
Abstract
Background
Neuroinflammation plays an important role in the pathogenesis of glaucoma. Tumor necrosis factor-alpha (TNF-α) is a major pro-inflammatory cytokine released from activated retinal glial cells in glaucoma. Here, we investigated how TNF-α induces retinal ganglion cell (RGC) hyperexcitability and injury.
Methods
Whole-cell patch-clamp techniques were performed to explore changes in spontaneous firing and evoked action potentials, and Na+ currents in RGCs. Both intravitreal injection of TNF-α and chronic ocular hypertension (COH) models were used. Western blotting, immunofluorescence, quantitative real-time polymerase chain reaction (q-PCR), and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) techniques were employed to investigate the molecular mechanisms of TNF-α effects on RGCs.
Results
Intravitreal injection of soluble TNF-α significantly increased the spontaneous firing frequencies of RGCs in retinal slices. When the synaptic transmissions were blocked, more than 90% of RGCs still showed spontaneous firing; both the percentage of cells and firing frequency were higher than the controls. Furthermore, the frequency of evoked action potentials was also higher than the controls. Co-injection of the TNF-α receptor 1 (TNFR1) inhibitor R7050 eliminated the TNF-α-induced effects, suggesting that TNF-α may directly act on RGCs to induce cell hyperexcitability through activating TNFR1. In RGCs acutely isolated from TNF-α-injected retinas, Na+ current densities were upregulated. Perfusing TNF-α in RGCs of normal rats mimicked this effect, and the activation curve of Na+ currents shifted toward hyperpolarization direction, which was mediated through p38 MAPK and STAT3 signaling pathways. Further analysis revealed that TNF-α selectively upregulated Nav1.6 subtype of Na+ currents in RGCs. Similar to observations in retinas of rats with COH, intravitreal injection of TNF-α upregulated the expression of Nav1.6 proteins in both total cell and membrane components, which was reversed by the NF-κB inhibitor BAY 11-7082. Inhibition of TNFR1 blocked TNF-α-induced RGC apoptosis.
Conclusions
TNF-α/TNFR1 signaling induces RGC hyperexcitability by selectively upregulating Nav1.6 Na+ channels, thus contributing to RGC apoptosis in glaucoma.
Similar content being viewed by others


Introduction
Glaucoma, the leading cause of irreversible blindness, is a retinal neurodegenerative disease, which affects over 60 million people around the world [1,2,3]. Progressive apoptotic death of retinal ganglion cells (RGCs) and degeneration of RGC axons and dendrites, resulting in visual field loss, are the fundamental pathogenesis of glaucoma [4,5,6,7,8]. Although aging is the chief risk factor for the development of glaucoma and sensitivity of ocular tissues to elevated IOP is an associated risk factor [9, 10]; however, reduction of IOP could not completely prevent the pathological progression of glaucoma [11, 12], suggesting that the mechanisms underlying RGC loss in glaucoma are complicated. Increasing evidence indicates that neuroinflammation induced by excessive pro-inflammatory factors, which are released from activated retinal glial cells, plays significant roles in RGC damage in glaucoma [13,14,15].
Tumor necrosis factor-alpha (TNF-α), a classical inflammation cytokine, exerts multiple functions in the nervous system by binding to two types of TNF-α receptor (TNFR1 and TNFR2) [16, 17]. Soluble TNF-α (17 kDa) preferentially binds to TNFR1, leading to neuroinflammation and cell death [18,19,20], while transmembrane TNF-α (22 kDa) primarily binds to TNFR2 that mediates neuroprotective effects [21, 22]. TNF-α also plays important roles in the pathogenesis of retinal diseases, such as glaucoma. Previous studies have demonstrated that TNF-α was involved in retinal axon loss and RGC death in glaucoma [23,24,25]. TNF-α-induced RGC death in glaucoma could be mediated by multiple pathways. TNF-α caused RGC loss by activation of death signaling, such as caspase 8 and oxidative stress [26]. In chronic ocular hypertension (COH) model, soluble TNF-α induced GluA2 subunit of AMPA receptor endocytosis and activated Ca2+-permeable GluA2-lacking AMPA receptors in RGCs, thus promoting RGC death [24]. Direct neutralization of soluble TNF-α in the retina of experimental glaucoma was able to reduce RGC death efficiently [24, 27].
In a previous study, we have shown that in a rat experimental glaucoma model, IOP elevation led to depolarized resting membrane potential in RGCs, and the cells displayed hyperexcitability, which was characterized by increased spontaneous firing. The hyperexcitability of RGCs could be attenuated by intravitreal pre-injecting the TNF-α antagonist XPro1595 and the nitric oxide (NO) antagonist L-NAME, indicating that inflammatory factors released from retinal glial cells may be involved [28]. Neuronal hyperexcitability has been demonstrated to be associated with cell apoptosis [29,30,31]. Changes in ion channels may contribute to the production of hyperexcitability, especially voltage-gated K+ and Na+ channels. Although it is reported that TNF-α could modulate outward K+ currents in RGCs [32], the mechanisms underlying TNF-α-induced hyperexcitability of RGCs in glaucoma are largely unknown. In this study, we show that TNF-α enhanced RGC excitability by upregulating Nav1.6 channels through activating TNFR1, thus contributing to RGC apoptosis.
Materials and methods
Animals
Male Sprague-Dawley rats (4 weeks old, weighing 100~110 g) were obtained from the SLAC Laboratory Animal Co., Ltd. (Shanghai, China) and housed under a 12-h light/dark cycle with enough food and water. All animal experiments were performed in accordance with the National Institutes of Health (NIH) guidelines for the Care and Use of Laboratory Animals and were approved by the Institutes of Brain Science at Fudan University. All the experiments described in this study were performed by researchers that were blind with respect to the treatments.
Intravitreal injection
Recombinant rat TNF-α (5 ng in 1 ml of 0.9% saline with 0.1% BSA, R&D systems, Minneapolis, MN, USA) (2 μl) was injected into the right vitreous cavity of the anesthetized rat with a micro-injector (Hamilton, Reno, NV, USA) under a stereoscopic microscope (Carl Zeiss). Given that the vitreous volume is ∼20 μl [33, 34], the concentration of TNF-α in the vitreous cavity is ∼0.5 ng/ml. 2 μl of 8-chloro-4-(phenylthio)-1-(trifluoromethyl)-[1,2,4]triazolo[4,3-a]quinoxaline (R7050, 10 μM, Tocris Bioscience, Ellisville, USA), BAY 11-7082 (10 μM), stattic (10 μM), or SB203580 (10 μM) (Selleck Chemical, Houston, TX) was intravitreally injected in the same manner. The eyes that received saline injection were used as controls. Details of the operation were described in our previous reports [28, 35].
Rat COH model
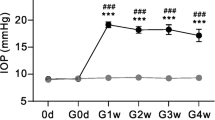
Rat COH model was produced in accordance with previous studies [28, 35]. In brief, the rat was anesthetized with 10% chloral hydrate (4 ml/kg, i.p.), and the operation eye (right eye) was further anesthetized with 0.4% oxybuprocaine hydrochloride eyedrop (Benoxil, Santen Pharmaceutical Co., Ltd., Osaka, Japan). The micro-magnetic beads (10 μl, diameter ≈ 10 μM, BioMag® Superparamagnetic Iron Oxide, Bangs Laboratories, Ins) were slowly injected into the anterior chamber under an OPMI VISU 140 microscope (Carl Zeiss, Jena, Germany). A small handheld magnet (0.45 Tesla) was used to distribute the microbeads evenly around the iridocorneal angle. The IOP was measured using a handheld digital tonometer (Tonolab, TioLat, Finland) under general anesthesia as described above. The average value of five consecutive measurements with a deviation of < 5% was accepted. The IOPs of both eyes were measured before surgery (baseline), immediately after operation (day 0), the day after the operation (day 1, COH1D), the third day (COH3D), and 1 and 2 weeks after the operation (COH1W, COH2W). All the measurements were performed at 9:00–10:00 a.m. to avoid the possible influence of circadian rhythms on IOP [36].
Preparation of retinal slices and isolated RGCs
Rat retinal slices (200 μm in thickness) were made on a Leica Vibrotome (VT1000S) and incubated in carbogen saturated (95% O2 and 5% CO2) artificial cerebral spinal fluid (ACSF) containing the following (in mM): 125 NaCl, 2.5 KCl, 25 NaHCO3, 1.25 NaH2PO4, 2.5 CaCl2, 1 MgCl2, and glucose 10 (pH 7.4) for 30 min at room temperature (22–24°C) before recording. To isolate RGCs, the retinas of anesthetized rats were removed quickly and incubated in the oxygenated Hank’s solution containing the following (in mM): NaCl 137, NaHCO3 0.5, NaH2PO4 1, KCl 3, CaCl2 2, MgSO4 1, HEPES 20, and glucose 16 (pH 7.4). After digesting in Hank’s solution with 1.6 U/ml papain (Worthington Biochemical, Freehold, NJ, USA) and 0.2 mg/ml L-cysteine for 26 min at 34°C, retinal neurons were obtained by mechanical dissociation. The detailed procedures were described in our previous reports [37, 38].
Patch-clamp recordings
Whole-cell patch-clamp experiments were performed using a patch-clamp amplifier (Axopatch 700B) and Digidata 1440A (Molecular Devices, Foster City, CA, USA) at room temperature. Patch pipettes (BF150-86-10 glass, Sutter Instrument Co., Novato, CA, USA) were pulled by a P-97 Flaming/Brown micropipette puller (Sutter Instrument) with a resistance of 5–8 MΩ after fire-polishing (Model MF-830, Narishige, Japan).
For spontaneous firing recordings in RGCs, the individual retinal slice was continuously superfused with oxygenated ACSF at a rate of 1–2 ml/min at room temperature. RGCs in retinal slices were identified with the help of infrared-differential interference contrast (IR-DIC) video microscopy (Olympus, Japan) as described [28, 38]. Patch pipette solution contained (in mM): 120 potassium d-gluconate, 1 EGTA, 10 HEPES, 4 ATP-Mg, 0.3 GTP-Na, 10 phosphocreatine, 0.1 CaCl2, 1 MgCl2, and Alexa Fluor 488 (pH 7.2 adjusted with KOH, 290–300 mOsm/L). To record Na+ currents, the bath solution was consisted of (in mM) 130 NaCl, 2 CaCl2, 1 MgCl2, 10 HEPES, 15 tetraethylammonium (TEA)-Cl, 10 4-aminopyridine (4-AP), and 10 glucose (pH 7.4 with NaOH, 300–310 mOsm/L with sucrose). The patch pipette solution contained (in mM) 130 CsCl, 10 NaCl, 5 HEPES, 8 EGTA, 10 TEA-Cl, 2 ATP-Mg, and 1 GTP-Na (pH 7.2 adjusted with CsOH, 290–300 mOsm/L with sucrose). Na+ currents were induced by a series of 50 ms depolarizing voltage pulses from a holding potential of −70 to +30 mV in increments of 10 mV. The inactivation curves of voltage-gated sodium channels (VGSCs) were detected by giving RGC a 200 ms pre-pulse from −70 mV (holding potential) to different voltages and then depolarizing to −10 mV. Both activation and inactivation curves were fitted by Boltzmann function.
Western blotting
Western blotting analysis was conducted following the procedures previously described [35]. Briefly, retinal total proteins or membrane proteins were extracted, and protein concentrations were determined with a bicinchoninic acid (BCA) assay kit (Pierce Biotechnology, IL, USA). Protein samples were then separated by SDS-PAGE gel in 6 or 10% and transferred to a PVDF membrane (Immobilon-P, Millipore, Billerica, MA, USA). The PVDF membrane was blocked for 6 h in 5% non-fat powdered milk and then incubated with primary antibody overnight at 4°C. In this study, the following primary antibodies were used: monoclonal mouse anti-β-actin (1:3000 dilution, Sigma-Aldrich, St. Louis, MO, USA), anti-TNF Receptor 1 (1:5000 dilution, Abcam, Cambridge, MA, USA), anti-TNF Receptor 2 (1:12000 dilution, Abcam), anti-Nav1.6 (1:600 dilution, Alomone Labs, Israel), and anti-GAPDH (1:1000 dilution, Cell Signaling Technology, MA, USA). The membranes were then incubated at room temperature for 1 h with donkey anti-mouse, rabbit, or goat IgG HRP (Jackson ImmunoResearch Labs, Wes Grove, PA, USA). The blots were visualized by an Odyssey near-infrared imaging scanner (FluorChem E System, Protein Simple, USA).
Immunohistochemistry
The protocol of immunofluorescence staining was followed as described in previous studies [35]. In short, dissected eyes were dehydrated with 10%, 20%, and 30% sucrose solutions gradually and fixed with 4% paraformaldehyde at 4°C for 12 h. Retinal slices at 14-μm thickness were vertically sectioned and blocked with a mixture of 3% bovine serum and PBS on chrome-alum-gelatin-coated slides (Thermo-Fisher Scientific, Pittsburgh, PA, USA). The slices were incubated with the primary antibodies: anti-NaV1.6 (1:600 dilution, Alomone Labs) and anti-Brn-3a (1:400 diluted, Santa Cruz Biotechnology, Santa Cruz, VA, USA) at 4°C for 48 h. For negative control, Nav1.6 antibody was pre-absorbed by Nav1.6 blocking peptide (Alomone Labs). Then, the slices were incubated with Cy3- or Alexa Fluor 488-labeled secondary antibodies (1:400 diluted, Sigma-Aldrich) for 2 h at room temperature. 150 μl of the DAPI working solution was added to each slide for detecting cell nuclei. After washing, the sections were covered with an anti-fade mounting medium and photographed with FluoView 1000 confocal microscope (Olympus, Tokyo, Japan).
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)
To detect neuronal apoptosis, the deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) assay was performed on whole flat-mounted retinas [35]. The DeadEnd Fluorometric TUNEL System G3250 kit (Promega, Madison, WI, USA) was used according to the manufacturer’s instructions. The whole retina was analyzed, and all TUNEL-positive signals that merged well with DAPI were counted. The fluorescence images were captured using the confocal microscope through a 20× objective (FluoView 1000, Olympus, Japan).
Quantitative real-time PCR
Total RNA was extracted from the whole retina using the TaKaRa MiniBEST Universal RNA Extraction Kit (#9767, Takara, Japan). Quantitative real-time polymerase chain reaction (q-PCR) was carried out using the PrimeScript™ RT reagent Kit with gDNA Eraser (#RR047A, Takara, Japan), and mRNA was measured using a TB Green® Premix Ex Taq™ II (#RR820A, Takara, Japan). The qPCR assays were performed on the QuantStudio 3 Real-Time PCR system (Thermo Fisher Scientific, USA). The relative mRNA levels were normalized from Ct values according to 2-△△ct calculation method. The sequences of primers used in this study are as follows: Nav1.1, forward 5′-GCG ATT ATG TGA CAA GCA TTT TG-3′, reverse 3′-CGG AGG GAG ATG AGC TTC AG-5′; Nav1.2, forward 5′-TTC ATG GCT TCC AAT CCC TCC-3′, reverse 3′-GGT GTC ACG TCA GTC TTC TCT-5′; Nav1.6, forward 5′-GCA AGC TCA AGA AAC CAC CC-3′, reverse 3′-CCG TAG ATG AAA GGC AAA CTC T-5′; β-actin, forward 5′-AGC CAT GTA CGT AGC CAT CC-3′, reverse 3′-CTC TCA GCT GTG GTG GTG AA-5′.
Reagents and drugs
R7050, d-(-)-2-amino-5-phosphonopentanoic acid (D-APV), bicuculline, CNQX, 4,9-anhydrotetrodotoxin (AHTTX), and TTX were purchased from Tocris (Tocris Bioscience, Ellisville, MO, USA). BAY 11-7082, SB203580 and stattic were from Selleck (Selleck Chemical), and the others were from Sigma-Aldrich (St. Louis, MO, USA).
Statistical analysis
Data were analyzed using GraphPad Prism (version 6.02, Graphpad Software Inc., USA), Clampfit 10.2 (Molecular Devices, Foster City, CA, USA), Igor 4.0 (WaveMetrics, Lake Oswego, OR, USA), and Origin 2018 (OriginLab, Northampton, MA, USA). In this work, the “n” represents cell number in electrophysiological experiments, or animal number in Western blotting, q-PCR, and TUNEL experiments. A Boltzmann function was used to fit the activation and inactivation curves. All experiments and measurements were performed in quadruplicate minimally and analyzed by t test, Brown-Mood test, Mann-Whitney test, or one-way ANOVA with Bonferroni’s post hoc test (multiple comparisons). Before performing t test or one-way ANOVA analysis, the data were analyzed using Shapiro-Wilk test or Brown-Forsythe test to evaluate the normality or the homogeneity of variance. Membrane potential and frequency of spontaneous firing were presented as a median and interquartile range, while the other data were expressed as mean ± SEM. The threshold for statistical significance was P < 0.05 in all tests.
Results
TNF-α/TNFR1 pathway contributes to RGC hyperexcitability
Since IOP elevation-induced hyperexcitability of RGCs in COH retinas could be reversed by TNF-α and NO blockers [28], we first examined whether TNF-α could induce RGC hyperexcitability in normal rats. Soluble TNF-α (5 ng/ml, 2 μl) was intravitreally injected, and retinal slices were made for electrophysiological recordings at 3 and 7 days (TNF-α3D and TNF-α7D) after the injections. In this study, all recordings in retinal slices were made on RGCs with a soma diameter of 10–15 μm, including both ON and OFF type cells, which were identified by their dendrite distribution in the IPL labeled by Alexa Fluor 488 and by negative current injections [38]. In previous studies [28, 38], we have shown that there were no significant differences between these two types of RGCs in both the frequency of spontaneous firing and the membrane potential under our recording conditions. Additionally, it was reported that at an early stage (2 weeks) of IOP elevation, dendritic pruning, and a transient increase in axon firing of RGCs in response to the preferred light stimulus were observed, which was independent of ON and OFF RGCs [6]. Therefore, data obtained from both types of RGCs were pooled in this study. As shown in Fig. 1a, RGCs displayed spontaneous firing under the current-clamped condition in normal rats (control, Ctr). Perfusion of the cocktail synaptic blockers, including bicuculline (10 μM), strychnine (10 μM), CNQX (10 μM), and D-APV (50 μM), almost completely blocked the spontaneous firing of the cell, similar to our previous report [38]. In the presence of cocktail synaptic blockers, current injection (+20 pA) could evoke the cell to fire action potentials (APs). In TNF-α injected retinas, the frequencies of spontaneous firing were significantly increased to 2.00 Hz (1.22, 3.87) (n = 11, P = 0.0018) and 3.40 Hz (2.66, 5.49) (n = 11, P < 0.001) in TNF-α3D and TNF-α7D groups, respectively, from the control value of 0.41 Hz (0.15, 0.97) (n = 12) (Fig. 1a, b). Although the membrane potentials (MP) of RGCs showed a trend toward depolarization in TNF-α injected retinas, there was no significant difference between control and TNF-α injected groups (Fig. 1a, c). When the synaptic transmissions were blocked, more than 90% of RGCs still showed spontaneous firing in both TNF-α3D and TNF-α7D groups, and the percentage of AP firing cells was higher than that of control (30.7%) (Fig. 1d, inset). Additionally, the frequencies of spontaneous firing were significantly increased (TNF-α3D: 0.25 Hz (0.13, 0.35), n = 11, P = 0.0054; TNF-α7D: 0.76 Hz (0.15, 1.59), n = 11, P = 0.0026), as compared to the control group (0.00 Hz (0.00, 0.08), n = 11) (Fig. 1d). Similarly, the frequencies of evoked AP were increased to 1.06 Hz (0.63, 3.04) (n = 11, P = 0.0353) and 4.22 Hz (1.24, 6.50) (n = 11, P = 0.0007) in TNF-α3D and TNF-α7D groups, respectively, from the control value of 0.23 Hz (0.05, 1.15) (n = 13) (Fig. 1e). Since soluble TNF-α-mediated effects are mainly through binding to TNFR1 [18,19,20], we examined whether TNF-α-induced RGC hyperexcitability is mediated by this receptor. Intravitreal co-injection of R7050 (10 μM, 2 μl), an inhibitor of TNFR1 signaling [39], blocked the TNF-α-induced increase in frequencies of spontaneous firing (Ctr: 0.41 Hz (0.15, 0.97), n = 12; TNF-α7D: 3.40 Hz (2.66, 5.49), n = 11, P < 0.001 vs. Ctr; R 7050+TNF-α7D: 0.95 Hz (0.30, 1.56), n = 14, P < 0.001 vs. TNF-α7D alone) (Fig. 1f–i). These results suggest that TNF-α may directly act on RGCs to induce cell hyperexcitability through activating TNFR1.

TNF-α induces hyperexcitability of RGCs through activating TNFR1. a Representative traces show spontaneous firing and evoked action potentials (AP) that were recorded in three different RGCs in rat retinal slices obtained from control (Ctr), 3 days (TNF-α3D) and 7 days (TNF-α7D) after intravitreal TNF-α injections. (a1) and (a2) are those from the recordings in a in a faster time scale. b, c Summary data show the changes in frequency (b) and membrane potential (MP) (c) of spontaneous firing in RGCs under different conditions. d Summary data show the changes in the frequency of spontaneous firing in RGCs under different conditions when synaptic transmissions were blocked. The insert shows the percentage of AP firing cells in different groups. e Summary data show the changes in the frequency of evoked AP under different conditions. n = 11 for each group. f–h Representative traces show spontaneous firing that was recorded in three different RGCs in rat retinal slices obtained from Ctr, TNF-α7D, and R7050+TNF-α7D groups, respectively. i Summary data show that the TNFR1 inhibitor R7050 eliminated TNF-α-induced increase of spontaneous firing frequency. n = 12, 11, 14, respectively. Data are represented as median and interquartile range. *P < 0.05, **P < 0.01, and ***P < 0.001 vs. Ctr; ###P < 0.001 vs. TNF-α7D
We also determined whether intravitreal injection of TNF-α may change the expression of TNF-α receptors. As shown in Fig. 2, in TNF-α-injected retinas, the protein levels of TNFR1 expression were significantly increased to 141.9 ± 8.2% of control (n = 7, P = 0.0147) and 143.9 ± 12.4% of control (n = 7, P = 0.0104) in TNF-α3D and TNF-α7D groups, respectively. As a positive control, the TNF-α protein levels were increased to 158.5 ± 11.7% of control in COH retinas at week 2 (COH2W) (n = 7, P < 0.001) (Fig. 2a, b). In contrast, the protein levels of TNFR2 expression kept unchanged in both TNF-α-injected and COH retinas (Fig. 2a, c). Furthermore, co-injection of R7050 abolished the TNF-α-induced upregulation of TNFR1 expression (TNF-α7D: 137.1 ± 11.1% of control, n = 7, P = 0.0147 vs. Ctr; R 7050+TNF-α7D: n = 7, 105.3 ± 7.8% of control, P = 0.0377 vs. TNF-α7D) (Fig. 2d, e).

TNF-α induces upregulation of TNFR1 expression. a Representative immunoblots show the changes of TNFR1 and TNFR2 protein levels in control (Ctr), TNF-α3D, TNF-α7D, and COH2W retinas. b, c Bar charts summarize the average densitometric quantification of TNFR1 (b) and TNFR2 (c) in different groups. n = 6 for each group. d, e Representative immunoblots and the relative protein levels of TNFR1 in control (Ctr), TNF-α7D, and R 7050+TNF-α7D retinas. n = 7 for each group. *P < 0.05, and ***P < 0.001 vs. Ctr; #P < 0.05 vs. TNF-α7D
TNF-α selectively upregulates Nav1.6 currents in RGCs
Previous studies have shown that TNF-α could regulate VGSCs in neurons, thus influencing neuronal excitability [40,41,42]. We tested the possibility that VGSCs are involved in TNF-α-induced RGC hyperexcitability. Whole cell Na+ currents were recorded in acutely isolated RGCs (Fig. 3a) from control and TNF-α intravitreally injected (TNF-α7D) rats. As compared with the controls, Na+ current densities in the TNF-α7D group were significantly and voltage-dependently increased (Fig. 3b, c). For example, at −30 mV peak Na+ current density was increased to −180.8 ± 14.7 pA/pF (n = 14; P = 0.0032) from the control value of −124.6 ± 4.45 pA/pF (n = 11) (Fig. 3d).

TNF-α voltage-dependently increases Na+ current density in RGCs. a Micrograph showing a typical acutely isolated RGC for recording. b Representative traces show Na+ currents recorded in acutely isolated RGCs from control (Ctr) and TNF-α7D retinas. The holding potentials of RGCs were set at −70 mV and the currents were evoked in the range of −70 to +30 mV with steps of 10 mV. c Current-voltage relationship curves of Na+ currents in RGCs of Ctr and TNF-α7D retinas. n = 11 and 14 for Ctr and TNF-α7D groups, respectively. d Summary data show the peak current densities of Na+ channels at −30 mV in RGCs of Ctr and TNF-α7D retinas. n = 11 and 14 for Ctr and TNF-α7D groups, respectively. *P < 0.05, **P < 0.01, and ***P < 0.001 vs. Ctr
Upregulation of Na+ currents induced by intravitreal injection of TNF-α may be mediated by directly regulating ion channels or/and increasing ion channel protein expression. We first tested the effect of TNF-α on VGSCs in acutely isolated RGCs from normal rats. Figure 4a shows the representative Na+ currents recorded in an RGC before and after TNF-α (5 ng/ml) application. TNF-α significantly and voltage-dependently enhanced peak Na+ current densities (Fig. 4b). In addition, the effect of TNF-α on Na+ currents was reversible. As shown in Fig. 4c, at −20 mV, stable current recordings could be kept for almost 8 min, perfusion of TNF-α reversibly increased the current amplitudes to 152.2 ± 10.5% of control (n = 11, P < 0.001), and washout pushed the currents almost to control levels (Fig. 4d, e). We also examined the changes of activation and inactivation curves of Na+ currents in RGCs before and after TNF-α application. As shown in Fig. 5a, the activation curve of Na + currents was shifted toward hyperpolarization direction after TNF-α application, with a V1/2 value being −41.20 ± 1.4 mV (n = 10, P = 0.0102) that was significantly different from control value (−38.12 ± 1.21 mV, n = 10) (Fig. 5b). However, TNF-α did not change the inactivation curve (control: V1/2 = −49.5 ± 2.1 mV, n = 7; TNF-α: V1/2 = −49.6 ± 2.3 mV, n = 7, P = 0.7648) (Fig. 5c, d). These results suggest that TNF-α could directly enhance Na+ currents and increase the activation probability of the channels.

TNF-α increases Na+ currents in RGCs of normal retinas. a Representative traces show Na+ currents recorded in an RGC before (control, Ctr) and after TNF-α (5 ng/ml) application. b Current-voltage relationship curves of Na+ currents in RGCs before (Ctr) and after TNF-α application. n = 11. c Time courses of Na+ currents at −20 mV in Ctr (n = 7) and TNF-α application groups (n = 8). d Representative Na+ current traces recorded in an RGC at −20 mV in the TNF-α group at different time points as shown in figure c panel. e Summary data show the changes in relative peak Na+ currents (at −20 mV) under different conditions. n = 11. *P < 0.05, **P < 0.01, and ***P < 0.001 vs. Ctr

TNF-α increases the activation probability of Na+ currents. a Activation curves of Na+ currents in RGCs before and after TNF-α application. b Bar chart summarizes the changes in the V1/2 of Na+ currents before and after TNF-α application. n = 12; *P < 0.05 vs. Ctr. c Representative Na+ currents recorded in an RGC before and after TNF-α application. The cell was given a 200 ms pre-pulse from a holding potential of −70 mV to different membrane potentials and then depolarized to −10 mV. d Inactivation curves of Na+ currents before and after TNF-α application. n = 6. Both the activation and inactivation curves were fitted with the Boltzmann equation
It was reported that p38 MAP kinase (p38 MAPK) and signal transducer and activator of transcription 3 (STAT3) signaling pathways were involved in TNF-α-mediated effects [43, 44]. The mechanisms underlying the TNF-α-induced enhancement of Na+ currents were explored. Figure 6a, b shows that TNF-α-induced upregulation of Na+ currents in RGCs was eliminated when the cells were pre-incubated with SB203580 (10 μM), a p38 MAPK inhibitor, for 30 min (100.4 ± 2.8% of control, n = 10, P = 0.8777). Similar results were obtained when RGCs were pre-incubated with Stattic (10 μM), a STAT3 inhibitor (99.4 ± 2.6% of control, n = 10, P = 0.8148) (Fig. 6c, d). These results indicate that TNF-α upregulates Na+ currents in RGCs through p38 MAPK and STAT3 signaling pathways.

p38 MAPK and STAT3 pathways mediate TNF-α-induced upregulation of Na+ currents in RGCs. a, b Sample traces recorded in an RGC at −20 mV show that pre-incubation of the p38 MAPK inhibitor SB203580 blocked the TNF-α-induced upregulation of peak Na+ currents (a), and summary data are shown in b. n = 10. c, d Sample traces recorded another RGC at −20 mV show that pre-incubation of the STAT3 inhibitor stattic blocked the TNF-α-induced upregulation of peak Na+ currents (c), and summary data are shown in d. n = 10
RGCs express functional Nav1.1, Nav1.2, and Nav1.6 Na+ channels, and Nav1.6 is the predominant one [45,46,47]. We examined which subtype(s) may be modulated by TNF-α. As shown in Fig. 7a, an RGC was pre-incubated with the Nav1.1 blocker ICA121421 (1 μM) and the Nav1.2 blocker phrixotoxin3 (10 nM). In the presence of these blockers, perfusion of TNF-α still increased Na+ currents. The average current amplitudes were increased to 128.3 ± 4.3% of control (n = 8, P < 0.001) (Fig. 7b). In contrast, when RGCs were pre-incubated with the Nav1.6 blocker 4,9-anhydrotetrodotoxin (AHTTX, 100 nM), perfusion of TNF-α no longer changed the currents (100.5 ± 4.7% of control, n = 6, P = 0.9168) (Fig. 7c, d). These results suggest that TNF-α may selectively enhance Nav1.6 currents.

TNF-α selectively upregulates Nav1.6 currents in rat RGCs. a Representative current traces recorded from an RGC show the changes of peak Nav1.6 currents before and after TNF-α application in the presence of the Nav1.1 blocker ICA12142 (1 μM) and the Nav1.2 blocker Phrixotoxin3 (10 nM). b Bar charts summarize the changes of Nav1.6 current amplitudes at −20 mV before and after TNF-α perfusion. Note that TNF-α enhanced Nav1.6 peak currents. n = 9. ***P < 0.001 vs. Ctr. c Representative traces show the changes in Nav1.1 and Nav1.2 mixed currents recorded in an RGC before and after TNF-α application in the presence of the Nav1.6 blocker AHTTX (100 nM). d Bar charts summarize the changes of mixed Nav1.1 and Nav1.2 current amplitudes before and after TNF-α application. n = 7
We then examined whether TNF-α injection may change the protein expression levels of Na+ channels. The qPCR analysis revealed that the mRNA levels of Nav1.6 were significantly increased at 3 days after the TNF-α injection, then declined at 7 days, while the mRNA levels of Nav1.1 and Nav1.2 were kept unchanged (Fig. 8a–c). Therefore, we examined Nav1.6 protein expression in TNF-α injected retinas. The total protein level of Nav1.6 (tNav1.6) in the TNF-α3D group was not changed as compared with the control (104.5 ± 5.7% of control, n = 7, P = 0.7949); however, it was increased to 126.5 ± 12.39% of control (n = 7, P = 0.0298) in the TNF-α7D group. As a positive control, the total protein level of Nav1.6 in COH2W was increased to 130.9 ± 8.9% of control (n = 7, P = 0.0118), which was reversed by pre-injecting the TNFR1 blocker R7050 (10 μM) (113.4 ± 6.1% of control, n = 7, P = 0.3053 vs. control) (Fig. 8d, e). Since Na+ channels expressed in the cell membrane are functional, the protein levels of Nav1.6 in the membrane component (mNav1.6) were further examined. Figure 8f, g clearly showed that intravitreal injection of TNF-α significantly upregulated mNav1.6 expression (TNF-α3D: 179.3 ± 6.9% of control, n = 4, P < 0.001; TNF-α7D: 143.9 ± 8.2% of control, n = 4, P = 0.0253). In COH retinas, mNav1.6 expression was also increased to 169.6 ± 14.6% of control, n = 4, P < 0.001). Consistently, double immunostaining showed that Nav1.6 was primarily expressed in the ganglion cell layer (GCL) and the inner plexiform layer (IPL), and intravitreal injection of TNF-α increased Nav1.6 expression, especially in the RGC axon-like structures (Fig. 8h).

TNF-α increases the expression of Nav1.6 in the retina. a–c Bar charts show the relative mRNA levels of Nav1.1, Nav1.2, and Nav1.6 in Ctr (a), TNF-α3D (b), and TNF-α7D (c) groups. n = 5 for each group. d Representative immunoblots show the changes in total Nav1.6 (tNav1.6) expression in control (Ctr), TNF-α3D, TNF-α7D, and COH2W without or with R7050 retinal extracts. e Bar charts summarize the average densitometric quantification of immunoreactive bands of tNav1.6 under different conditions as shown in d. n = 6 for each group. All data are normalized to their corresponding β-actin and then to Ctr. f Representative immunoblots show the changes in Nav1.6 expression in membrane components (mNav1.6) in control (Ctr), TNF-α3D, TNF-α7D, and COH2W retinal extracts. n = 4 for each group. All data are normalized to their corresponding GAPDH and then to Ctr. *P < 0.05 and ***P < 0.001 vs. Ctr. h Immunofluorescence labeling showing Nav1.6 expression in rat retinal vertical slices taken from the normal saline-injected retina (Ctr, h1-h3) and TNF-α-injected retinas on 7 days after the injection (TNF-α7D, h4-h6). h7-h9: double immunofluorescence staining showing Nav1.6 expression when the Nav1.6 antibody was pre-absorbed with its blocking peptide (BP). Note that the enhanced expression of Nav1.6 in the axon-like structures in the TNF-α7D group. Scale bars = 20 μm. GCL, ganglion cell layer; IPL, inner plexiform layer; INL, inner nuclear layer; OPL, outer plexiform layer; ONL, outer nuclear layer
Signaling pathways involved in TNF-α-induced increase in Nav1.6 expression were also examined. Unexpectedly, p38 MAPK and STAT3 signaling pathways were not involved in the upregulation of Nav1.6 expression since intravitreal injections of the p38 MAPK inhibitor SB203580 or the STAT3 inhibitor stattic failed to block the TNF-α-induced effects on Nav1.6 expression (SB203580+TNF-α7D: n =4, P = 0.6528 vs. TNF-α7D alone; stattic+TNF-α7D: n =4, P = 0.8004 vs. TNF-α7D alone) (Fig. 9a, b). In contrast, intravitreal injection of the NF-κB inhibitor BAY 11-7082 (10 μM) could efficiently block the upregulated expression of Nav1.6 induced by TNF-α (99.9 ± 4.6% of control, n =4, P = 0.0053 vs. 121.9 ± 4.0% of control in TNF-α7D alone, n = 4) (Fig. 9c).

NF-κB signaling pathway mediates TNF-α-induced upregulation of Nav1.6 expression. a Representative immunoblots show the changes in tNav1.6 expression in control (Ctr), TNF-α7D, and SB203580+TNF-α7D retinal extracts (upper panel), and summary data are shown in the lower panel. b Representative immunoblots (upper panel) and summary data (lower panel) of tNav1.6 protein levels in Ctr, TNF-α7D, and stattic+TNF-α7D groups. c Representative immunoblots (upper panel) and summary data (lower panel) of tNav1.6 protein levels in Ctr, TNF-α7D, and BAY 11-7082+TNF-α7D groups. All data are normalized to their corresponding β-actin and then to Ctr. n = 4 for each group. *P < 0.05, **P < 0.01, and ***P < 0.001 vs. Ctr; ##P < 0.01 vs. TNF-α7D
TNF-α induces RGC apoptosis
Finally, we explored the TNF-α-induced effects on RGC survival by using TUNEL-staining method. Representative TUNEL staining images captured from whole flat-mounted retinas under different conditions are shown in Fig. 10a. Intravitreal injection of TNF-α remarkably increased the number of TUNEL-positive RGCs to 254.4 ± 15.6 (n = 7, P < 0.001) and 202.0 ± 18.4 (n= 7, P < 0.001) in TNF-α3D and TNF-α7D groups, respectively, which could be reversed by co-injection of the TNFR1 blocker R7050 (R7050+TNF-α3D: 86.4 ± 13.8, n = 5, P < 0.001 vs. TNF-α3D group; R7050+TNF-α7D: 75.8 ± 9.6, n = 5, P < 0.001 vs. TNF-α7D group) (Fig. 10b). Similarly, pre-injection of R7050 (10 μM) could partially reduce the number of TUNEL-positive RGCs in COH retinas (128.8 ± 3.8, n = 5, P = 0.0295 vs. control; P = 0.0024 vs. COH2W group) (Fig. 10b).

Inhibition of TNFR1 reduces RGC apoptosis in TNF-α injected and COH retinas. a Representative images of TUNEL staining detection of apoptotic RGCs in Ctr (a1), TNF-α3D (a4), R7050+TNF-α3D (a7), TNF-α7D (a10), R7050+TNF-α7D (a13), COH2W (a16), and R7050+COH2W (a19) groups. R7050 (10 μM, 2 μl) was intravitreally injected 1 day before TNF-α injection or COH operation. All images were taken from the whole-flat mounted retinas in the regions at angle 0°. a2, a5, a8, a11, a14, a17, and a20 were DAPI images. Merged images of TUNEL and DAPI were showed in a3, a6, a9, a12, a15, a18, and a21. Scale bar = 50 μm for all images. b Bar charts summarize the average number of TUNEL-positive signals in whole flat-mounted retinas in different groups. n = 5–7. *P < 0.05, **P < 0.01, and ***P < 0.001 vs. Ctr; ###P < 0.001
Discussion
TNF-α-induced neuronal hyperexcitability contributes to RGC injury
It is commonly known that neuroinflammation plays a key role in the pathogenesis of glaucoma although the detailed mechanisms have not been completely elucidated [13, 48]. As one of the major inflammatory factors, TNF-α is released from the activated glial cells in glaucomatous retinas [49,50,51]. In this study, we demonstrated that TNF-α could induce RGC hyperexcitability, as evidenced by increased spontaneous firing and evoked AP frequencies of the cells, thus contributing to RGC injury in glaucoma. Changes in spontaneous firing may have resulted from the balance between excitatory and inhibitory inputs. When the synaptic transmissions were blocked, most of RGCs (> 90%) still showed spontaneous firing in the TNF-α-injected retinas, while a lesser number of RGCs fired AP spontaneously with a very low frequency in the control retinas, suggesting that TNF-α have a direct action on RGCs. Indeed, a number of studies have reported the enhanced excitability of RGCs in glaucomatous retinas [6, 28, 52, 53]. In an experimental glaucoma model, we have shown that IOP elevation resulted in RGC hyperexcitability, which may be mediated by inflammatory factors [28]. The present work provided further evidence that TNF-α was indeed involved. It should be noted that IOP elevation also induced a depolarized resting membrane potential in RGCs, in addition to hyperexcitability [28], while TNF-α had no significant effect on the membrane potential of RGCs. These results suggest that more factors may mediate RGC hyperexcitability in glaucoma, such as activated EphB/ephrinB signaling, group I metabotropic glutamate receptors, and G-protein-coupled dopamine receptors [28, 38, 54], which remains to be further investigated. It is noteworthy that there was evidence showing that IOP elevation in a mouse model induced a reduction in the frequency of spontaneous and light-evoked firing in RGCs when the IOP was elevated for 15 and 30 days [55]. For this apparent inconsistence, we speculated that it may be, at least in part, due to the recordings made at different stages of IOP elevation. Our recordings were made at an earlier stage (3–7 days after TNF-α injection). Progressive IOP elevation may result in RGC dendritic damage and loss of excitatory synapses at 15 and 30 days [55], thus reducing spontaneous firing. Actually, the spontaneous firing of rat RGCs was dependent upon presynaptic inputs [38]. Moreover, Risner et al. showed that a transient increase of light-induced firing frequency in RGCs was observed at 2 weeks after IOP elevation, and the frequency was decreased at 4 weeks [6].
In addition, Margolis and Detwiler reported that ON and OFF RGCs showed distinct spiking properties in the mouse retina: resting activity of ON RGCs was dependent on tonic excitatory inputs, whereas OFF RGCs exhibited properties of pacemaker neurons and continued to fire in the absence of synaptic inputs [56]. However, previous studies and our present results showed that in rat retina, the frequency of spontaneous firing and the membrane potential did not show significant differences between ON and OFF subtypes of RGCs [28, 38, 57]. To explain this inconsistency, we speculated that ON and OFF RGCs in mice might receive different synaptic inputs and have distinct intrinsic properties.
It should be noted that in dissociated RGCs, most dendrites and part of the axon of RGC may be destroyed during the isolated process. In this study, we usually chose the RGC with axon for recordings. Since dendrites were destroyed in these cells, some ion channels mainly expressed in dendrites, such as hyperpolarization-activated cation channel (Ih) and inwardly rectifying potassium channel (Kir) [38], were lost, which may change the excitability of RGCs. However, in this study, all recordings made in the dissociated RGCs were performed to record VGSC currents. Considering the fact that Nav1.6 VGSCs are mainly expressed in the soma and the axon, especially axon initial segment (AIS), it may be no significant influence on the results.
In the central nervous system (CNS), pleiotropic effects of TNF-α are mainly mediated by two signaling pathways, soluble TNF-α/TNFR1, and transmembrane TNF-α/TNFR2, which may lead to opposing outcomes, deleterious, or beneficial effects for neurons [18,19,20,21,22, 58]. Although we cannot precisely separate the functional outcomes of these two TNF-α signaling, it is possible that under pathological conditions, the dominant pro-inflammatory receptor TNFR1 plays an important role in neuronal injury. In this study, we showed that TNF-α-induced RGC hyperexcitability was mediated through activating TNFR1. Firstly, the TNFR1 inhibitor R7050 completely abolished the TNF-α-induced increase in spontaneous firing of RGCs in retinal slices. Secondly, the protein levels of TNFR1 in TNF-α injected and COH retinas were significantly upregulated, while TNFR2 expression was kept unchanged. Consistently, in glaucomatous patients, increased TNF-α concentration in aqueous humor and TNFR1 expression in the retina were observed, but TNFR2 expression levels showed unchanged [50]. However, it is noteworthy that activation of TNFR2 expressed in Müller cells promoted the production of TNF-α and may cause positive feedback to aggravate neuroinflammation in glaucoma [43, 59]. Therefore, the effects of TNFR2 in glaucoma remain to be addressed in the future. Furthermore, pre-injection of R7050 blocked the TNF-α-induced increase in the number of TUNEL-positive RGCs and partially reduced the number of apoptotic RGCs induced by IOP elevation, suggesting that TNF-α/TNFR1 signaling was indeed involved in RGC hyperexcitability and RGC injury in experimental glaucoma. These results are consistent with previous reports that neuronal hyperexcitability is associated with cell apoptosis [29,30,31, 60]. It should be noted that TNF-α-induced RGC hyperexcitability could increase Ca2+ influx through voltage-gated Ca2+ channels, together with activated Ca2+-permeable GluA2-lacking AMPA receptors [24], resulting in intracellular Ca2+ overload and triggering cellular death signaling [26].
TNF-α induces RGC hyperexcitability by selectively upregulating Nav1.6 currents
VGSCs are essential for AP generation and conduction. Modulation of Na+ channels may change neuronal excitability [61, 62]. We found that TNF-α-induced RGC hyperexcitability was mediated by upregulating Na+ currents, as evidenced by the following facts. Firstly, intravitreal injection of TNF-α significantly and voltage-dependently increased Na+ current density in RGCs. Secondly, external application of TNF-α reversibly increased Na+ currents. Thirdly, TNF-α shifted the activation curves of Na+ currents toward hyperpolarizing direction, suggesting that TNF-α resulted in Na+ channel activation easier. These results are consistent with observations in dorsal root ganglion (DRG) neurons that TNF-α promoted neuronal excitability by increasing sodium channel currents densities, thus contributing to neuropathic pain hypersensitivity [42, 63,64,65].
One of the major findings in this study is that TNF-α induced RGC hyperexcitability was mediated by selectively upregulating Nav1.6 currents although RGCs express the other subtypes of Na+ channels, such as Nav1.1 and Nav1.2 [45,46,47]. Nav1.6 channel is one of the important subtypes of Na+ channels that determine neuronal excitability in the CNS [46, 66]. Even though the TTX-resistant Nav1.8 subtype of Na+ channels was found expressed in mouse RGCs [67, 68], our previous study has shown that in rat RGCs, Nav1.6 is a predominate subtype of Na+ channels, while TTX-resistant Na+ channels seem unlikely expressed in rat RGCs [47]. In the present study, we showed that TNF-α regulated Nav1.6 channels through two pathways. The first one is that TNF-α directly upregulated the currents through intracellular p38 MAPK and STAT3 signaling pathways [43, 44]. It should be noted that p38 MAPK and STAT3 are two important transcriptional regulation signaling molecules. They may transcriptionally activate Na1.6 expression, thus enhancing Nav1.6 currents. However, we found that blocking p38 MAPK and STAT3 signaling pathways completely eliminated the acute TNF-α application-induced enhancement of Na+ currents, but did not influence the increased Nav1.6 protein expression due to longer TNF-α exposure (7 days), suggesting that these two signaling pathways may directly modulate Na+ channels in a transcriptional-independent manner. Indeed, previous studies reported that p38 MAPK was involved in the upregulation of different subtypes of Na+ currents [42, 69,70,71], and activation of STAT3 directly increased the activity of electrogenic Na+/HCO3- cotransporter 1 [72]. However, it was also reported that activation of p38 MAPK reduced the amplitude of peak Nav1.6 currents in the DRG-derived cell line ND7/23 and hippocampal neurons [71, 73]. It is possible that p38 MAPK may mediate different effects in different cells. The detailed mechanisms remain to be elucidated in our future study. The second one is that TNF-α upregulated the Nav1.6 expression in RGCs through NF-ĸB. NF-κB is one of the most important transcriptional factors. TNF-α has been reported to upregulate the expression of subtypes of Na+ channels, such as Nav1.7 and Nav1.3 [74, 75]. It is interesting that STAT3, another transcriptional factor, could promote the transcription and expression of Nav1.7-1.9 in DRG neurons [76], and TNF-α-activated STAT3 also facilitated Nav1.6 expression by increasing the histone H4 acetylation in Scn8a promoter in DRGs [64]. However, our data showed that inhibition of STAT3 failed to block the TNF-α-induced upregulation of Nav1.6 expression. Instead, STAT3 mediated the direct increase of Na+ currents by TNF-α in acutely isolated RGCs. In addition, immunohistochemical experiments showed that TNF-α-induced increase in Nav1.6 expression was mainly found in the axon-like structures. It was reported that Nav1.6 expressed in the distal of the AIS of cortical pyramidal neurons promoted action potential initiation [77]. Therefore, we speculated that upregulated expression of Nav1.6 in the TNF-α-injected retinas may contribute to RGCs hyperexcitability.
Nav1.6 is a potential therapeutic target in glaucoma
Neuroinflammation plays an important role in the pathogenesis of multiple neurodegenerative diseases, including glaucoma, Alzheimer’s disease (AD), and Parkinson’s disease (PD) [78,79,80,81]. Nav1.6 is highly expressed in the nodes of Ranvier and AIS of neurons [47, 82], and neurons that expressed Nav1.6 show a higher excitability [83]. Abnormal Nav1.6 expression is closely related to various neuronal diseases [84,85,86]. In this study, we showed that TNF-α-mediated Nav1.6 upregulation was a major factor for RGC hyperexcitability and injury in glaucoma, suggesting that sodium channel blockers selectively targeting on Nav1.6 may be a potential therapeutic strategy in the treatment of glaucoma. Indeed, specific inhibition of Nav1.6 has showed to effectively relieve neuronal hyperexcitability and neuropathic pain [87, 88]. NBI 921352 (known as XEN 901), a novel small molecule Nav1.6 inhibitor, has been approved by FDA to initiate the phase 2 clinical trial in epilepsy patients [89].
Conclusions
Taken together, all these results demonstrated that proinflammatory cytokine TNF-α induced the hyperexcitability of RGCs by enhancing Nav1.6 currents and protein expression through activating TNFR1, thus contributing to RGC apoptosis in glaucoma. Our results revealed a novel mechanism of TNF-α in glaucoma and provided a promising therapeutic target.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Abbreviations
- AHTTX:
-
4,9-Anhydrotetrodotoxin
- AP:
-
Action potential
- ACSF:
-
Artificial cerebral spinal fluid
- BCA:
-
Bicinchoninic acid
- COH:
-
Chronic ocular hypertension
- DRG:
-
Dorsal root ganglion
- d-APV:
-
d-(-)-2-amino-5-phosphonopentanoic acid
- IOP:
-
Intraocular pressure
- GCL:
-
Ganglion cell layer
- INL:
-
Inner nuclear layer
- IPL:
-
Inner plexiform layer
- OPL:
-
Outer plexiform layer
- ONL:
-
Outer nuclear layer
- R7050:
-
8-Chloro-4-(phenylthio)-1-(trifluoromethyl)-[1,2,4]triazolo[4,3-a]quinoxaline
- RGCs:
-
Retinal ganglion cells
- TNF-α:
-
Tumor necrosis factor-alpha
- TNFR1:
-
TNF-α receptor 1
- TNFR2:
-
TNF-α receptor 2
- TUNEL:
-
Terminal deoxynucleotidyl transferase dUTP nick end labeling
- VGSCs:
-
Voltage-gated sodium channels
References
Kingman S. Glaucoma is second leading cause of blindness globally. Bull World Health Organ. 2004;82(11):887–8. /S0042-96862004001100019.
Quigley HA, Broman AT. The number of people with glaucoma worldwide in 2010 and 2020. Br J Ophthalmol. 2006;90(3):262–7 https://doi.org/10.1136/bjo.2005.081224.
Tham YC, Li X, Wong TY, Quigley HA, Aung T, Cheng CY. Global prevalence of glaucoma and projections of glaucoma burden through 2040 a systematic review and meta-analysis. Ophthalmology. 2014;121(11):2081–90 https://doi.org/10.1016/j.ophtha.2014.05.013.
Calkins DJ. Critical pathogenic events underlying progression of neurodegeneration in glaucoma. Prog Retin Eye Res. 2012;31(6):702–19 https://doi.org/10.1016/j.preteyeres.2012.07.001.
Whitmore AV, Libby RT, John SW. Glaucoma: thinking in new ways - a role for autonomous axonal self-destruction and other compartmentalised processes? Prog Retin Eye Res. 2005;24(6):639–62 https://doi.org/10.1016/j.preteyeres.2005.04.004.
Risner ML, Pasini S, Cooper ML, Lambert WS, Calkins DJ. Axogenic mechanism enhances retinal ganglion cell excitability during early progression in glaucoma. Proc Natl Acad Sci U S A. 2018;115(10):E2393–402 https://doi.org/10.1073/pnas.1714888115.
Almasieh M, Wilson AM, Morquette B, Cueva Vargas JL, Di Polo A. The molecular basis of retinal ganglion cell death in glaucoma. Prog Retin Eye Res. 2012;31(2):152–81 https://doi.org/10.1016/j.preteyeres.2011.11.002.
Johnson EC, Deppmeier LM, Wentzien SK, Hsu I, Morrison JC. Chronology of optic nerve head and retinal responses to elevated intraocular pressure. Invest Ophthalmol Vis Sci. 2000;41(2):431–42.
Leske MC, Wu SY, Hennis A, Honkanen R, Nemesure B, BESs Study Group. Risk factors for incident open-angle glaucoma: the Barbados Eye Studies. Ophthalmology. 2008;115(1):85–93 https://doi.org/10.1016/j.ophtha.2007.03.017.
Liu B, McNally S, Kilpatrick JI, Jarvis SP, O'Brien CJ. Aging and ocular tissue stiffness in glaucoma. Surv Ophthalmol. 2018;63(1):56–74 https://doi.org/10.1016/j.survophthal.2017.06.007.
Pfeiffer N, Lamparter J, Gericke A, Grus FH, Hoffmann EM, Wahl J. Neuroprotection of medical IOP-lowering therapy. Cell Tissue Res. 2013;353(2):245–51 https://doi.org/10.1007/s00441-013-1671-1.
Tian K, Shibata-Germanos S, Pahlitzsch M, Cordeiro MF. Current perspective of neuroprotection and glaucoma. Clin Ophthalmol. 2015;9:2109–18 https://doi.org/10.2147/OPTH.S80445.
Baudouin C, Kolko M, Melik-Parsadaniantz S, Messmer EM. Inflammation in glaucoma: from the back to the front of the eye, and beyond. Prog Retin Eye Res. 2020;100916 https://doi.org/10.1016/j.preteyeres.2020.100916.
Wilson GN, Inman DM, Dengler-Crish CM, Smith MA, Crish SD. Early pro-inflammatory cytokine elevations in the DBA/2J mouse model of glaucoma. J Neuroinflammation. 2015;12(1):176. https://doi.org/10.1186/s12974-015-0399-0.
Ten Berge JC, Fazil Z, van den Born I, Wolfs RCW, Schreurs MWJ, Dik WA, et al. Intraocular cytokine profile and autoimmune reactions in retinitis pigmentosa, age-related macular degeneration, glaucoma and cataract. Acta Ophthalmol. 2019;97(2):185–92 https://doi.org/10.1111/aos.13899.
Moss ML, Jin SL, Milla ME, Bickett DM, Burkhart W, Carter HL, et al. Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-alpha. Nature. 1997;385(6618):733–6 https://doi.org/10.1038/385733a0.
Subedi L, Lee SE, Madiha S, Gaire BP, Jin M, Yumnam S, et al. Phytochemicals against TNF alpha-mediated neuroinflammatory diseases. Int J Mol Sci. 2020;21(3):764 https://doi.org/10.3390/ijms21030764.
Alvarez A, Cacabelos R, Sanpedro C, Garcia-Fantini M, Aleixandre M. Serum TNF-alpha levels are increased and correlate negatively with free IGF-I in Alzheimer disease. Neurobiol Aging. 2007;28(4):533–6 https://doi.org/10.1016/j.neurobiolaging.2006.02.012.
Clausen BH, Wirenfeldt M, Høgedal SS, Frich LH, Nielsen HH, Schroder HD, et al. Characterization of the TNF and IL-1 systems in human brain and blood after ischemic stroke. Acta Neuropathol Commun. 2020;8(1):81 https://doi.org/10.1186/s40478-020-00957-y.
Sivakumar V, Foulds WS, Luu CD, Ling EA, Kaur C. Retinal ganglion cell death is induced by microglia derived pro-inflammatory cytokines in the hypoxic neonatal retina. J Pathol. 2011;224(2):245–60 https://doi.org/10.1002/path.2858.
Lee JC, Park CW, Shin MC, Cho JH, Lee HA, Kim YM, et al. Tumor necrosis factor receptor 2 is required for ischemic preconditioning-mediated neuroprotection in the hippocampus following a subsequent longer transient cerebral ischemia. Neurochem Int. 2018;118:292–303 https://doi.org/10.1016/j.neuint.2018.05.008.
Pegoretti V, Baron W, Laman JD, Eisel ULM. Selective modulation of TnF-TnFRs signaling: insights for multiple sclerosis treatment. Front Immunol. 2018;9:925 https://doi.org/10.3389/fimmu.2018.00925.
Sawada H, Fukuchi T, Tanaka T, Abe H. Tumor necrosis factor-alpha concentrations in the aqueous humor of patients with glaucoma. Invest Ophthalmol Vis Sci. 2010;51(2):903–6 https://doi.org/10.1167/iovs.09-4247.
Cueva Vargas JL, Osswald IK, Unsain N, Aurousseau MR, Barker PA, Bowie D, et al. Soluble tumor necrosis factor alpha promotes retinal ganglion cell death in glaucoma via calcium-permeable AMPA receptor activation. J Neurosci. 2015;35(35):12088–102. https://doi.org/10.1523/JNEUROSCI.1273-15.2015.
Roh M, Zhang Y, Murakami Y, Thanos A, Lee SC, Vavvas DG, et al. Etanercept, a widely used inhibitor of tumor necrosis factor-α (TNF-α), prevents retinal ganglion cell loss in a rat model of glaucoma. PLoS One. 2012;7(7):e40065. https://doi.org/10.1371/journal.pone.0040065.
De Groef L, Salinas-Navarro M, Van Imschoot G, Libert C, Vandenbroucke RE, Moons L. Decreased TNF levels and improved retinal ganglion cell survival in MMP-2 null mice suggest a role for MMP-2 as TNF Sheddase. Mediat Inflamm. 2015;2015:108617 https://doi.org/10.1155/2015/108617.
Liu ST, Zhong SM, Li XY, Gao F, Li F, Zhang ML, et al. EphrinB/EphB forward signaling in Muller cells causes apoptosis of retinal ganglion cells by increasing tumor necrosis factor alpha production in rat experimental glaucomatous model. Acta Neuropathol Commun. 2018;6(1):111 https://doi.org/10.1186/s40478-018-0618-x.
Zhao Y, Li Q, Li XY, Cui P, Gao F, Zhu K, et al. Involvement of mGluR I in EphB/ephrinB reverse signaling activation induced retinal ganglion cell apoptosis in a rat chronic hypertension model. Brain Res. 2018;1683:27–35 https://doi.org/10.1016/j.brainres.2018.01.017.
Ashpole NM, Song W, Brustovetsky T, Engleman EA, Brustovetsky N, Cummins TR, et al. Calcium/calmodulin-dependent protein kinase II (CaMKII) inhibition induces neurotoxicity via dysregulation of glutamate/calcium signaling and hyperexcitability. J Biol Chem. 2012;287(11):8495–506 https://doi.org/10.1074/jbc.M111.323915.
Turovsky EA, Turovskaya MV, Kononov AV, Zinchenko VP. Short-term episodes of hypoxia induce posthypoxic hyperexcitability and selective death of GABAergic hippocampal neurons. Exp Neurol. 2013;250:1–7 https://doi.org/10.1016/j.expneurol.2013.09.006.
Zucchini S, Buzzi A, Barbieri M, Rodi D, Paradiso B, Binaschi A, et al. Fgf-2 overexpression increases excitability and seizure susceptibility but decreases seizure-induced cell loss. J Neurosci. 2008;28(49):13112–24. https://doi.org/10.1523/JNEUROSCI.1472-08.2008.
Diem R, Meyer R, Weishaupt JH, Bahr M. Reduction of potassium currents and phosphatidylinositol 3-kinase-dependent Akt phosphorylation by tumor necrosis factor-alpha rescues axotomized retinal ganglion cells from retrograde cell death in vivo. J Neurosci. 2001;21(6):2058–66 https://doi.org/10.1523/JNEUROSCI.21-06-02058.2001.
Remtulla S, Hallett PE. A schematic eye for the mouse, and comparisons with the rat. Vis Res 1985;25(1):21-31. https://doi.org/10.1016/0042-6989(85)90076-8
Yu DY, Cringle SJ. Oxygen distribution in the mouse retina. Invest Ophthalmol Vis Sci. 2006;47(3):1109–12 https://doi.org/10.1167/iovs.05-1118.
Xu LJ, Gao F, Cheng S, Zhou ZX, Li F, Miao Y, et al. Activated ephrinA3/EphA4 forward signaling induces retinal ganglion cell apoptosis in experimental glaucoma. Neuropharmacology. 2020;178:108228 https://doi.org/10.1016/j.neuropharm.2020.108228.
Krishna R, Mermoud A, Baerveldt G, Minckler DS. Circadian rhythm of intraocular pressure: a rat model. Ophthalmic Res. 1995;27(3):163–7 https://doi.org/10.1159/000267660.
Li Q, Wu N, Cui P, Gao F, Qian WJ, Miao Y, et al. Suppression of outward K(+) currents by activating dopamine D1 receptors in rat retinal ganglion cells through PKA and CaMKII signaling pathways. Brain Res. 2016;1635:95–104 https://doi.org/10.1016/j.brainres.2016.01.039.
Li Q, Cui P, Miao Y, Gao F, Li XY, Qian WJ, et al. Activation of group I metabotropic glutamate receptors regulates the excitability of rat retinal ganglion cells by suppressing Kir and I h. Brain Struct Funct. 2017;222(2):813–30 https://doi.org/10.1007/s00429-016-1248-3.
Lucas-Ruiz F, Galindo-Romero C, Salinas-Navarro M, Gonzalez-Riquelme MJ, Vidal-Sanz M, Agudo BM. Systemic and intravitreal antagonism of the TNFR1 signaling pathway delays axotomy-induced retinal ganglion cell loss. Front Neurosci. 2019;13:1096 https://doi.org/10.3389/fnins.2019.01096.
He XH, Zang Y, Chen X, Pang RP, Xu JT, Zhou XA, et al. TNF-alpha contributes to up-regulation of Nav1.3 and Nav1.8 in DRG neurons following motor fiber injury. Pain. 2010;151(2):266–79 https://doi.org/10.1016/j.pain.2010.06.005.
Zhang XL, Ding HH, Xu T, Liu M, Ma C, Wu SL, et al. Palmitoylation of delta-catenin promotes kinesin-mediated membrane trafficking of Nav1.6 in sensory neurons to promote neuropathic pain. Sci Signal. 2018;11(523):eaar4394 https://doi.org/10.1126/scisignal.aar4394.
Li Q, Qin L, Li JH. Enhancement by TNF-alpha of TTX-resistant Na-V current in muscle sensory neurons after femoral artery occlusion. Am J Phys Regul Integr Comp Phys. 2020;318(4):R772–80 https://doi.org/10.1152/ajpregu.00338.2019.
Bai Y, Dergham P, Nedev H, Xu J, Galan A, Rivera JC, et al. Chronic and acute models of retinal neurodegeneration TrkA activity are neuroprotective whereas p75NTR activity is neurotoxic through a paracrine mechanism. J Biol Chem. 2010;285(50):39392–400 https://doi.org/10.1074/jbc.M110.147801.
Sabio G, Davis RJ. TNF and MAP kinase signalling pathways. Semin Immunol. 2014;26(3):237–45 https://doi.org/10.1016/j.smim.2014.02.009.
Fjell J, Dib-Hajj S, Fried K, Black JA, Waxman SG. Differential expression of sodium channel genes in retinal ganglion cells. Brain Res Mol Brain Res 1997;50(1-2):197-204. https://doi.org/10.1016/s0169-328x(97)00187-3
Van Wart A, Matthews G. Expression of sodium channels Nav1.2 and Nav1.6 during postnatal development of the retina. Neurosci Lett. 2006;403(3):315–7 https://doi.org/10.1016/j.neulet.2006.05.019.
Yin N, Yang YL, Cheng S, Wang HN, Hu X, Miao Y, et al. Dopamine D2 receptor-mediated modulation of rat retinal ganglion cell excitability. Neurosci Bull. 2020;36(3):230–42 https://doi.org/10.1007/s12264-019-00431-3.
Williams PA. Marsh-Armstrong N, Howell GR, The Lasker/IRRF initiative on astrocytes and glaucomatous neurodegeneration participants. Neuroinflammation in glaucoma: A new opportunity. Exp Eye Res. 2017;157:20–7 https://doi.org/10.1016/j.exer.2017.02.014.
Soto I, Howell GR. The complex role of neuroinflammation in glaucoma. Cold Spring Harb Perspect Med. 2014;4(8):a017269 https://doi.org/10.1101/cshperspect.a017269.
Yang X, Luo C, Cai J, Powell DW, Yu D, Kuehn MH, et al. Neurodegenerative and inflammatory pathway components linked to TNF-alpha/TNFR1 signaling in the glaucomatous human retina. Invest Ophthalmol Vis Sci. 2011;52(11):8442–54 https://doi.org/10.1167/iovs.11-8152.
Wei X, Cho K-S, Thee EF, Jager MJ, Chen DF. Neuroinflammation and microglia in glaucoma: time for a paradigm shift. J Neurosci Res. 2019;97(1):70–6 https://doi.org/10.1002/jnr.24256.
McGrady NR, Risner ML, Vest V, Calkins DJ. TRPV1 tunes optic nerve axon excitability in glaucoma. Front Physiol. 2020;11:249 https://doi.org/10.3389/fphys.2020.00249.
Tao X, Sabharwal J, Wu SM, Frankfort BJ. Intraocular pressure elevation compromises retinal ganglion cell light adaptation. Invest Ophthalmol Vis Sci. 2020;61(12):15 https://doi.org/10.1167/iovs.61.12.15.
Cui P, Li XY, Zhao Y, Li Q, Gao F, Li LZ, et al. Activation of dopamine D1 receptors enhances the temporal summation and excitability of rat retinal ganglion cells. Neuroscience. 2017;355:71–83 https://doi.org/10.1016/j.neuroscience.2017.04.046.
Della Santina L, Inman DM, Lupien CB, Horner PJ, Wong RO. Differential progression of structural and functional alterations in distinct retinal ganglion cell types in a mouse model of glaucoma. J Neurosci. 2013;33(44):17444–57 https://doi.org/10.1523/JNEUROSCI.5461-12.2013.
Margolis DJ, Detwiler PB. Different mechanisms generate maintained activity in ON and OFF retinal ganglion cells. J Neurosci. 2007;27(22):5994–6005 https://doi.org/10.1523/JNEUROSCI.0130-07.2007.
Zhou X, Li G, Yang B, Wu J. Quercetin enhances inhibitory synaptic inputs and reduces excitatory synaptic inputs to OFF- and ON-type retinal ganglion cells in a chronic glaucoma rat model. Front Neurosci. 2019;13:672 https://doi.org/10.3389/fnins.2019.00672.
Probert L. TNF and its receptors in the CNS: the essential, the desirable and the deleterious effects. Neuroscience. 2015;302:2–22 https://doi.org/10.1016/j.neuroscience.2015.06.038.
Lebrun-Julien F, Bertrand MJ, De Backer O, Stellwagen D, Morales CR, Di Polo A, et al. ProNGF induces TNF alpha-dependent death of retinal ganglion cells through a p75(NTR) non-cell-autonomous signaling pathway. Proc Natl Acad Sci U S A. 2010;107(8):3817–22 https://doi.org/10.1073/pnas.0909276107.
Pasantes-Morales H, Tuz K. Volume changes in neurons: hyperexcitability and neuronal death. Contrib Nephrol. 2006;152:221–40 https://doi.org/10.1159/000096326.
Goldfarb M, Schoorlemmer J, Williams A, Diwakar S, Wang Q, Huang X, et al. Fibroblast growth factor homologous factors control neuronal excitability through modulation of voltage-gated sodium channels. Neuron. 55(3):449–63 https://doi.org/10.1016/j.neuron.2007.07.006.
Wang Y, Du X, Bin R, Yu S, Xia Z, Zheng G, et al. Genetic variants identified from epilepsy of unknown etiology in Chinese children by targeted exome sequencing. Sci Rep. 2017;7(1):40319. https://doi.org/10.1038/srep40319.
de Macedo FHP, Aires RD, Fonseca EG, Ferreira RCM, Machado DPD, Chen L, et al. TNF-alpha mediated upregulation of NaV1.7 currents in rat dorsal root ganglion neurons is independent of CRMP2 SUMOylation. Mol Brain. 2019;12(1):117 https://doi.org/10.1186/s13041-019-0538-0.
Ding HH, Zhang SB, Lv YY, Ma C, Liu M, Zhang KB, et al. TNF-alpha/STAT3 pathway epigenetically upregulates Nav1.6 expression in DRG and contributes to neuropathic pain induced by L5-VRT. J Neuroinflammation. 2019;16(1):29 https://doi.org/10.1186/s12974-019-1421-8.
Leo M, Argalski S, Schafers M, Hagenacker T. Modulation of voltage-gated sodium channels by activation of tumor necrosis factor receptor-1 and receptor-2 in small DRG neurons of rats. Mediat Inflamm. 2015;2015:124942 https://doi.org/10.1155/2015/124942.
Mojumder DK, Frishman LJ, Otteson DC, Sherry DM. Voltage-gated sodium channel alpha-subunits Na(v)1.1, Na(v)1.2, and Na(v)1.6 in the distal mammalian retina. Mol Vis. 2007;13:2163–82.
O'Brien BJ, Caldwell JH, Ehring GR, Bumsted O'Brien KM, Luo S, Levinson SR. Tetrodotoxin-resistant voltage-gated sodium channels Na(v)1.8 and Na(v)1.9 are expressed in the retina. J Comp Neurol. 2008;508(6):940–51 https://doi.org/10.1002/cne.21701.
Smith BJ, Côté PD, Tremblay F. Contribution of Nav1.8 sodium channels to retinal function. Neuroscience. 2017;340:279–90 https://doi.org/10.1016/j.neuroscience.2016.10.054.
Chen W, Sheng J, Guo J, Gao F, Zhao X, Dai J, Wang G, Li K Tumor necrosis factor-alpha enhances voltage-gated Na(+) currents in primary culture of mouse cortical neurons. J Neuroinflammation 2015;12:126. https://doi.org/10.1186/s12974-015-0349-x, 1
Jin X, Gereau RW 4th. Acute p38-mediated modulation of tetrodotoxin-resistant sodium channels in mouse sensory neurons by tumor necrosis factor-alpha. J Neurosci. 2006;26(1):246–55 https://doi.org/10.1523/JNEUROSCI.3858-05.2006.
Wittmack EK, Rush AM, Hudmon A, Waxman SG, Dib-Hajj SD. Voltage-gated sodium channel Nav1.6 is modulated by p38 mitogen-activated protein kinase. J Neurosci. 2005;25(28):6621–30 https://doi.org/10.1523/JNEUROSCI.0541-05.2005.
Giannaki M, Schrödl-Häußel M, Khakipoor S, Kirsch M, Roussa E. STAT3-dependent regulation of the electrogenic Na+/HCO3- cotransporter 1 (NBCe1) functional expression in cortical astrocytes. J Cell Physiol. 2021;236(3):2036–50 https://doi.org/10.1002/jcp.29990.
Gasser A, Cheng X, Gilmore ES, Tyrrell L, Waxman SG, Dib-Hajj SD. Two Nedd4-binding motifs underlie modulation of sodium channel Nav1.6 by p38 MAPK. J Biol Chem. 2010;285(34):26149–61 https://doi.org/10.1074/jbc.M109.098681.
Huang Y, Zang Y, Zhou L, Gui W, Liu X, Zhong Y. The role of TNF-alpha/NF-kappa B pathway on the up-regulation of voltage-gated sodium channel Nav1.7 in DRG neurons of rats with diabetic neuropathy. Neurochem Int. 2014;75:112–9 https://doi.org/10.1016/j.neuint.2014.05.012.
Zang Y, He XH, Xin WJ, Pang RP, Wei XH, Zhou LJ, et al. Inhibition of NF-kappaB prevents mechanical allodynia induced by spinal ventral root transection and suppresses the re-expression of Nav1.3 in DRG neurons in vivo and in vitro. Brain Res. 2010;1363:151–8 https://doi.org/10.1016/j.brainres.2010.09.048.
Zhang F, Wang Y, Liu Y, Han H, Zhang D, Fan X, et al. Transcriptional regulation of voltage-gated sodium channels contributes to GM-CSF-induced pain. J Neurosci. 2019;39(26):5222–33 https://doi.org/10.1523/JNEUROSCI.2204-18.2019.
Hu W, Tian C, Li T, Yang M, Hou H, Shu Y. Distinct contributions of Na(v)1.6 and Na(v)1.2 in action potential initiation and backpropagation. Nat Neurosci. 2009;12(8):996–1002 https://doi.org/10.1038/nn.2359.
Adornetto A, Russo R, Parisi V. Neuroinflammation as a target for glaucoma therapy. Neural Regen Res. 2019;14(3):391–4 https://doi.org/10.4103/1673-5374.245465.
Decourt B, Lahiri DK, Sabbagh MN. Targeting tumor necrosis factor alpha for Alzheimer’s disease. Curr Alzheimer Res. 2017;14(4):412–25 https://doi.org/10.2174/1567205013666160930110551.
Ransohoff RM. How neuroinflammation contributes to neurodegeneration. Science. 2016;353(6301):777–83 https://doi.org/10.1126/science.aag2590.
Risner ML, McGrady NR, Pasini S, Lambert WS, Calkins DJ. Elevated ocular pressure reduces voltage-gated sodium channel NaV1.2 protein expression in retinal ganglion cell axons. Exp Eye Res. 2020;190:107873 https://doi.org/10.1016/j.exer.2019.107873.
Trimmer JS, Rhodes KJ. Localization of voltage-gated ion channels in mammalian brain. Annu Rev Physiol. 2004;66(1):477–519. https://doi.org/10.1146/annurev.physiol.66.032102.113328.
Xu DE, Zhang WM, Yang ZZ, Zhu HM, Yan K, Li S, et al. Amyloid precursor protein at node of Ranvier modulates nodal formation. Cell Adhes Migr. 2014;8(4):396–403 https://doi.org/10.4161/cam.28802.
Chen L, Huang J, Benson C, Lankford KL, Zhao P, Carrara J, et al. Sodium channel Nav1.6 in sensory neurons contributes to vincristine-induced allodynia. Brain. 2020;143(8):2421–36 https://doi.org/10.1093/brain/awaa208.
Craner MJ, Hains BC, Lo AC, Black JA, Waxman SG. Co-localization of sodium channel Nav1.6 and the sodium-calcium exchanger at sites of axonal injury in the spinal cord in EAE. Brain. 2004;127(Pt 2):294–303 https://doi.org/10.1093/brain/awh032.
Patel RR, Barbosa C, Brustovetsky T, Brustovetsky N, Cummins TR. Aberrant epilepsy-associated mutant Nav1.6 sodium channel activity can be targeted with cannabidiol. Brain. 2016;139(Pt 8):2164–81 https://doi.org/10.1093/brain/aww129.
Hargus NJ, Nigam A, Bertram EH 3rd, Patel MK. Evidence for a role of Nav1.6 in facilitating increases in neuronal hyperexcitability during epileptogenesis. J Neurophysiol. 2013;110(5):1144–57 https://doi.org/10.1152/jn.00383.2013.
Li L, Shao J, Wang J, Liu Y, Zhang Y, Zhang M, et al. MiR-30b-5p attenuates oxaliplatin-induced peripheral neuropathic pain through the voltage-gated sodium channel Nav1.6 in rats. Neuropharmacology. 2019;153:111–20 https://doi.org/10.1016/j.neuropharm.2019.04.024.
Bialer M, Johannessen SI, Koepp MJ, Levy RH, Perucca E, Perucca P, et al. Progress report on new antiepileptic drugs: a summary of the Fifteenth Eilat Conference on New Antiepileptic Drugs and Devices (EILAT XV). I. Drugs in preclinical and early clinical development. Epilepsia. 2020;61(11):2365–85 https://doi.org/10.1111/epi.16726.
Acknowledgements
We thank Dr. Xiong-Li Yang for the helpful discussion and critical comments on the manuscript.
Funding
This work was supported by grants from the National Natural Science Foundation of China (81790642, 31872765), the Shanghai Municipal Science and Technology Major Project (No.2018SHZDZX01), ZJ Lab, and the Shanghai Center for Brain Science and Brain-Inspired Technology.
Author information
Authors and Affiliations
Contributions
ZW and XHS conceived and designed the experiments; SC, HNW, LJX, FL, and YM performed the experiments; SC, BL, XHS, and ZW analyzed the data; SC, XHS, and ZW wrote the manuscript. The authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All experiments described in this study were carried out in accordance with the National Institutes of Health (NIH) guidelines for the Care and Use of Laboratory Animals and were approved by the Institutes of Brain Science at Fudan University.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Cheng, S., Wang, HN., Xu, LJ. et al. Soluble tumor necrosis factor-alpha-induced hyperexcitability contributes to retinal ganglion cell apoptosis by enhancing Nav1.6 in experimental glaucoma. J Neuroinflammation 18, 182 (2021). https://doi.org/10.1186/s12974-021-02236-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12974-021-02236-6