
Abstract
Inflammation may contribute to multiple brain pathologies. One cause of inflammation is lipopolysaccharide/endotoxin (LPS), the levels of which are elevated in blood and/or brain during bacterial infections, gut dysfunction and neurodegenerative diseases, such as Parkinson’s disease. How inflammation causes neuronal loss is unclear, but one potential mechanism is microglial phagocytosis of neurons, which is dependent on the microglial P2Y6 receptor. We investigated here whether the P2Y6 receptor was required for inflammatory neuronal loss. Intraperitoneal injection of LPS on 4 successive days resulted in specific loss of dopaminergic neurons (measured as cells staining with tyrosine hydroxylase or NeuN) in the substantia nigra of wild-type mice, but no neuronal loss in cortex or hippocampus. This supports the hypothesis that neuronal loss in Parkinson’s disease may be driven by peripheral LPS. By contrast, there was no LPS-induced neuronal loss in P2Y6 receptor knockout mice. In vitro, LPS-induced microglial phagocytosis of cells was prevented by inhibition of the P2Y6 receptor, and LPS-induced neuronal loss was reduced in mixed glial–neuronal cultures from P2Y6 receptor knockout mice. This supports the hypothesis that microglial phagocytosis contributes to inflammatory neuronal loss, and can be prevented by blocking the P2Y6 receptor, suggesting that P2Y6 receptor antagonists might be used to prevent inflammatory neuronal loss in Parkinson’s disease and other brain pathologies involving inflammatory neuronal loss.
Similar content being viewed by others


Background
Inflammation contributes to the pathology and neuronal loss of various CNS diseases [1,2,3,4], but the mechanisms are unclear, and thus means to prevent inflammatory neuronal loss are underdeveloped. The inflammatory response of the brain is mainly mediated by microglia, which are resident brain macrophages that when inflamed can kill neurons by multiple mechanisms, including by phagocytosing neurons [1,2,3]. Dead neurons do not accumulate significantly in neurodegenerative diseases, and one potential cause of this is that neurons are phagocytosed when alive [5, 6]. Phagocytosis of live cells normally results in death of the engulfed cells, a type of cell death termed phagoptosis, i.e., cell death by phagocytosis [7]. We have shown that activated microglia become highly phagocytic and cause neuronal loss by phagocytosis of stressed-but-viable neurons [8,9,10,11].
One cause of inflammation is lipopolysaccharide/endotoxin (LPS), derived from the cell wall of gram-negative bacteria, which activates inflammation via Toll-like receptor 4 (TLR4) on immune cells, such as microglia [12]. A variety of conditions, such as liver cirrhosis, gingivitis, sepsis and Alzheimer’s disease elevate plasma LPS levels, and eventually cause neurodegeneration [12]. One cause of elevated LPS is infection with gram-negative bacteria, and bacterial infections are common in patients with neurodegenerative diseases and can both exacerbate clinical symptoms and accelerate disease progression [13, 14]. Recently, it was reported that plasma LPS levels were substantially elevated in a subset of Parkinson’s disease patients, particularly those at high risk of dementia [15]. Gut dysfunction is one of the earliest symptoms of Parkinson’s disease, and may result in translocation of LPS from the gut (where levels are very high) into the blood (where levels are normally very low) [12, 15, 16]. Addition of LPS (at levels found in disease) to the blood of healthy volunteers results in rapid body and brain inflammation with microglial activation [17]. This all supports the hypothesis that LPS and microglial activation contribute to neurodegeneration [12], but further evidence and mechanisms are required before potential treatments can be devised.
Microglial phagocytosis of neurons requires the P2Y6 receptor (P2Y6R), encoded by the P2ry6 gene, and activated by extracellular uridine diphosphate (UDP) [18, 19]. In the brain, P2Y6R is almost exclusively expressed by microglia [18, 20, 21]. The human P2RY6 gene is highly homologous to the mouse gene [22, 23], and GWAS data indicate that variants of the human gene are associated with cognitive performance [24], consistent with a role in brain development or neuroprotection. P2ry6 gene expression levels were elevated in monocytes from Parkinson’s disease patients, and expression was increased in a microglial cell line treated with LPS [25]. Microglial P2Y6R has been found to mediate microglial phagocytosis of kainite-treated neurons, which released UTP/UDP that activated P2Y6R on microglia, inducing engulfment of these neurons [18]. However, in that work, it was assumed that the neurons releasing UDP were dying, and it was not determined whether inhibiting P2Y6R prevented neuronal loss [18]. We found that an inhibitor of P2Y6R (MRS2578) prevented neuronal loss induced by LPS injected into brain or cell cultures [9], but we did not test whether: (i) this was mediated by P2Y6R, and (ii) lack of P2Y6R was protective against peripheral LPS. The work described here seeks to determine whether the inflammatory neuronal loss induced by peripheral LPS can be prevented by blocking the microglial P2Y6 receptor. The work shows that peripheral LPS specifically induces neuronal loss in the substantia nigra, and this neuronal loss is fully prevented by P2ry6 knockout. This suggests that microglial phagocytosis contributes to inflammatory neuronal loss, and that blocking P2Y6R might be neuroprotective in Parkinson’s disease.
Methods
Animals
P2ry6 knockout (P2ry6−/−) mice were kindly provided by Bernard Robaye (ULB Brussels) and maintained on a C57Bl/6 background (Charles River Laboratories). P2ry6−/− mice and wild-type (WT) littermates were used to establish homozygous WT and P2ry6−/− sub-lines. In offspring from these sub-lines, littermates were randomly assigned to control and LPS treatment groups. Details of experimental animals used for each study are given in Table 1.
Intraperitoneal injection of LPS
LPS from Salmonella abortus equi (S-form; Enzo Life Sciences) or PBS (control) was injected intraperitoneally at a dose of 1 µg/g/day in a total volume of 100 µl at the same time of day on 4 subsequent days. Animals were sacrificed 14 days after the final LPS injection.
Transcardial perfusion and tissue sectioning
Mice were given terminal anaesthesia (150 µl Euthatal i.p.) and, once unresponsive to pain, perfused transcardially, through a 25-gauge needle, with 20 ml PBS, pH 7.4 followed by 60 ml 4% PFA, pH 7.4 using a perfusion pump with flow rate of 4 ml/min. Following perfusion, brains were removed and post-fixed overnight in the same solution, and cryoprotected by immersion in 10–30% sucrose solution until sectioning. Brain sections were cut to 20 µm thickness using a Compresstome VF-200 vibratome (Precisionary Instruments), collected on Superfrost Plus slides (Thermo Fisher) and dried overnight. Serial coronal sections (25 µm) through the whole brain were collected using a sliding microtome and placed in PBS as free-floating sections.
Immunostaining of brain slices
All steps were carried out at room temperature unless indicated otherwise. Brain slices were re-hydrated for 1 h in PBS and heat-mediated antigen retrieval was carried out at 95 °C for 20 min in citrate buffer (10 mM sodium citrate, 0.05% Tween 20, pH 6.0). Following washes in PBS (6 × 10 min), slices were permeabilized in PBS with 0.5% Triton X-100 for 10 min followed by 1 h incubation in blocking solution (20% normal goat serum in PBS). Slices were then incubated in primary antibody solution (5% normal goat serum in PBS plus appropriate primary antibody) at 4 °C overnight. Following washes in PBS (6 × 10 min), slices were incubated with secondary antibody for 2 h, washed (6 × 10 min, PBS) and mounted using Vectashield mounting medium with DAPI (Vector Laboratories). Primary antibodies used were Anti-NeuN (Millipore, mouse monoclonal, 1:500 dilution), anti-IbaI (Wako, rabbit polyclonal, 1:500 dilution) and anti-Tyrosine Hydroxylase (Merck, rabbit polyclonal, 1:500 dilution). Secondary antibodies were Alexa-Fluor 488 anti-mouse, Alexa-Fluor 568 anti-rabbit and Alexa-Fluor 633 anti-rabbit (all ThermoFisher, goat, 1:1000 dilution). Imaging was carried out on an Olympus FV1000 upright laser-scanning confocal microscope with a 60×, 1.35 NA oil immersion objective using 488, 559 and 635 nm laser lines.
Image analysis using ImageJ 1.49 software
All image analysis was carried out using ImageJ 1.49 software. All manual counting and quantification was performed blinded to genotype and treatment condition. Neuronal numbers were quantified for the CA1 and CA3 regions of the hippocampus as well as the outer layer of the motor cortex. Neuronal density within these regions was quantified by counting of neuronal cell bodies within areas of fixed size. CA1 and CA3 regions were ‘straightened’ in Fiji [26], allowing rectangles of constant size to be placed at the same position relative to identifiable brain structures. CA1 and CA3 width was determined at three fixed points along their length using line intensity profiles of the NeuN stain. Four brain sections were analyzed per animal, with both right and left sides of the hippocampus included in the analysis. For quantification of neuronal density in motor cortex, regions of interest of fixed size were placed randomly in anatomically matched sections and NeuN+ cells counted manually. Three areas were counted on each side of the midline for a total of four sections per animal. For quantification of cell densities (neuron and microglia) in SN, left and right sides of four anatomically matched sections were analyzed for each animal. Dopaminergic cells were counted as tyrosine-hydroxylase-positive cells in the entire substantia nigra pars compacta (SNpc). For the same area, NeuN+ neurons and Iba1+ microglia were counted and cell counts were normalized to the area.
Cell line experiments
BV-2 are a murine microglial cell line, immortalised by a v-raf/v-myc-carrying retrovirus [27]. BV-2 (passage < 30) were maintained in DMEM supplemented with 10% FBS (glial medium). At confluence, cells were harvested using 0.5% trypsin in PBS, seeded at 4 × 104 cells/well (microscopy) or 5 × 104 cells/well (flow cytometry) in DMEM supplemented with 0.5% FBS (0.5% glial medium), and left to adhere for 24 h prior to treatment.
PC12 are a rat pheochromocytoma cell line [28], that can be differentiated into dopaminergic neuron-like cells [29]. PC12 were maintained in RPMI medium supplemented with 10% horse serum and 5% FBS (PC12 medium). To generate differentiated PC12, cells were seeded on collagen (4 μg/well) at 5 × 104 cells/well in RPMI supplemented with 0.5% horse serum and 100 ng/ml NGF, and left to differentiate for 3 days.
BV-2 cells were seeded at 1.25 × 104 cells/well on TAMRA-stained (50 µM) differentiated PC12 cells. Mixed cultures were stimulated with LPS (100 ng/ml) after 24 h, and MRS2578 (1 µM) was added daily where indicated. After a further 72 h, cultures were stained with Hoechst 33342, propidium iodide and IB4 and imaged. The number of TAMRA-positive BV-2 cells (BV-2 having phagocytosed PC12) was quantified.
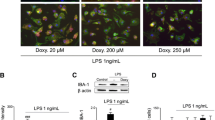
Cytokine and chemokine release from microglia
Primary microglia were isolated from mixed glial cultures from wild-type and P2ry6−/− mice, and treated with ± 100 ng/ml lipopolysaccharide for 16 h, then the extracellular cell supernatant was centrifuged at 10,000 RCF to remove cellular debris. Supernatant was then assayed using an ELISA for 62 mouse cytokines and chemokines as per the manufacturer’s instructions (abcam, ab133995). Densitometric measurements were quantified using ImageJ, whereby intensity values were normalised between membranes using positive control spots. The fold change ± LPS for P2ry6+/+ and P2ry6−/− microglia was tested for significance using unpaired t tests for each cytokine/chemokine, followed by a Holm–Šídák multiple comparisons test. There were no significant differences for any cytokine/chemokine: VEGF, VCAM-1, thrombopoietin, sTNF RI, sTNF RII, TNF α, TIMP-1, TECK, TCA-3, TARC, SDF-1 α, SCF, RANTES, P-selectin, PF-4, MIP-3 α, MIP-3 β, MIP-2, MIP-1 γ, MIP-1 α, MIG, M-CSF, MCP-5, MCP1, lymphotactin, L-selectin, LIX, leptin, leptin R, KC, IL-17, IL-13, IL-12 p70, IL-12 p40/70, IL-10, IL-9, IL-6, IL-4, IL-3 Rb, IL-3, IL-2, IL-1β, IL-1α, IGFBP-6, IGFBP-5, IGFBP-3, IFN-γ, GM-CSF, GCSF, fractalkine, Fas ligand, eotaxin-2, eotaxin, CXCL16, CTACK, CRG-2, CD40, CD30 T, CD30 L, BLC and Axl (based on three independent experiments).
Primary cell culture experiments
Primary mixed neuronal/glial cultures were prepared from cerebella of postnatal days 3–5 WT and P2ry6−/− mouse pups as previously described [30]. Cells were treated with 500 ng/ml of lipopolysaccharide for 3 days. To assess cell viability after LPS treatment, we measured the rate of reduction of MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium) to formazan by the cells. Thus, mixed neuronal/glial cultures were incubated with MTT (0.58 mg/ml) for 2 h at 37 °C. Afterward, the converted dye was liberated from the cells and solubilized by addition of dimethyl sulfoxide (DMSO), and the absorbance intensity of λ = 590 nm light was measured.
Statistical analysis
All statistical analysis was carried out using GraphPad Prism 6 and the statistical tests are indicated in the figure legends.
Results
We tested whether P2Y6R knockout (P2ry6−/−) protected against the neuronal loss induced by peripheral inflammation. Mice received doses of 1 µg/g LPS intraperitoneally (i.p.) on 4 successive days, a treatment which has been shown to result in significant loss of neurons in the substantia nigra [31]. As expected, LPS-treated wild-type animals showed a significant reduction in the total number of tyrosine hydroxylase (TH)-positive dopaminergic neurons in the substantia nigra pars compacta (SNpc) (Fig. 1a, b). The total number of NeuN-positive neurons in this area was also significantly reduced in LPS-treated wild-type (WT) animals compared to vehicle-treated controls (Fig. 1c). However, the proportion of NeuN+ neurons lost (21%) was less than the proportion of dopaminergic neurons lost (29%), suggesting that dopaminergic neurons were somewhat more vulnerable to peripheral LPS treatment.

P2ry6 knockout mice are protected against peripheral LPS-induced neuronal loss. WT and P2ry6−/− mice were injected i.p. with LPS on 4 successive days and 14 days later the substantia nigra was sectioned and stained to quantify neuronal and microglial density. a Representative images of anatomically matched, anti-tyrosine hydroxylase (TH) stained sections of substantia nigra pars compacta (SNpc) in LPS-treated and vehicle-treated WT and P2ry6−/− mice. Scale bar: 100 µm. b TH-positive dopaminergic neurons per section in SNpc. c Relative densities of NeuN-positive neurons in SNpc. c Relative densities of Iba-positive microglia (or other macrophages) in SNpc. Each data point represents one animal and error bars represent mean ± SEM. Statistical analysis was performed using two-way ANOVA with post hoc Tukey’s multiple comparison test. *p < 0.05, **p < 0.01, ****p < 0.0001
In contrast to wild-type mice, P2ry6−/− mice had no significant LPS-induced neuronal loss, measured by tyrosine hydroxylase or NeuN (Fig. 1b, c). Thus, genetic ablation of P2Y6R completely prevents neuronal loss in this inflammatory model that is particularly relevant to PD [12, 32]. Neither genotype nor LPS had any significant effect on microglial density in the substantia nigra (Fig. 2d), indicating that P2Y6R ablation did not prevent neuronal loss by preventing microglial proliferation, (which is also one measure of microglia activation).

Peripheral LPS treatment does not alter neuronal densities in hippocampus or prefrontal cortex. a Mouse brain slice stained for nuclei (with DAPI, green) and for neuronal nuclei (with anti-NeuN antibodies, magenta). Regions of interest for quantification in CA1, CA3 and cortex are indicated (scale-bar 500 μm). NeuN+ neuronal densities in anatomically matched sections of b CA1 hippocampus, c CA3 hippocampus or d motor cortex of wild-type (WT) and P2ry6−/− mice following i.p. injection of PBS (vehicle) or LPS. Mean width of e CA1 hippocampus and f CA3 hippocampus of same mice. Each data point represents one animal and error bars represent mean ± SEM. Statistical analysis was performed using two-way ANOVA, followed by Sidak-adjusted post-hoc tests, but there were no statistically significant differences
LPS treatment did not cause any change in the density of NeuN-positive neurons in hippocampal areas CA1 and CA3 or motor cortex (Fig. 2a–d), or any change in the width of CA1 or CA3 (Fig. 2e, f). This indicates selective vulnerability (or exposure) of the SNpc to peripheral LPS treatment, consistent with the hypothesis that elevated peripheral LPS may contribute to neuronal loss in Parkinson’s disease [12].
To help elucidate mechanisms in vitro, we co-cultured BV-2 microglia with differentiated PC12 dopaminergic neurons [33], and found that LPS induced microglial phagocytosis of neurons, which was prevented by a specific inhibitor of P2Y6R (1 μM MRS278) (Fig. 3a, b). This supports the ideas that LPS can increase microglial phagocytosis of neurons, and that inhibition of P2Y6R can prevent this and, therefore, might have therapeutic potential.

P2Y6R inhibition or knockout prevents LPS-induced phagocytosis and neuronal loss in culture. a Bright field and fluorescent images of differentiated TAMRA-stained PC12 (red), co-cultured for 3 days with IB4-labelled BV-2 (green), nuclei stained with Hoechst 33342 (blue) ± LPS. BV-2 phagocytosis of PC12 is visible as punctate TAMRA staining in IB4-stained cells (white arrowheads). Representative images; scale bar = 50 µm. b Quantification of BV-2 phagocytosis of PC12 cells ± LPS ± MRS2578 after 3 days. c Mixed neuronal–glial cultures from cerebella of wild-type (WT) or P2ry6−/− mice were treated for 3 days with ± 500 ng/ml LPS, then cell viability was measured by MTT assay. Data represent mean ± SEM of at least three independent experiments. Statistical analysis was performed using one-way ANOVA with post hoc Tukey’s multiple comparison test. ***p < 0.001 compared to vehicle-treated control (first bar). +++p < 0.001 compared to LPS-treated vehicle control (second bar). #p < 0.05 compared to LPS-treated wild-type condition (second bar)
To test whether P2ry6 knockout affects the LPS-induced activation of microglia, primary microglia were isolated from wild-type and P2ry6−/− mice, and treated ± LPS for 16 h, then cytokine release into the culture medium was measured using an ELISA for 62 mouse cytokines and chemokines. However, there were no significant differences between wild-type and P2ry6−/− microglia in the fold change in cytokine/chemokine release induced by LPS (see “Methods”).
We have previously shown that LPS induces neuronal loss in primary glial–neuronal cultures via microglial phagocytosis [8,9,10,11], so we tested here whether this neuronal loss required the engulfment receptor P2Y6R. We found that addition of LPS caused a loss of viability in cultures from wild-type (WT) mice, but this was reduced in cultures from P2Y6R knockout (P2ry6−/−) mice (Fig. 3c). Thus, P2Y6R knockout is neuroprotective against LPS both in vitro and in vivo.
Discussion
We tested whether P2ry6 knockout affected LPS-induced neuronal loss in vivo and in vitro. We used a chronic peripheral LPS (intraperitoneal injection) model that has previously been described to induce dopaminergic neuronal loss in the substantia nigra, as indicated by loss of cells staining with TH, NeuN and/or MAP2 [31, 34]. The LPS also induced inflammatory activation of the microglia (for the first few days after LPS), including particularly the expression of phagocytic genes, such as complement C3, and the neuronal loss was prevented by knockout of C3 [31, 34, 35]. This model may be directly relevant to human PD, as intestinal permeability increases early in PD, potentially allowing leakage of bacterial products, such as LPS into the blood and peritoneum, and blood LPS levels increase in some PD patients [12, 13, 15, 36]. One source of this LPS is the gut microbiome, which is disturbed in PD and PD models, and may affect PD progression [36, 37].
We observed robust loss of dopaminergic neurons in the substantia nigra in wild-type mice treated with LPS; however, P2ry6 knockout mice were protected against this neuronal loss. Interestingly, in wild-type mice, we found no evidence for LPS-induced neuronal loss in the cortex or the hippocampus of wild-type and P2ry6 knockout mice. Thus, this peripheral inflammation-driven model of dopaminergic neuronal loss in the substantia nigra appears to model the selective vulnerability of this neuronal population observed in PD, and lack of P2Y6R protects against this neuronal loss. Consistent with this, we previously reported that the P2Y6R inhibitor MRS2578 prevented neuronal loss in the striatum of rats injected with LPS [9]. Similarly, Oliveira-Giacomelli et al. [38] have recently reported that MRS2578 prevented dopaminergic neuronal loss in the substantia nigra of rats injected with 6-hydroxydopamine, a model of PD, indicating the potential of P2Y6R inhibitors to reduce neuronal loss in PD. Furthermore, Yang et al. [25] reported that LPS increased P2Y6R expression by microglia, and that P2Y6R expression was increased in peripheral blood monocytes from PD patients relative to healthy controls, indicating elevated P2Y6R expression in PD patients, possibly due to elevated LPS, and potentially inducing P2Y6R-mediated phagocytosis.
We measured loss of neurons in the SNpc using tyrosine hydroxylase (TH) as a marker of dopaminergic neurons, and “neuronal nuclei” (NeuN) as a marker of total neurons. However, Cannon and Greenamyre [39] reported that a proportion of TH+ cells in the SNpc did not express detectable NeuN, and Ünal-Çevik et al. [40] reported that some cortical neurons lost detectable NeuN hours after brain ischaemia but had intact nuclei, implying that live neurons could lose NeuN staining. However, they attributed this to caspase-3 activation in these cells, and translocation of NeuN to the cytoplasm; thus, they concluded that counting NeuN+ cells could still be used to measure neuronal loss/death. TH expression can also vary in neurons [41] and can fall in Parkinson’s disease [42], but there are very few reports of neurons losing detecting TH staining while remaining live neurons, and as TH is required for dopamine synthesis, cells without TH can not be dopaminergic neurons [41]. Our finding that both TH+ and NeuN+ cells in the SNpc are reduced by LPS and this is prevented by P2Y6R knockout, supports the conclusion that the reduction in neuronal numbers is due to cell loss mediated by P2Y6R, but we cannot rule out that some of the neurons dedifferentiated and this was prevented by P2Y6R knockout. Similarly, as we did not directly measure microglial phagocytosis of neurons in vivo, we cannot be sure that the neuronal loss was due to microglial phagocytosis. However, this interpretation would be consistent with the mechanism of LPS-induced neuronal loss previously characterized in vitro and in vivo, and consistent with the P2Y6R being required for microglial phagocytosis of neurons [8, 9, 18].
In vitro, LPS induced BV-2 microglial phagocytosis of dopaminergic PC12 cells that was prevented by inhibition of P2Y6R, consistent with our previous findings [33]. P2ry6 knockout protected against neuronal loss induced by LPS in primary glial–neuronal cultures. We have previously shown that this neuronal loss induced by LPS is mediated by microglial phagocytosis of stressed-but-viable neurons [8,9,10,11], and the results here are consistent with this being mediated by the engulfment receptor P2Y6R.
Overall, our findings indicate that the P2Y6 receptor contributes to neuronal loss under inflammatory conditions. This suggests that a P2Y6R antagonist might prevent neuronal loss in Parkinson’s and other diseases with inflammatory neuronal loss.
Availability of data and materials
The data sets supporting the conclusions of this article are included within the article.
Abbreviations
- CA1:
-
Cornu Ammonis subfield 1
- CA3:
-
Cornu Ammonis subfield 3
- i.p.:
-
Intraperitoneal
- ko:
-
Knockout
- LPS:
-
Lipopolysaccharide
- P2Y6R:
-
Pyrimidinergic receptor P2Y6
- SNpc:
-
Substantia nigra pars compacta
- UDP:
-
Uridine diphosphate
- wt:
-
Wild-type
References
Salter MW, Stevens B. Microglia emerge as central players in brain disease. Nat Med. 2015;23:1018–27.
Vilalta A, Brown GC. Neurophagy, the phagocytosis of live neurons and synapses by glia, contributes to brain development and disease. FEBS J. 2018;285:3566–75.
Nichols MR, St-Pierre MK, Wendeln AC, Makoni NJ, Gouwens LK, Garrad EC, Sohrabi M, Neher JJ, Tremblay ME, Combs CK. Inflammatory mechanisms in neurodegeneration. J Neurochem. 2019;149:562–81.
Jayaraj RL, Azimullah S, Beiram R, Jalal FY, Rosenberg GA. Neuroinflammation: friend and foe for ischemic stroke. J Neuroinflamm. 2019;16:142.
Fricker M, Tolkovsky AM, Borutaite V, Coleman M, Brown GC. Neuronal cell death. Physiol Rev. 2018;98:813–80.
Butler CA, Popescu AS, Kitchener EJA, Allendorf DH, Puigdellívol M, Brown GC. Microglial phagocytosis of neurons in neurodegeneration, and its regulation. J Neurochem. 2021;158:621–39.
Brown GC, Neher JJ. Microglial phagocytosis of live neurons. Nat Rev Neurosci. 2014;15:209–16.
Neher JJ, Neniskyte U, Zhao JW, Bal-Price A, Tolkovsky AM, Brown GC. Inhibition of microglial phagocytosis is sufficient to prevent inflammatory neuronal death. J Immunol. 2011;186:4973–83.
Neher JJ, Neniskyte U, Hornik T, Brown GC. Inhibition of UDP/P2Y6 purinergic signaling prevents phagocytosis of viable neurons by activated microglia in vitro and in vivo. Glia. 2014;62:1463–75.
Fricker M, Oliva-Martín MJ, Brown GC. Primary phagocytosis of viable neurons by microglia activated with LPS or Aβ is dependent on calreticulin/LRP phagocytic signalling. J Neuroinflamm. 2012;9:196.
Fricker M, Neher JJ, Zhao JW, Théry C, Tolkovsky AM, Brown GC. MFG-E8 mediates primary phagocytosis of viable neurons during neuroinflammation. J Neurosci. 2012;32:2657–66.
Brown GC. The endotoxin hypothesis of neurodegeneration. J Neuroinflamm. 2019;16:180.
Perry VH, Cunningham C, Holmes C. Systemic infections and inflammation affect chronic neurodegeneration. Nat Rev Immunol. 2007;7:161–7.
Liccardo D, Marzano F, Carraturo F, Guida M, Femminella GD, Bencivenga L, Agrimi J, Addonizio A, Melino I, Valletta A, Rengo C, Ferrara N, Rengo G, Cannavo A. Potential bidirectional relationship between periodontitis and alzheimer’s disease. Front Physiol. 2020;11:683.
Wijeyekoon RS, Kronenberg-Versteeg D, Scott KM, Hayat S, Kuan WL, Evans JR, Breen DP, Cummins G, Jones JL, Clatworthy MR, Floto RA, Barker RA, Williams-Gray CH. Peripheral innate immune and bacterial signals relate to clinical heterogeneity in Parkinson’s disease. Brain Behav Immun. 2020;87:473–88.
Forsyth CB, Shannon KM, Kordower JH, Voigt RM, Shaikh M, Jaglin JA, Estes JD, Dodiya HB, Keshavarzian A. Increased intestinal permeability correlates with sigmoid mucosa alpha-synuclein staining and endotoxin exposure markers in early Parkinson’s disease. PLoS ONE. 2011;6:e28032.
Sandiego CM, Gallezot JD, Pittman B, et al. Imaging robust microglial activation after lipopolysaccharide administration in humans with PET. Proc Natl Acad Sci USA. 2015;112:12468–73.
Koizumi S, Shigemoto-Mogami Y, Nasu-Tada K, Shinozaki Y, Ohsawa K, Tsuda M, Joshi BV, Jacobson KA, Kohsaka S, Inoue K. UDP acting at P2Y6 receptors is a mediator of microglial phagocytosis. Nature. 2007;446:1091–5.
Anwar S, Pons V, Rivest S. Microglia purinoceptor P2Y6: an emerging therapeutic target in CNS diseases. Cells. 2020;9:E1595.
Moore DJ, Chambers JK, Wahlin JP, Tan KB, Moore GB, Jenkins O, Emson PC, Murdock PR. Expression pattern of human P2Y receptor subtypes: a quantitative reverse transcription-polymerase chain reaction study. Biochim Biophys Acta. 2001;1521:107–19.
Spangenberg E, Severson PL, Hohsfield LA, Crapser J, Zhang J, Burton EA, Zhang Y, Spevak W, Lin J, Phan NY, Habets G, Rymar A, Tsang G, Walters J, Nespi M, Singh P, Broome S, Ibrahim P, Zhang C, Bollag G, West BL, Green KN. Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer’s disease model. Nat Commun. 2019;10:3758.
Communi D, Parmentier M, Boeynaems JM. Cloning, functional expression and tissue distribution of the human P2Y6 receptor. Biochem Biophys Res Commun. 1996;222:303–8.
Maier R, Glatz A, Mosbacher J, Bilbe G. Cloning of P2Y6 cDNAs and identification of a pseudogene: comparison of P2Y receptor subtype expression in bone and brain tissues. Biochem Biophys Res Commun. 1997;240:298–302.
Lee JJ, Wedow R, Okbay A, et al. Gene discovery and polygenic prediction from a genome-wide association study of educational attainment in 1.1 million individuals. Nat Genet. 2018;50:1112–21.
Yang X, Lou Y, Liu G, Wang X, Qian Y, Ding J, Chen S, Xiao Q. Microglia P2Y6 receptor is related to Parkinson’s disease through neuroinflammatory process. J Neuroinflamm. 2017;14:38.
Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez J-Y, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. Fiji: an open-source platform for biological-image analysis. Nat Meth. 2012;9:676–82.
Bocchini V, et al. An immortalized cell line expresses properties of activated microglial cells. J Neurosci Res. 1992;31:616–21.
Greene L, Tischler A. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc Natl Acad Sci USA. 1976;73:2424–8.
Greene L. Nerve growth factor prevents the death and stimulates the neuronal differentiation of clonal PC12 pheochromocytoma cells in serum-free medium. J Cell Biol. 1978;78:747–55.
Carrillo-Jimenez A, Puigdellívol M, Vilalta A, Venero JL, Brown GC, StGeorge-Hyslop P, Burguillos MA. Effective knockdown of gene expression in primary microglia with siRNA and magnetic nanoparticles without cell death or inflammation. Front Cell Neurosci. 2018;12:313.
Bodea LG, Wang Y, Linnartz-Gerlach B, Kopatz J, Sinkkonen L, Musgrove R, Kaoma T, Muller A, Vallar L, Di Monte DA, Balling R, Neumann H. Neurodegeneration by activation of the microglial complement-phagosome pathway. J Neurosci. 2014;34:8546–56.
Tufekci KU, Genc S, Genc K. The endotoxin-induced neuroinflammation model of Parkinson’s disease. Parkinsons Dis. 2011;2011:487450.
Hornik TC, Vilalta A, Brown GC. Activated microglia cause reversible apoptosis of pheochromocytoma cells, inducing their cell death by phagocytosis. J Cell Sci. 2016;129:65–79.
Beier EE, Neal M, Alam G, Edler M, Wu LJ, Richardson JR. Alternative microglial activation is associated with cessation of progressive dopamine neuron loss in mice systemically administered lipopolysaccharide. Neurobiol Dis. 2017;108:115–27.
Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, Huang D, Kidd G, Dombrowski S, Dutta R, Lee JC, Cook DN, Jung S, Lira SA, Littman DR, Ransohoff RM. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci. 2006;9(7):917–24.
Perez-Pardo P, Dodiya HB, Engen PA, Forsyth CB, Huschens AM, Shaikh M, Voigt RM, Naqib A, Green SJ, Kordower JH, Shannon KM, Garssen J, Kraneveld AD, Keshavarzian A. Role of TLR4 in the gut-brain axis in Parkinson’s disease: a translational study from men to mice. Gut. 2019;68(5):829–43.
Gorecki AM, Preskey L, Bakeberg MC, Kenna JE, Gildenhuys C, MacDougall G, Dunlop SA, Mastaglia FL, Akkari PA, Koengten F, Anderton RS. Altered gut microbiome in Parkinson’s disease and the influence of lipopolysaccharide in a human α-synuclein over-expressing mouse model. Front Neurosci. 2019;13:839.
Oliveira-Giacomelli Á, Albino MC, de Souza HDN, Corrêa-Velloso J, de Jesus Santos AP, Baranova J, Ulrich H. P2Y6 and P2X7 receptor antagonism exerts neuroprotective/neuroregenerative effects in an animal model of Parkinson’s disease. Front Cell Neurosci. 2019;13:476.
Cannon JR, Greenamyre JT. NeuN is not a reliable marker of dopamine neurons in rat substantia nigra. Neurosci Lett. 2009;464(1):14–7.
Unal-Cevik I, Kilinç M, Gürsoy-Ozdemir Y, Gurer G, Dalkara T. Loss of NeuN immunoreactivity after cerebral ischemia does not indicate neuronal cell loss: a cautionary note. Brain Res. 2004;1015(1–2):169–74.
White RB, Thomas MG. Moving beyond tyrosine hydroxylase to define dopaminergic neurons for use in cell replacement therapies for Parkinson’s disease. CNS Neurol Disord Drug Targets. 2012;11(4):340–9.
Kastner A, Hirsch EC, Herrero MT, Javoy-Agid F, Agid Y. Immunocytochemical quantification of tyrosine hydroxylase at a cellular level in the mesencephalon of control subjects and patients with Parkinson’s and Alzheimer’s disease. J Neurochem. 1993;61(3):1024–34.
Acknowledgements
The authors would like to thank Dr. Bernard Robaye (ULB Brussels) for kindly providing the P2Y6R knockout mice.
Funding
This work was supported by: the Medical Research Council UK [MR/L010593]. This project has received funding from the Innovative Medicines Initiative 2 Joint Undertaking under Grant Agreement No 115976. This Joint Undertaking receives support from the European Union’s Horizon 2020 research and innovation programme and EFPIA.
Author information
Authors and Affiliations
Contributions
Study design: SM, GCB. Experimental work: SM, FWVT, AV, TCH, MP. Data analysis: SM, FWVT, AV, MP, GCB. Manuscript: SM, MP, GCB. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All animal work was carried out in accordance with the Animals (Scientific Procedures) Act 1986 Amendment Regulations 2012 following ethical review by the University of Cambridge Animal Welfare and Ethical Review Body (AWERB), and the Home Office.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Milde, S., van Tartwijk, F.W., Vilalta, A. et al. Inflammatory neuronal loss in the substantia nigra induced by systemic lipopolysaccharide is prevented by knockout of the P2Y6 receptor in mice. J Neuroinflammation 18, 225 (2021). https://doi.org/10.1186/s12974-021-02280-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12974-021-02280-2