, Willems L. In vitro and in vivo oncogenic potential of bovine leukemia virus G4 protein. J Virol. 1998;72(3):2554–9.
Lefèbvre L, Vanderplasschen A, Ciminale V, Heremans H, Dangoisse O, Jauniaux JC, et al. Oncoviral bovine leukemia virus G4 and human T-cell leukemia virus type 1 p13II accessory proteins interact with farnesyl pyrophosphate synthetase. J Virol. 2002;76(3):1400–14.
Hiraragi H, Michael B, Nair A, Silic-Benussi M, Ciminale V, Lairmore M. Human T-lymphotropic virus type 1 mitochondrion-localizing protein p13II sensitizes Jurkat T cells to Ras-mediated apoptosis. J Virol. 2005;79(15):9449–57.
Semmes OJ, Majone F, Cantemir C, Turchetto L, Hjelle B, Jeang KT. HTLV-I and HTLV-II Tax: differences in induction of micronuclei in cells and transcriptional activation of viral LTRs. Virology. 1996;217(1):373–9.
Endo K, Hirata A, Iwai K, Sakurai M, Fukushi M, Oie M, et al. Human T-cell leukemia virus type 2 (HTLV-2) Tax protein transforms a rat fibroblast cell line, but lss efficiently than HTLV-1 Tax. J Virol. 2002;76(6):2648–53.
Acknowledgements
Laçin Yapindi and Tetiana Hutchison are thanked for their helpful comments during the development of this manuscript.
Funding
This work was supported by a grant (1R15CA202265-01A1) from the National Cancer Institute/National Institutes of Health to RH.
Author information
Authors and Affiliations
Contributions
RH conceived and wrote the review the final manuscript. The author read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The author declares no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Harrod, R. Silencers of HTLV-1 and HTLV-2: the pX-encoded latency-maintenance factors. Retrovirology 16, 25 (2019). https://doi.org/10.1186/s12977-019-0487-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12977-019-0487-9