
Abstract
Background
Neuroinflammation may contribute to psychiatric symptoms in older people, in particular in the context of Alzheimer’s disease (AD). We sought to identify systemic and central nervous system (CNS) inflammatory alterations associated with neuropsychiatric symptoms (NPS); and to investigate their relationships with AD pathology and clinical disease progression.
Methods
We quantified a panel of 38 neuroinflammation and vascular injury markers in paired serum and cerebrospinal fluid (CSF) samples in a cohort of cognitively normal and impaired older subjects. We performed neuropsychiatric and cognitive evaluations and measured CSF biomarkers of AD pathology. Multivariate analysis determined serum and CSF neuroinflammatory alterations associated with NPS, considering cognitive status, AD pathology, and cognitive decline at follow-up visits.
Results
NPS were associated with distinct inflammatory profiles in serum, involving eotaxin-3, interleukin (IL)-6 and C-reactive protein (CRP); and in CSF, including soluble intracellular cell adhesion molecule-1 (sICAM-1), IL-8, 10-kDa interferon-γ-induced protein, and CRP. AD pathology interacted with CSF sICAM-1 in association with NPS. Presenting NPS was associated with subsequent cognitive decline which was mediated by CSF sICAM-1.
Conclusions
Distinct systemic and CNS inflammatory processes are involved in the pathophysiology of NPS in older people. Neuroinflammation may explain the link between NPS and more rapid clinical disease progression.
Similar content being viewed by others
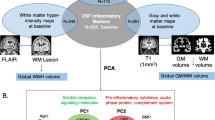


Background
Neuropsychiatric symptoms (NPS) are frequent in older people and may represent the clinical manifestation of a variety of acute and chronic cerebral pathologies. NPS are particularly common among patients with Alzheimer’s disease (AD) [1, 2], and may precede the clinical stages of dementia [3, 4] and mild cognitive impairment (MCI) [5]. In the context of AD, early occurring NPS increase clinical disease severity and caregiver burden, and have been associated with more rapid cognitive and functional decline [2, 5,6,7].
Increased neuroinflammatory activity and altered cytokine levels in the CNS have been related to psychiatric and behavioral syndromes. The activation of the central innate immune response by cytokines initiates a behavioral response known as sickness behavior [8] which is characterized by the co-occurrence of anxiety, depression, and apathy. In animal models, this increase in cytokine levels is also associated with increased neurodegeneration [9,10,11]. In humans, chronic inflammatory diseases and altered cytokine levels are associated with NPS such as depression, anxiety and apathy [12,13,14,15]. In older people, both systemic and CNS inflammation have been associated with NPS in general [16], and in relation to neurocognitive disorders in particular [17,18,19,20]. In the context of AD, neuroinflammatory markers present in peripheral blood may indicate cerebral pathology or systemic inflammation relevant for the core AD processes [19, 21] and be associated with disease severity [22]. However, the specific inflammatory markers associated with NPS and their possible contribution to the clinical course remain largely unknown.
In this study we analyzed a large panel of neuroinflammatory markers at systemic and CNS levels, and hypothesized that specific circulating blood and CSF neuroinflammatory marker profiles are associated with the occurrence of NPS in general and of individual syndromes such as depression, anxiety and apathy in older subjects with normal cognition or with beginning cognitive decline. A further objective was to determine how these inflammatory profiles change depending on the presence of cerebral AD pathology and/or cognitive impairment. Finally, we verified if the identified inflammatory signatures were associated with clinical disease progression at follow-up visits.
Methods
Study population
We enrolled 87 community dwelling individuals, aged 55 or older, from a brain aging study conducted in the Department of Psychiatry and the Department of Clinical Neurosciences, University Hospital of Lausanne, Switzerland. Cognitively impaired participants were recruited among memory clinic outpatients and displayed no major psychiatric or neurological disorders, nor substance abuse or severe or unstable physical illness that may contribute to cognitive impairment, including history or clinical signs of inflammation. All subjects in this group had clinical diagnoses of either MCI or mild dementia, and a Clinical Dementia Rating (CDR [23]) score ≥ 0.5 and displayed memory impairment and/or impairment in other cognitive domains such as executive tasks or language skills [24]. Cognitively healthy participants were recruited through journal announcements or word of mouth and had no history, symptoms or signs of relevant psychiatric or neurological disease and no cognitive impairment. An overall clinical, neurological and comprehensive neuropsychological assessment and the administration of informant questionnaires was performed for all participants as previously described [19]. Additional clinical and neuropsychological follow-up evaluations were performed at 18 and 36 months using the same methods and tests whenever possible.
Study procedures
Neuropsychological cognitive and neuropsychiatric assessments
The neuropsychological assessment and the administration of informant questionnaires to the participant’s proxy was previously described [19]. Briefly, cognitive performance was assessed with the Clinical Dementia Rating (CDR), CDR Sum of Boxes (CDR-SoB) and Mini-Mental State Examination (MMSE). The neuropsychological assessment also included the Buschke Double Memory Test [25], the Stroop Test [26], and the Trail Making Tests A and B [27]. Functional assessment included the activities of daily living (ADL) and instrumental ADL (IADL) tests [28]. All participants with CDR ≥ 0.5 were considered cognitively impaired and those with CDR = 0 as cognitively healthy. The Neuropsychiatric Inventory questionnaire (NPI-Q) [29] was administered to assess neuropsychiatric symptoms. Twelve categories, ten behavioral and two neurovegetative (Night-time behavior and Appetite/Eating), were scored for their severity ranging from 0 to 3. Total NPI-Q score was obtained by adding the 12 scores. Participants were considered to have NPS if the total NPI-Q score was > 0.
Biochemical sample collection and handling
Lumbar and venous punctures were conducted during the same visit between 8:30 and 10 a.m. after an overnight fast yielding 10–12 ml of CSF and 40 ml of blood, respectively, were performed, spun down at 4 °C, immediately aliquoted, and snap frozen at − 80 °C until assayed [19]. Study personnel blinded to clinical data performed biochemical and genetic analyses.
Biochemical measures
The CSF albumin index (Qalb) as a marker of blood–brain barrier (BBB) function along with the Apolipoprotein E (APOE) genotype were determined as previously described [30].
Cerebrospinal fluid AD biomarkers
CSF beta-amyloid 1–42 (Aβ1–42), total-tau (Tau), and tau phosphorylated at threonine 181 (pTau181) concentrations were measured using commercially available ELISA kits (Fujirebio, Gent, Belgium). A pTau181/Aβ1–42 ratio > 0.0779 was defined as an AD CSF profile as previously described [19]. Briefly, this value was determined using center data obtained from the study cohort and was the fitted value that that maximized the Youden index in a ROC analysis predicting CDR category [24].
Inflammation marker measurements
Quantitative analysis of 38 neuroinflammatory biomarkers in both CSF and serum was achieved using a sandwich immunoassay (Meso Scale Diagnostics, Rockville, USA) as previously described [30]. Biomarkers with more than 5% missing data or below level of quantification were filtered out, resulting in a selection of 28 analytes. These included basic fibroblast growth factor, C-reactive protein (CRP), eotaxin-1, eotaxin-3, interferon-γ (IFN-γ), interleukin-12, interleukin-15 (IL-15), interleukin-16 (IL-16), interleukin-6 (IL-6), interleukin-7, interleukin-8 (IL-8), 10-kDa interferon-γ-induced protein (IP-10), monocyte chemoattractant protein 1 (MCP-1), monocyte chemoattractant protein 4 (MCP-4), macrophage-derived chemokine (MDC), macrophage inflammatory protein 1α (MIP-1α), macrophage inflammatory protein 1β, phosphorylated insulin-like growth factor-1 receptor, serum amyloid A, soluble fms-like tyrosine kinase-1 (sFLT-1), soluble intercellular adhesion molecule-1 (sICAM-1), circulating vascular cell adhesion molecule-1 (sVCAM-1), thymus and activation-regulated chemokine (TARC), angiopoietin-1 receptor (TIE-2), tumor necrosis factor-α, vascular endothelial growth factor (VEGF), vascular endothelial growth factor C and vascular endothelial growth factor D precursor (VEGF-D). Prior to analysis, outliers within all measured markers exceeding the cutoff value of mean ± 3 × SD were replaced by the cutoff value. Neuroinflammatory marker measurements were also logn-transformed prior to correlation and regression analyses to approach Gaussian distribution. Normal distribution was subsequently assessed using the Kolmogorov–Smirnov test that identified 6 neuroinflammatory biomarkers in CSF (IL-7, MCP-4, MDC, MIP-1α, SAA, TARC) and 5 in serum (eotaxin-3, IL-6, MIP-1α, TIE-2, VEGF-C) not normally distributed.
Statistical and analytical approaches
Descriptive statistics for the cohort were performed using t-tests comparing participants with and without NPS for continuous variables and Chi-square tests for categorical variables. Correlations between NPI-Q total score and neuroinflammatory markers were assessed with Spearman’s rho. Independence of variables used in regression models was tested with variance inflation factor (VIF). No variable entered in these models had VIF above 7, with a majority below 3, demonstrating absence of multicollinearity and allowing regression models to be used. Benjamini–Hochberg correction of P-value for multiple testing was applied for all analyses using a false-discovery rate of 0.25. Statistical data analysis was performed with IBM SPSS Statistics software version 25. All statistical models performed in this study were verified for possible overfitting using the Hosmer–Lemeshow test for goodness-of-fit. Models with a Hosmer–Lemeshow Chi-squared value yielding a P-value > 0.05 were rejected and the previous iteration was considered instead.
Association between NPS and neuroinflammation markers
In order to identify neuroinflammatory marker combinations in CSF or serum associated with NPS, we used binary regression models with NPI-Q > 0 or NPI-Q = 0 as dependent variable while entering either all CSF or all serum markers in the models. We explored the effect of the following confounders: cognitive status (CDR = 0 or CDR > 0), CSF AD biomarker profiles (pTau181/Aβ1–42 ratio) and BBB function (Qalb) by entering them into the model before considering inflammatory marker concentrations. All models used a forward selection method based on the significance of the score statistic to select the smallest possible number of molecules associated with the occurrence of NPS in each case.
Associations of single neuropsychiatric syndromes with neuroinflammation markers
To determine inflammatory marker signatures associated with the presence of individual syndromes (NPI-Q > 0 for each individual category) linked to sickness behavior and associated with neuroinflammation in the literature, i.e., depression, apathy and anxiety, we used binary logistic regression models corrected for cognitive status (CDR = 0 or CDR > 0) as above. These models used a forward selection method based on the significance of the score statistic to select the smallest number of molecules associated with each individual symptom.
Interactions between cognitive status, AD pathology, and the associations of inflammatory markers with NPS
The interactions between an AD CSF profile, neuroinflammatory markers and the presence of NPS were tested by creating and interaction variable: pTau181/Aβ1–42 ratio × neuroinflammatory marker measurements. This was then used in a binary regression model with NPS occurrence as dependent variable. This approach tested whether a positive CSF biomarker profile for AD modified the effect of each individual neuroinflammatory on the occurrence of NPS.
Association of neuroinflammation markers with progression of disease severity
NPI-Q scores at baseline were correlated with progression of disease severity as measured by changes in CDR-SoB at follow-up clinical assessments at 18 and 36 months in the whole cohort and in participants with a positive NPI-Q score using Spearman rho’s. Linear regression was used to test associations between CSF neuroinflammatory markers associated with NPS, and cognitive decline computed by change in CDR-SoB over time (CDR-SoB difference at follow-up/time between baseline and follow-up) corrected for baseline CDR-SoB assessment. Finally, we tested the association of the interaction between NPI-Q total score and sICAM-1 on CDR-SoB change using a binary logistic regression model using cognitive decline at 18 or 36 months (0 = no CDR-SoB change; 1 = CDR-SoB change) as dependent variable. We entered NPI-Q total score, sICAM-1 concentration and the interaction between NPI-Q score and sICAM-1 concentration (NPI-Q score × sICAM-1 levels) as variables into this model.
Results
Characteristics of the cohort
Demographic, clinical, and biological characteristics of participants with or without NPS (NPI-Q = 0 or NPI-Q > 0) are given in Table 1. The frequency of anxiety, apathy and depression measured by the NPI-Q is presented in Additional file 1: Table S1, by group with or without NPS as well as for individuals with CDR = 0 and NPI-Q > 0. Anxiety was the most common symptom in cognitively healthy participants. Longitudinal distribution of cognitive status (CDR = 0 or CDR > 0) and the occurrence of NPS within the cohort are shown in Additional file 1: Table S2. In both cases, larger changes in distribution were observed after 36 months. In addition, 67% of the participants with NPS at baseline presented NPS after 18 months (90% after 36 months) suggesting a persisting phenotype. In the whole cohort, total NPI-Q score was positively correlated with both CDR and CDR-SoB and negatively correlated with MMSE (data not shown).
Neuroinflammatory signatures associated with the presence of NPS
The measured concentrations of all individual CSF and serum neuroinflammatory markers are shown in Additional file 1: Tables S3 and S4, respectively. We found 5 markers in CSF (IL-8, IL-15, sFLT-1, sICAM-1 and sVCAM-1, Additional file 1: Fig. S1A) and 1 in serum (MIP-1α, Additional file 1: Fig. S1B) displaying significantly different concentrations between participants with or without NPS. The concentrations of a similar set of molecules in CSF (sICAM-1, sVCAM-1, sFLT-1, IL-8 and IL-15, MCP-1 and MCP-4) showed positive correlations with total NPI-Q score in the whole cohort (Additional file 1: Fig. S1C). In serum, the concentration of MIP-1α showed a significant positive correlation with NPI-Q total score, while CRP and VEGF concentrations negatively correlated with NPI-Q total score (Additional file 1: Fig. S1D).
Using binary regression models, we identified a combination of four neuroinflammatory markers in CSF that best predicted the occurrence of NPS: CRP, IP-10, sICAM-1 and IL-8 (Fig. 1). This CSF neuroinflammatory signature of NPS was unchanged when considering either baseline cognitive status or the presence of an AD CSF profile. The pTau181/Aβ1–42 ratio was also associated with NPS in this context (β-coefficient = 1.626 and P-value = 0.013). Each of the selected individual CSF neuroinflammatory markers was also associated with the volume of a specific subset of brain regions (Additional file 1: Table S5). In serum, the best predicting combination was: Eotaxin-3, IL-6 and CRP (Fig. 1). When considering baseline cognitive status only CRP (β-coefficient = − 0.752 and P-value = 0.042) remained associated with the presence of NPS; and both CRP (β-coefficient = − 0.784 and P-value = 0.033), and VEGF-D (β-coefficient = − 2.22 and P-value = 0.044) were associated with the presence of NPS when considering the CSF AD biomarker profile. In parallel, we also investigated the effects of BBB function on these associations by correcting our model for Qalb. In this model, the CSF inflammatory markers associated with the occurrence of NPS were CRP, IP-10 and sICAM-1. In serum, MIP-1α, VEGF were associated with the occurrence of NPS (data not shown). When added to a reference model built using APOE status, sex, age, years of education and cognitive status at baseline (Additional file 1: Table S6); the identified CSF signature of sICAM-1, IP-10, IL-8 and CRP together significantly contributed to improve prediction of NPS (Additional file 1: Fig. S2, AUC = 0.908, P value = 0.0058).

Binary logistic regression models revealing associations between neuroinflammatory molecules and neuropsychiatric symptoms (NPS). Associations of CSF (left) and serum (right) neuroinflammatory marker concentrations with the occurrence of NPS. Individual β-coefficients for each significantly associated neuroinflammatory molecule are shown. *P value < 0.05; **P value < 0.01; ***P value < 0.001. CRP C-reactive protein, IP-10 10-kDa IFN-γ induced protein, sICAM-1 soluble intracellular adhesion molecule-1, IL-8 Interleukin-8, IL-6 Interleukin-6
Neuroinflammatory markers associated with single neuropsychiatric syndromes
We also identified specific CSF and serum neuroinflammatory markers associated with the occurrence of each depression, apathy and anxiety measured by NPI-Q (Fig. 2). In CSF, sICAM-1 was associated with the presence of all three symptoms and CRP was associated with the presence of anxiety only. In serum, we identified associations with the levels of IL-8, MIP-1β, TARC and VEGF-D, and the occurrence of depression.

Associations of CSF (left) and serum (right) neuroinflammatory markers with anxiety, depression and apathy. Standardized β-coefficients obtained by regression models are shown. *P value < 0.05; **P value < 0.01; ***P value < 0.001. CRP C-reactive protein, IP-10 10-kDa IFN-γ induced protein, sICAM-1 soluble intracellular adhesion molecule-1, IL-8 Interleukin-8, MIP-1β macrophage inflammatory protein 1β, TARC thymus and activation-regulated chemokine, VEGF-D, vascular endothelial growth factor (D precursor)
Interaction between neuroinflammation, NPS and AD pathology
Since the CSF concentrations of CRP, IP-10, sICAM-1, and a positive CSF biomarker profile for AD at baseline were associated with a positive NPI-Q score, we tested whether the interaction between neuroinflammation markers and a positive CSF biomarker profile for AD was also associated with NPS. Using binomial regression models with interaction variables (pTau181/Aβ1–42 ratio × individual neuroinflammatory markers), we found that only the interaction between the pTau181/Aβ1–42 ratio and CSF sICAM-1 was associated with the presence of NPS (Nagelkerke R2 = 0.363; coefficient = 3.386; P value = 0.027).
NPS, neuroinflammation and progression of clinical disease severity
NPI-Q at baseline was correlated with clinical severity progression as measured by changes in CDR-SoB at follow-up clinical assessments at 18 and 36 months (n = 87 and 85, respectively, Fig. 3A) in the whole cohort and in participants with a positive NPI-Q score. We also tested in a linear regression model the associations between the concentrations of molecules identified in the CSF signature in all models and disease severity progression. Only sICAM-1 was positively associated with disease severity progression at 18 and 36 months (Fig. 3B). A moderation model testing the interaction between NPI-Q score and sICAM-1 concentration revealed that the association between NPI-Q and CDR-SoB change at 18 months is affected by sICAM-1 concentration directly (Fig. 3C).

Associations of neuroinflammatory markers with progression of disease severity. A Correlations between NPI-Q total score at baseline and disease severity progression as measured by change in CDR-SoB (ΔCDR-SoB) over time. B Associations between neuroinflammatory markers and cognitive status at baseline and changes of disease severity in the whole cohort measured by change in CDR-SoB (ΔCDR-SoB) at 18 and 36 months. Standardized β-coefficients and obtained by linear regression models are shown. C Moderation model evaluating the contribution of NPI-Q score, CSF sICAM-1 at baseline and their interaction to disease severity progression at 18 and 36 months (ΔCDR-SoB). β-Coefficients obtained by binary logistic regression models are shown. CDR-SoB, Clinical Dementia Rating Sum of Boxes; CRP, C-reactive protein; sICAM-1, soluble intracellular adhesion molecule-1; IP-10, 10-kDa IFN-γ induced protein; NPI-Q, Neuropsychiatric Inventory Questionnaire score; Inter., Interaction Between NPI-Q score and sICAM-1 concentration (NPI-Q score × sICAM-1 levels)
Discussion
Assessing a large panel of neuroinflammation and vascular injury markers in paired blood and CSF samples we identified distinct systemic and CNS inflammatory signatures associated with the presence of neuropsychiatric symptoms, including altered levels of eotaxin-3, IL-6 and CRP in serum, and sICAM-1, IP-10, IL-8 and CRP in CSF. While NPS were more common in participants with cognitive impairment, the associations between CSF inflammation markers and NPS were independent of the presence of cognitive impairment at baseline. A significant interaction with AD pathology was observed for the association of NPS with CSF sICAM-1 only. Finally, the presence of NPS at baseline was associated with more rapid cognitive decline at follow-up visits, and this association was mediated by increased sICAM-1 in CSF.
While several CSF inflammation marker changes were associated with NPS, a combination of sICAM-1, IP-10, IL-8 and CRP levels best predicted the occurrence of NPS, with these molecules being independently associated with the NPS phenotype. This finding is in accordance with the concept that cytokines form a complex interplaying network enhancing and/or suppressing the production of each other, and together associating with clinical manifestation [31]. In the context of cognitive decline, sICAM-1 and CRP have previously been associated with AD pathology [19, 32]. IP-10 levels have been reported to be normal in AD [33], but have been associated with NPS and in particular with depression in one study [34]. CSF levels of IL-8 have previously been associated with both delirium and anxiety [35, 36]. A previous study has associated CRP levels with NPS [37]. CRP increases in the acute phase of immune response, but can also have a protective role by regulating inflammatory reactions via action on the complement system [38]. Taken together, the molecular signature of NPS we have identified is in line with most previous reports on single markers. The CSF signature of NPS was independent of the presence of cognitive impairment indicating that CNS neuroinflammation leading to NPS may also occur in the absence, or before the onset of cognitive decline. Furthermore, this signature was not dependent on BBB integrity, suggesting that the related inflammatory process originates within the CNS.
In serum, eotaxin-3, IL-6 and CRP levels together best predicted the presence of NPS. Very few previous studies have investigated, in older people in the context of cognitive decline or AD, associations of NPS with inflammation mediators from circulating blood. High serum concentrations of IL-6 and TNFα have been associated with the occurrence of depression and anxiety in patients with clinically diagnosed AD dementia [39]. Circulating CRP levels have also been associated with increased depression scores in an elderly non-demented population [40]. However, there is also conflicting evidence showing no association of CRP with depression in elderly people with AD-type dementia [39]. To our knowledge, no previous studies have linked serum eotaxin-3 levels with NPS. The serum inflammatory marker signature seems to depend on both BBB integrity and the presence of an AD CSF profile suggesting that both contribute to the association between systemic inflammation association and NPS. Previous studies have reported associations between inflammation mediators in circulating blood and atrophy in specific brain regions [41,42,43], suggesting neuroinflammation-driven neurodegeneration within these individual regions. A possible explanation may be that following BBB breakdown related to aging [44] or AD pathology [30], a cross-talk may occur between inflammation in the CNS and systemic inflammatory processes [45]. Systemic inflammation may contribute to CNS inflammation which may further result in cerebral dysfunction and neuronal injury, and the clinical manifestation of both cognitive impairment and NPS [18]. This was suggested for example for circulating IL-6 which may increase the concentration of sICAM-1 in the CNS, resulting in BBB damage and increased glial activation [46, 47]. On the other hand, systemic inflammation may also reflect cerebral pathology [21] and neuroinflammation related to core AD pathology [19].
This study extends previous evidence of associations between single CSF inflammatory molecules and depression and anxiety [35, 48,49,50] and reports evidence on apathy. Specifically, we observed a positive association between CSF sICAM-1 levels and the occurrence of anxiety, depression, and apathy. This suggests a role for this neuroinflammatory marker in the manifestation of these symptoms. Indeed, an association between sICAM-1 and depression has previously been reported [51]. On the other hand, some of the inflammatory molecules we investigated were specifically associated with a single symptom. In CSF, CRP was associated only with anxiety while in serum, IL-8, MIP-1α, TARC and VEGF-D were associated only with depression. This indicates distinct neuroinflammatory profiles for these syndromes. Some of these associations have previously been reported, such as serum IL-8 with anxiety [35]. Overall, the associations between neuroinflammation and individual syndromes observed in our study suggest that while in general neuroinflammation can result in a wide spectrum of neuropsychiatric manifestations, specific neuroinflammatory mediators could play a more important role in specific syndromes.
In the present study, the observed association between CSF neuroinflammatory markers and NPS could be driven by the large proportion of participants with AD pathology in the subgroup with NPS. However, when we accounted for the presence of cerebral core AD pathology in our models, the associations between CSF inflammatory markers and NPS was not altered. This indicates the CSF inflammatory signature of NPS is at least partially independent of AD. This is clinically relevant, as these inflammatory processes may cause NPS also in the absence of AD pathology, and therefore may be considered a distinct pathophysiological aspect from AD.
Baseline NPI-Q score was correlated with progression in the clinical severity at follow-up visits, in line with previous observations [2, 5, 7]. Also, higher CSF sICAM-1 concentration was associated with more rapid progression of disease severity, i.e., more rapid cognitive and related functional decline. Furthermore, the interaction analysis showed that the association between the presence of NPS at baseline and clinical disease progression is mediated by CSF sICAM-1 levels. Together with the observed CSF sICAM-1 in associations with all single neuropsychiatric syndromes, this finding suggests a key role of sICAM-1 for the pathogenesis of NPS and the association of NPS with clinical disease progression.
sICAM-1 was further associated with the volume of the hippocampus and 3rd ventricle (reflecting atrophy of surrounding areas). These regions have been associated with both neurodegeneration in AD and inflammation in previous works [52, 53]. In addition, CSF sICAM-1 was previously found to correlate with measures of both neurodegeneration and tau hyperphosphorylation in the CSF [19]. Together, this suggests that regional neurodegeneration and related neuroinflammation with increased sICAM-1 levels represent underlying pathology of the occurring NPS, in particular of anxiety, depression and apathy. Considering previous studies showing that brain regions associated with NPS [54] are also associated with more rapid disease progression in AD [55], our findings suggest that the interaction between sICAM-1 driven neuroinflammation and AD pathology may occur within the hippocampus and the areas surrounding the 3rd ventricle. Therefore, increased sICAM-1 related inflammation in the CNS appears to be both involved in neuronal injury and tau pathology and contribute to NPS, and in particular in the context of AD (Fig. 4).

Graphical representation of the role of CNS neuroinflammation, in NPS, functional cognitive decline and regional brain atrophy. Significant associations or interactions are represented by arrows with standardized coefficients indicated alongside. The dashed arrow represents the interaction of sICAM-1 on the association between AD pathology and NPS. The dotted line represents an association demonstrated in a previous study [19]. Associations between neuroinflammation and NPS are described in detail in Fig. 1 and are independent of BBB function and cognitive status. *P value < 0.05; **P value < 0.01; ***P value < 0.001; C-reactive protein; IP-10, 10-kDa IFN-γ induced protein; sICAM-1, soluble intracellular adhesion molecule-1; A, correlation coefficient
This is to our knowledge the first study to assess a large panel of inflammatory markers in paired serum and CSF samples to determine inflammatory signatures of NPS in older people while considering cognitive status and biomarkers of AD pathology. The size of our cohort does not allow for stratification into subgroups according to the A/T/(N) classification framework, limiting further exploration of the relationship between the identified neuroinflammatory marker profiles and AD pathology and how neuroinflammation could play differential roles in different pathological presentations. A further limitation of this study is related to the fact that subjects included in this cohort had no psychiatric disorder or severe psychiatric symptoms that could interfere with cognition, and that candidates with more marked neuropsychiatric symptoms were not considered. However, both elderly subjects with normal cognition and with early stages of cognitive decline were considered, allowing to evaluate the relevance for clinical disease progression over time. Apathy had a relatively low frequency in our cohort and some relationships with this syndrome could have been missed. Furthermore, we did not include subjects with more advanced stages of dementia, where the pathogenesis of NPS may differ from early stages of cognitive decline. Whether the identified signatures can be used in clinical practice as markers of neuroinflammation-related NPS and as targets for intervention needs to be further investigated in independent samples.
Conclusions
We have identified specific CSF and serum inflammatory signatures associated with NPS that indicate distinct contributions of systemic and CNS inflammation to the pathophysiology of NPS. This study is the first to relate inflammation markers to NPS in older people with normal cognition or with cognitive impairment while considering AD pathology as indicated by CSF biomarkers. While the CSF signature appears to indicate inflammatory processes that originate within the CNS and are independent of the core AD pathology, the serum signature may represent systemic inflammatory activity which may contribute to pathological processes in the CNS that clinically manifest as NPS. The inflammatory signatures of NPS indicate syndrome-specific underlying processes opening the perspective of targeted interventions to reduce NPS and their long-term consequences.
Availability of data
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- Aβ1–42:
-
Beta-amyloid 1–42
- AD:
-
Alzheimer’s disease
- AUC:
-
Area under the curve
- BBB:
-
Blood–brain barrier
- CDR:
-
Clinical Dementia Rating
- CDR-SoB:
-
CDR Sum of Boxes
- CRP:
-
C-reactive protein
- CSF:
-
Cerebrospinal fluid
- CNS:
-
Central nervous system
- IFN-γ:
-
Interferon-γ
- IL:
-
Interleukin
- IP-10:
-
10-KDa interferon-γ-induced protein
- MCP-1,4:
-
Monocyte chemoattractant protein 1,4
- MDC:
-
Macrophage-derived chemokine
- MIP-1α:
-
Macrophage inflammatory protein 1α
- MMSE:
-
Mini-Mental State Examination
- NPI-Q:
-
Neuropsychiatric Inventory Questionnaire
- NPS:
-
Neuropsychiatric symptoms
- pTau181:
-
Tau phosphorylated at threonine 181
- Qalb:
-
CSF albumin index
- ROC:
-
Receiver operating characteristic
- SAA:
-
Serum amyloid A
- sFLT-1:
-
Soluble fms-like tyrosine kinase-1
- sICAM-1:
-
Soluble intracellular cell adhesion molecule-1
- TARC:
-
Thymus and activation-regulated chemokine
- Tau:
-
Total-tau
- TIE-2:
-
Angiopoietin-1 receptor
- VEGF:
-
Vascular endothelial growth factor
- VEGF-D:
-
Vascular endothelial growth factor D precursor
- VIF:
-
Variance inflation factor
References
Youn JC, Lee DY, Jhoo JH, Kim KW, Choo IH, Woo JI. Prevalence of neuropsychiatric syndromes in Alzheimer’s disease (AD). Arch Gerontol Geriatr. 2011;52(3):258–63.
Rozum WJ, Cooley B, Vernon E, Matyi J, Tschanz JT. Neuropsychiatric symptoms in severe dementia: associations with specific cognitive domains the Cache County Dementia Progression Study. Int J Geriatr Psychiatry. 2019;34:1087–94.
Wise EA, Rosenberg PB, Lyketsos CG, Leoutsakos JM. Time course of neuropsychiatric symptoms and cognitive diagnosis in National Alzheimer’s Coordinating Centers volunteers. Alzheimer’s Dement (Amsterdam, Netherlands). 2019;11:333–9.
Liew TM. Neuropsychiatric symptoms in early stage of Alzheimer’s and non-Alzheimer’s dementia, and the risk of progression to severe dementia. Age Ageing. 2021;50(5):1709–18.
Burhanullah MH, Tschanz JT, Peters ME, Leoutsakos JM, Matyi J, Lyketsos CG, et al. Neuropsychiatric symptoms as risk factors for cognitive decline in clinically normal older adults: the cache county study. Am J Geriatr Psychiatry. 2019;28:64–71.
Missotten P, Squelard G, Ylieff M, Di Notte D, Paquay L, de Lepeleire J, et al. Relationship between quality of life and cognitive decline in dementia. Dement Geriatr Cogn Disord. 2008;25(6):564–72.
Vetrano DL, Rizzuto D, Calderon-Larranaga A, Onder G, Welmer AK, Bernabei R, et al. Trajectories of functional decline in older adults with neuropsychiatric and cardiovascular multimorbidity: a Swedish cohort study. PLoS Med. 2018;15(3):e1002503.
Dantzer R. Cytokine, sickness behavior, and depression. Immunol Allergy Clin N Am. 2009;29(2):247–64.
Combrinck MI, Perry VH, Cunningham C. Peripheral infection evokes exaggerated sickness behaviour in pre-clinical murine prion disease. Neuroscience. 2002;112(1):7–11.
Cunningham C, Wilcockson DC, Campion S, Lunnon K, Perry VH. Central and systemic endotoxin challenges exacerbate the local inflammatory response and increase neuronal death during chronic neurodegeneration. J Neurosci. 2005;25(40):9275–84.
Cunningham C, Campion S, Lunnon K, Murray CL, Woods JF, Deacon RM, et al. Systemic inflammation induces acute behavioral and cognitive changes and accelerates neurodegenerative disease. Biol Psychiatry. 2009;65(4):304–12.
Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9(1):46–56.
Rengasamy M, Zhong Y, Marsland A, Chen K, Douaihy A, Brent D, et al. Signaling networks in inflammatory pathways and risk for suicidal behavior. Brain Behav Immunity Health. 2020;7:100122.
Irving P, Barrett K, Nijher M, Lusignan SD. Prevalence of depression and anxiety in people with inflammatory bowel disease and associated healthcare use: population-based cohort study. Evid Based Ment Health. 2021;24:102–9.
Obermanns J, Krawczyk E, Juckel G, Emons B. Analysis of cytokine levels, T regulatory cells and serotonin content in patients with a depressive disorder. Eur J Neurosci. 2021;53:3476–89.
Capuron L, Castanon N. Role of inflammation in the development of neuropsychiatric symptom domains: evidence and mechanisms. Curr Top Behav Neurosci. 2017;31:31–44.
Holmes C, Cunningham C, Zotova E, Woolford J, Dean C, Kerr S, et al. Systemic inflammation and disease progression in Alzheimer disease. Neurology. 2009;73(10):768–74.
Popp J. Delirium and cognitive decline: more than a coincidence. Curr Opin Neurol. 2013;26(6):634–9.
Popp J, Oikonomidi A, Tautvydaite D, Dayon L, Bacher M, Migliavacca E, et al. Markers of neuroinflammation associated with Alzheimer’s disease pathology in older adults. Brain Behav Immun. 2017;62:203–11.
Amin J, Boche D, Clough Z, Teeling J, Williams A, Gao Y, et al. Peripheral immunophenotype in dementia with Lewy bodies and Alzheimer’s disease: an observational clinical study. J Neurol Neurosurg Psychiatry. 2020;91(11):1219–26.
Morgan AR, Touchard S, Leckey C, O’Hagan C, Nevado-Holgado AJ, Barkhof F, et al. Inflammatory biomarkers in Alzheimer’s disease plasma. Alzheimer’s Dement. 2019;15:776–87.
Licastro F, Pedrini S, Caputo L, Annoni G, Davis LJ, Ferri C, et al. Increased plasma levels of interleukin-1, interleukin-6 and alpha-1-antichymotrypsin in patients with Alzheimer’s disease: peripheral inflammation or signals from the brain? J Neuroimmunol. 2000;103(1):97–102.
Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology. 1993;43(11):2412–4.
Mathys J, Gholamrezaee M, Henry H, von Gunten A, Popp J. Decreasing body mass index is associated with cerebrospinal fluid markers of Alzheimer’s pathology in MCI and mild dementia. Exp Gerontol. 2017;100:45–53.
Buschke H, Sliwinski MJ, Kuslansky G, Lipton RB. Diagnosis of early dementia by the Double Memory Test: encoding specificity improves diagnostic sensitivity and specificity. Neurology. 1997;48(4):989–97.
Stroop JR. Studies of interference in serial verbal reactions. J Exp Psychol. 1935;18(6):643–62.
Reitan RM. Validity of the trail making test as an indicator of organic brain damage. Percept Mot Skills. 1958;8(3):271–6.
Lawton MP, Brody EM. Assessment of older people: self-maintaining and instrumental activities of daily living. Gerontologist. 1969;9(3 Part 1):179–86.
Cummings JL, Mega M, Gray K, Rosenberg-Thompson S, Carusi DA, Gornbein J. The Neuropsychiatric Inventory: comprehensive assessment of psychopathology in dementia. Neurology. 1994;44(12):2308–14.
Bowman GL, Dayon L, Kirkland R, Wojcik J, Peyratout G, Severin IC, et al. Blood-brain barrier breakdown, neuroinflammation, and cognitive decline in older adults. Alzheimer’s Dement. 2018;14(12):1640–50.
Becher B, Spath S, Goverman J. Cytokine networks in neuroinflammation. Nat Rev Immunol. 2017;17(1):49–59.
Ray S, Britschgi M, Herbert C, Takeda-Uchimura Y, Boxer A, Blennow K, et al. Classification and prediction of clinical Alzheimer’s diagnosis based on plasma signaling proteins. Nat Med. 2007;13(11):1359–62.
Janelidze S, Mattsson N, Stomrud E, Lindberg O, Palmqvist S, Zetterberg H, et al. CSF biomarkers of neuroinflammation and cerebrovascular dysfunction in early Alzheimer disease. Neurology. 2018;91(9):e867–77.
Rivera-Rivera Y, García Y, Toro V, Cappas N, López P, Yamamura Y, et al. Depression correlates with increased plasma levels of inflammatory cytokines and a dysregulated oxidant/antioxidant balance in HIV-1-infected subjects undergoing antiretroviral therapy. J Clin Cell Immunol. 2014;5(6):1000276.
Janelidze S, Suchankova P, Ekman A, Erhardt S, Sellgren C, Samuelsson M, et al. Low IL-8 is associated with anxiety in suicidal patients: genetic variation and decreased protein levels. Acta Psychiatr Scand. 2015;131(4):269–78.
Sajjad MU, Blennow K, Knapskog AB, Idland A-V, Chaudhry FA, Wyller TB, et al. Cerebrospinal fluid levels of interleukin-8 in delirium, dementia, and cognitively healthy patients. J Alzheimers Dis. 2020;73(4):1363–72.
Capuron L, Schroecksnadel S, Féart C, Aubert A, Higueret D, Barberger-Gateau P, et al. Chronic low-grade inflammation in elderly persons is associated with altered tryptophan and tyrosine metabolism: role in neuropsychiatric symptoms. Biol Psychiatry. 2011;70(2):175–82.
Bíró A, Rovó Z, Papp D, Cervenak L, Varga L, Füst G, et al. Studies on the interactions between C-reactive protein and complement proteins. Immunology. 2007;121(1):40–50.
Holmes C, Cunningham C, Zotova E, Culliford D, Perry VH. Proinflammatory cytokines, sickness behavior, and Alzheimer disease. Neurology. 2011;77(3):212–8.
van den Biggelaar AHJ, Gussekloo J, de Craen AJM, Frölich M, Stek ML, van der Mast RC, et al. Inflammation and interleukin-1 signaling network contribute to depressive symptoms but not cognitive decline in old age. Exp Gerontol. 2007;42(7):693–701.
Han JH, Wong KS, Wang YY, Fu JH, Ding D, Hong Z. Plasma level of sICAM-1 is associated with the extent of white matter lesion among asymptomatic elderly subjects. Clin Neurol Neurosurg. 2009;111(10):847–51.
Satizabal CL, Zhu YC, Mazoyer B, Dufouil C, Tzourio C. Circulating IL-6 and CRP are associated with MRI findings in the elderly: the 3C-Dijon Study. Neurology. 2012;78(10):720–7.
Gongvatana A, Correia S, Dunsiger S, Gauthier L, Devlin KN, Ross S, et al. Plasma cytokine levels are related to brain volumes in HIV-infected individuals. J Neuroimmune Pharmacol. 2014;9(5):740–50.
Yousef H, Czupalla CJ, Lee D, Chen MB, Burke AN, Zera KA, et al. Aged blood impairs hippocampal neural precursor activity and activates microglia via brain endothelial cell VCAM1. Nat Med. 2019;25(6):988–1000.
Dayon L, Cominetti O, Wojcik J, Galindo AN, Oikonomidi A, Henry H, et al. Proteomes of paired human cerebrospinal fluid and plasma: relation to blood-brain barrier permeability in older adults. J Proteome Res. 2019;18(3):1162–74.
Miguel-Hidalgo JJ, Nithuairisg S, Stockmeier C, Rajkowska G. Distribution of ICAM-1 immunoreactivity during aging in the human orbitofrontal cortex. Brain Behav Immun. 2007;21(1):100–11.
Closhen D, Bender B, Luhmann HJ, Kuhlmann CR. CRP-induced levels of oxidative stress are higher in brain than aortic endothelial cells. Cytokine. 2010;50(2):117–20.
Wang AK, Miller BJ. Meta-analysis of cerebrospinal fluid cytokine and tryptophan catabolite alterations in psychiatric patients: comparisons between schizophrenia, bipolar disorder, and depression. Schizophr Bull. 2018;44(1):75–83.
Enache D, Pariante CM, Mondelli V. Markers of central inflammation in major depressive disorder: a systematic review and meta-analysis of studies examining cerebrospinal fluid, positron emission tomography and post-mortem brain tissue. Brain Behav Immun. 2019;81:24–40.
Felger JC, Haroon E, Patel TA, Goldsmith DR, Wommack EC, Woolwine BJ, et al. What does plasma CRP tell us about peripheral and central inflammation in depression? Mol Psychiatry. 2020;25(6):1301–11.
van Dooren FE, Schram MT, Schalkwijk CG, Stehouwer CD, Henry RM, Dagnelie PC, et al. Associations of low grade inflammation and endothelial dysfunction with depression—The Maastricht Study. Brain Behav Immun. 2016;56:390–6.
Barron AM, Tokunaga M, Zhang MR, Ji B, Suhara T, Higuchi M. Assessment of neuroinflammation in a mouse model of obesity and beta-amyloidosis using PET. J Neuroinflamm. 2016;13(1):221.
Bartos A, Gregus D, Ibrahim I, Tintera J. Brain volumes and their ratios in Alzheimer s disease on magnetic resonance imaging segmented using Freesurfer 6.0. Psychiatry Res Neuroimaging. 2019;287:70–4.
Boublay N, Schott AM, Krolak-Salmon P. Neuroimaging correlates of neuropsychiatric symptoms in Alzheimer’s disease: a review of 20 years of research. Eur J Neurol. 2016;23(10):1500–9.
Whitwell JL. Progression of atrophy in Alzheimer’s disease and related disorders. Neurotox Res. 2010;18(3–4):339–46.
Acknowledgements
The authors wish to thank Richard Kirkland from Prometheus Laboratories for performing measurements and Dr. Cornelia Pocnet and Dr. Leonardo Zullo for their comments and critical assessment of the manuscript.
Funding
This work was supported by grants from the Swiss National Research Foundation (SNF 320030_141179), Synapsis Foundation—Alzheimer Research Switzerland (Grant number 2017-PI01) and funding from the Nestlé Institute of Health Sciences.
Author information
Authors and Affiliations
Contributions
CC and JP contributed to study design and wrote the article. CC performed data modeling, statistical analyses, data interpretation and made figures. JR and BM provided regional volumetric data. GB and LD provided neuroinflammatory biomarker assessment and critical review of the manuscript. JP supervised and coordinated the study, contributed to writing the article and provided data interpretation and a critical review of the whole manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The local ethics committee of canton Vaud (Switzerland) approved this study (No. 171/2013), and all participants or their legal representatives provided written informed consent.
Consent for publication
This work is original, has not already been published in a journal, and is not currently under consideration by another journal. The authors agree to the terms of the BioMed Central Copyright and License Agreement and gave consent for publication in the Journal of Neuroinflammation.
Competing interests
JP received consultation honoraria from the Nestlé Institute of Health Sciences, Ono Pharma, OM Pharma, and from Fujirebio Europe. LD is an employee of the Société des Produits Nestlé SA The other authors declare no potential conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Neuropsychiatric symptom distribution within the study cohort. Table S2. Longitudinal distribution of cognitive and neuropsychiatric status within the cohort. Table S3. Concentrations (in pg/ml) of neuroinflammatory markers in CSF. Table S4. Concentrations (in pg/ml) of neuroinflammatory markers in serum. Table S5. Associations of volumetric data with neuroinflammatory marker concentration. Table S6. Reference model. Figure S1. Concentrations and correlations of neuroinflammatory markers with NPI-Q score in the whole cohort. Figure S2. Predictive model of the presence of neuropsychiatric symptoms.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Clark, C., Richiardi, J., Maréchal, B. et al. Systemic and central nervous system neuroinflammatory signatures of neuropsychiatric symptoms and related cognitive decline in older people. J Neuroinflammation 19, 127 (2022). https://doi.org/10.1186/s12974-022-02473-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12974-022-02473-3