
Abstract
Background
Anti-N-methyl-D-aspartate receptor (NMDAR) encephalitis is a recently discovered autoimmune syndrome associated with psychosis, dyskinesia, and seizures. However, the underlying mechanisms of this disease remain unclear, in part because of a lack of suitable animal models.
Methods
This study describes a novel female C57BL/6 mouse model of anti-NMDAR encephalitis that was induced by active immunization against NMDARs using an amino terminal domain (ATD) peptide from the GluN1 subunit (GluN1356–385).
Results
Twelve weeks after immunization, the immunized mice showed significant memory loss. Furthermore, antibodies from the cerebrospinal fluid of immunized mice decreased the surface NMDAR cluster density in hippocampal neurons which was similar to the effect induced by the anti-NMDAR encephalitis patients’ antibodies. Immunization also impaired long-term potentiation at Schaffer collateral–CA1 synapses and reduced NMDAR-induced calcium influx.
Conclusion
We established a novel anti-NMDAR encephalitis model using active immunization with peptide GluN1356–385 targeting the ATD of GluN1. This novel model may allow further research into the pathogenesis of anti-NMDAR encephalitis and aid in the development of new therapies for this disease.
Similar content being viewed by others

Background
Anti-N-methyl-D-aspartate receptor (NMDAR) encephalitis is an autoimmune disorder of the central nervous system (CNS) that predominantly affects young females. The typical clinical manifestations include progressive development of neurologic and psychiatric symptoms, including abnormal movements, seizures, impaired memory, and behavior disorders [1]. Nearly 80% of patients show CSF laboratory abnormalities, presenting as mild lymphocytic pleocytosis and normal or moderately increased protein levels and 60 % of patients have specific CSF oligoclonal bands [2, 3].
It is believed that the ectopic expression of NMDARs contributes to triggering the immune response [4]. Antigens are taken up by antigen-presenting cells that travel to regional lymph nodes, and plasma cells produce antibodies (ABs) that later react with NMDARs in the brain, impairing blood-brain barrier permeability [5]. NMDARs mediate glutamatergic synaptic transmission and play a prominent role in synaptic plasticity. Previous studies have suggested that the pathogenicity of autoimmune ABs is a key mechanism of anti-NMDAR encephalitis [1, 6]. Human CSF-derived NMDAR ABs were shown to downregulate NMDAR levels in both in vitro and in vivo studies [7, 8]. In addition, reduced NMDAR expression can result in increased extracellular glutamate and thus affect the pons/medullary respiratory center [7].
Passive immunization in mice, using intrathecal infusion of CSF from affected humans, leads to behaviors of depression-like behaviors, anhedonia, and memory deficits. Although these studies provide compelling evidence of the pathogenicity of antibodies, it is unable to show the process of autoantibody production in vivo [9, 10]. Thus, an animal model of active immunity is needed that more closely mimics disease progression.
Previous investigations have highlighted the extracellular amino-terminal domain (ATD) of the GluN1 subunit, especially the N368/G369 region, as essential for immunoreactivity [11, 12]. Therefore, ATD peptides were used to immunize mice. Twelve weeks later, the mice demonstrated behavioral changes and AB infiltration, which was most prominent in the hippocampus. The presence of GluN1 ABs and their effect on NMDARs were also confirmed.
Methods
Study design and mice immunization
The study aimed to investigate the effects of active immunization with NMDAR peptides in normal adult mice. C57BL/6 mice (10 weeks old, female) were immunized with different GluN1 extracellular peptides emulsified in an equal volume of Complete Freund’s Adjuvant (CFA) supplemented with Mycobacterium tuberculosis H37Ra (4 mg/mL) at a final peptide concentration of 1 mg/mL. Mice were immunized subcutaneously on the back with 200 μg of peptide in the emulsion mixture and received two booster injections with peptide emulsion at 4 and 8 weeks after the first immunization. Mice in control group received emulsion mixture of CFA and equal volume of phosphate-buffered saline (PBS). All mice were intraperitoneally injected with 200 ng of pertussis toxin (List Biological Laboratories) on the day of the last immunization and 48 h later.
To detect antibody titer, cerebrospinal fluid and serum were obtained from three mice and tested every other week. Behavioral tests and histological staining were performed 12 weeks after the first immunization.
Patient sample collection
We collected CSF from patients with high titers of anti-GluN1 ABs (> 1:300) during routine clinical examinations. All patients fulfilled the clinical diagnostic criteria for anti-NMDAR encephalitis, revised in 2016 [13]. The study protocol was approved by the ethics committee of Nanfang Hospital, Southern Medical University, and written informed consent was obtained from each participant or their guardian.
Antibodies purification
CSF ABs from patients or immunized mice were purified using protein G Sepharose columns and were then used to treat neurons or brain slices. Considering the low amount of CSF obtained from an individual mouse, we pooled the CSF of 40 mice for further procedures. For the purification process, 2 mL of sample diluted with PBS was incubated in a chromatography spin column (Thermo Scientific) of protein G Sepharose beads for 30 min. After three washes with PBS, the samples were eluted with elution buffer, dialyzed against PBS, concentrated in stock solutions of 4 mg/mL, and stored at – 80 °C until use.
Preparation and staining of GluN1-expressing HEK cells
Human embryonic kidney 293 (HEK293) cells were transiently transfected with NMDAR subunit genes (NR1/NR2A; DsRed2-labeled) as previously described [14]. Twenty-four hours later, cells were fixed on coverslips with acetone and incubated overnight at 4 °C with the purified ABs from patients or CSF from immunized mice (starting at a 1:1 ratio) in PBS containing 5 % bovine serum albumin (BSA). After washing with PBS, the cells were labeled with fluorescein isothiocyanate (FITC)-conjugated anti-human IgG (ab7149, Abcam) or FITC-conjugated anti-mouse IgG (ab6785, Abcam) and observed under a fluorescence microscope (BX51, Olympus, Japan).
Site-directed mutagenesis
A point mutation was generated using the Stratagene QuikChange Mutagenesis kit (210518, Agilent) according to the manufacturer’s instructions. The following primers were designed for the N368Q point mutation: Forward: 5′-gggatgacatgggtaccttggtagatg cccacttgca-3′; Reverse: 5′-tgcaagtgggcatctaccaaggtacccatgtcatccc-3′.
Primary neuronal cultures
Hippocampal neurons from embryonic day 18 rat brains were prepared and maintained as previously described [15]. The hippocampi were dissociated with papain at 37 °C for 30 min, and then separated with a fire-polished Pasteur pipette. After centrifugation at 300×g, the cells were resuspended in Neurobasal medium. Cells were counted and plated onto poly-D-lysine-treated 24-well plates. After 6 h, the supernatant was removed and replaced with 500 μL of fresh culture medium. Cells were then cultured for 14 days for subsequent experiments.
Immunocytochemistry
Immunocytochemistry was performed to detect autoantibody 12 weeks after immunization with GluN1356–385 peptide or a control peptide GluN1369–386 which was unable to induce specific ABs. The mouse brains were cut into 15 mm slices, permeabilized with 0.5% Triton X-100, preincubated with 10% normal goat serum, and incubated with fluorescent secondary ABs (1:200, anti-Mouse Alexa Fluor 488, A-11029) to detect autoantibodies. The slices were imaged using a confocal microscope (LSM 880, Carl Zeiss, Germany). To stain surface NMDARs, primary hippocampal neurons were incubated with ABs (5 μg/mL) derived from the CSF of patients or immunized mice. After 18 h of incubation, the AB-bound surface receptors were incubated with fluorescently conjugated secondary ABs (1:200, anti-Mouse Alexa Fluor 488, A-11029; 1:200, anti-Human Alexa Fluor 488, A-11013; Invitrogen) at room temperature for 1 h. Cells were then fixed with methanol at – 20 °C for 5 min, permeabilized with (0.5%) Triton X-100 for 10 min, blocked in 10% normal goat serum for 30 min and incubated with anti-PSD95 primary AB (1:200, Synaptic Systems, 124-003) at 4 °C overnight to label postsynaptic densities, and visualized after staining with Alexa Fluor 647 (1:200, Invitrogen, A-21244) for 1 h at room temperature.
Electrophysiological recording
Brain slice preparation and electrophysiological recording were performed as previously described [16]. Mouse hippocampal slices (300 μm) were prepared using a vibratome (VT1000S, Leica, Germany). The slices were kept at 30 °C for at least 60 min before experiments in artificial CSF (ACSF; NaCl 124 mM, KCl 2.5 mM, MgSO4 2.0 mM, NaH2PO4 1.25 mM, NaHCO3 26 mM, CaCl2 2 mM, and glucose 10 mM; pH 7.3), bubbled with a mixture of 95% O2 and 5% CO2. Field excitatory postsynaptic potentials (fEPSP) were evoked in the CA1 stratum radiatum by stimulating Schaffer collaterals with a two-concentric bipolar stimulating electrode, and were recorded with ACSF-filled glass pipettes. LTP was induced by applying theta burst stimulation. Purified ABs were diluted in ACSF (100 μg/mL) and applied by switching the perfusion from the control ACSF to the AB-containing ACSF. For each recording, the baseline synaptic transmission was monitored for 10 min before AB perfusion, and AB-containing ACSF was continuously washed out with ACSF after theta burst stimulation until the end of the experiment. For immunized mice, fEPSP were evoked in brain slices in the same way and recorded for 20 min as the baseline prior to the induction of LTP. The acquired data were analyzed with pClamp 10 software (Axon Instruments, USA).
Calcium imaging
Hippocampal neurons were incubated with 20 μg/mL of patient or mouse ABs at 37 °C for 18 h. To detect calcium flux, the cells were loaded with Fura-2 (1 μM, Invitrogen, F1221) and incubated at 37 °C for 15–30 min, followed by a 15–30-min incubation at room temperature. After washing in Tyrode’s solution, the cells were transferred into wells containing NBQX (10 μM, Tocris, 0373). NMDA (10 μM, Sigma, M3262) was used to stimulate NMDAR-mediated calcium influx. Imaging was performed using an inverted fluorescence microscope (Eclipse TE2000-U, Nikon, Japan) with a charge-coupled device camera. Image series of images were acquired at 800 ms intervals for 40 s at an excitation wavelength of 470 nm. All data were obtained from five independent samples. The fluorescence intensity at each timepoint was measured using ImageJ software (NIH).
Behavioral assessments
Twelve weeks after immunization, mice were tested using a series of behavioral experiments. Behavioral testing was conducted at uniform times, from 09:00 to 12:00, by researchers blinded to the group allocations. Behavioral parameters were recorded using a video tracking system (Smart 3.0, Panlab, Spain). Details of behavioral tests are described in the Supplementary Methods.
Western blot analyses
Minute Plasma Membrane Protein Isolation Kit (SM-005, Invent Biotechnologies) was used to extract membrane and total cell proteins from the hippocampus and primary neurons according to the manufacturer's protocol. Forty micrograms of the membrane protein or total cell protein sample was mixed with sodium dodecyl sulfate (SDS) sample buffer and boiled at 98 °C for 10 min. Samples were separated by electrophoresis on 10% SDS-polyacrylamide gels and transferred to polyvinylidene fluoride membranes. After blocking with 5% BSA, the membranes were incubated with anti-GluN1 ABs (1:500, Synaptic Systems, 114-011) and anti-β-actin ABs (1:2000, Proteintech, 20536-1) overnight at 4 °C, washed three times and then incubated with horseradish peroxidase-conjugated anti-mouse or anti-rabbit secondary ABs (1:2000; Proteintech) for 1 h at room temperature. The immunocomplexes were detected using enhanced chemiluminescence (Thermo Scientific).
Statistical analyses
Data are presented as mean ± SEM. Independent samples t tests or Mann–Whitney U tests were used as appropriate for each experiment. Kruskal-Wallis test was used to analyze nonparametric data. All analyses were performed using SPSS V24.0 (IBM, USA) and the histograms were plotted using GraphPad Prism 6.0 (GraphPad Software, USA) or ImageJ. A value of P < 0.05 was considered statistically significant.
Results
Detection of antigen specificity of peptide-induced autoantibodies
To induce an active immune mouse model of anti-NMDAR encephalitis, mice were immunized by subcutaneous injection with 200 μg of peptide emulsified in Complete Freund’s Adjuvant (Fig. 1a). CSF from peptide-immunized mice was collected 14 days after immunization. The CSF ABs were purified and detected in HEK293 cells transfected with GluN1 subunits (DsRed2-labeled). We found that only the GluN1356–385 peptide could induce CSF autoantibodies that were specific for GluN1 (Fig. 1b).

Characterization of ABs from the CSF of mice immunized with peptides of the GluN1 subunit. a The peptide sequences used in the immunization experiment. b Immunofluorescence detection of GluN1 ATD peptide induced ABs against GluN1 protein using HEK293 cells
Binding properties of GluN1356–385 peptide-induced ABs and their effects on NMDARs in vitro
Binding of ABs to GluN1 from human patients can be prevented by a single amino acid mutation in the ATD of GluN1 (N368Q) [11]. We therefore generated a GluN1 subunit construct with a mutation at amino acid 368 (N368Q) and tested the GluN1-specific clones for their reactivity in transfected HEK293 cells transfected with the mutant construct. The results showed that binding to the mutant was eliminated for the GluN1356–385 ABs (Fig. 2a).

Epitope analysis of GluN1356-385 ABs and their effects in vitro. a ABs were applied to HEK293 cells transfected with wild-type GluN1 or a construct with mutated amino acid 368 (N368Q). b, e Hippocampal neurons were immunostained for surface GluN1and PSD95 after treatment for 24 h with patients’, GluN1 ABs, or control ABs and imaged with confocal microscopy. c, d Western blot analysis of GluN1 on membrane fraction and total cells. f Electrophysiological effects of patients’ AB and GluN1356–385 AB on tetanus-induced LTP on brain slices (f, left) (n = 5). Example traces of fEPSPs recorded before (black) and after (red) TBS (f, left, top). Normalized slope of field excitatory postsynaptic potentials in hippocampal slices after ABs treatment (f, right). g Effects of patients’ AB and GluN1356–385 AB on NMDAR-mediated calcium influx (n = 5). *P < 0.05; **P < 0.01
The binding of the GluN1356–385 ABs to GluN1 led to the question of whether GluN1356–385 ABs mediate the internalization of NMDARs in neurons. We therefore incubated primary murine hippocampal neurons with purified human or mouse CSF ABs. AB binding resulted in a marked downregulation of NMDAR-positive synaptic clusters (Fig. 2b, e). Western blots of GluN1 also revealed its significantly reduced amount in the membrane fractions of GluN1356–385 AB-treated neurons, indicating a profound loss of synaptic NMDARs (Fig. 2c, d).
Inhibitory effect of ABs on LTP in the hippocampal CA1 region and NMDAR-mediated calcium influx
The synaptic plasticity of neurons is closely related to learning and memory. We next tested whether tetanus-induced LTP was affected at Schaffer collateral–CA1 synapses in hippocampal slices. In this experiment, CSF from patients or immunized mice was applied for 10 min before theta burst stimulation was applied. Neither of the CSF types affected baseline transmission. The magnitude of LTP at 30 min post-tetanus was significantly smaller with ABs from patients’ CSF (104.0 ± 11.1%, n = 5, P < 0.005) and GluN1356–385 ABs (110.7 ± 8.1%, n = 5, P < 0.01) than in the control group (151.7 ± 6.2%, n = 5) (Fig. 2f). Consistent with these electrophysiological observations, calcium imaging experiments using patients’ ABs or GluN1356–385 ABs revealed a marked reduction in NMDAR-induced calcium influx, affecting the total amount of calcium influx (patients’ ABs, n = 5, P < 0.05; GluN1356–385 ABs, n = 5, P < 0.05) (Fig. 2g).
Localization of the GluN1 356-385 ABs and their effects in vivo
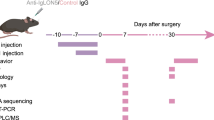
A functional antibody suggests that it may induce related pathological changes in mice. To verify this, we designed a 12-week experiment to explore whether peptide immunization can induce symptoms in mice. Considering the AB-mediated internalization of NMDARs is a reversible process [7], when the ABs disappear, NMDAR function may be restored. To induce high concentrations of autoantibodies targeting NMDARs, we repeatedly immunized mice with GluN1356–385 peptide, with three immunizations at 4-week intervals (Fig. 3a). This procedure maintained the titer of autoantibodies within the CSF and serum of mice (Fig. 3b).

Localization of GluN1356-385 ABs and their effects in vivo. a The experimental design for active immunization of mice at different time points. b CSF and serum AB titers of mice during 12 weeks of immunization (n = 3). c Immunostaining of mice IgG in brain slices of mice immunized with GluN1356–385 peptides. d Western blot analysis of GluN1 on membrane fraction and total cell lysates from the hippocampi of immunized mice (n = 5). e Normalized slope of fEPSP in a hippocampal slice of immunized mice (right). Example traces of fEPSPs recorded before (black) and after (red) TBS (left). *P < 0.05
Twelve weeks after immunization, the brains of GluN1356–385 peptide-immunized mice were used for immunofluorescence experiments staining for IgG. IgG were detected and mainly distributed in the hippocampus and cerebellum (Fig. 3c). We collected the hippocampal tissues of the immunized mice and extracted membrane proteins to detect the GluN1 expression. Western blotting showed that the membrane amount of GluN1 was significantly reduced after immunization with GluN1356–385, indicating a decrease of synaptic NMDARs on the cell surface (Fig. 3d). We further determined whether induced LTP was affected after GluN1356–385 immunization. The magnitude of LTP in mice immunized with GluN1356–385 (112.0 ± 13.2%, n = 5) was significantly reduced compared with the control group (143.1 ± 12.3%, n = 5, P = 0.04). (Fig. 3e).
Behavioral changes of mice after immunization with GluN1356–385 peptide
Symptoms of memory deficits and schizophrenia-like changes are the main presentations that occur in patients with anti-NMDAR encephalitis [4]. To verify the effects of ABs on neurological function and behavioral phenotypes in mice, we performed a series of experiments investigating memory, anhedonia, depressive-like behavior, anxiety, aggression, and locomotor activity in immunized mice. Twelve weeks after immunization, we observed decreased exploration time of the novel object and a lower discrimination index in the GluN1356–385 group in the novel object recognition test (NORT) (n = 12, P = 0.03) (Fig. 4a), in which the immunized mice spent more time exploring the familiar object. The three-chamber experiment revealed that GluN1356–385 peptide-immunized mice made fewer visits to strangers than CFA-immunized control mice (n = 15, P = 0.01) (Fig. 4b). In contrast, there were no differences between the mice in tests of anxiety or depressive behavior tests (open field test and elevated plus-maze) (Fig. 4c, d).

Behavioral changes of mice 12 weeks after immunization with GluN1356–385 peptide. a Exploration time and Discrimination Index from the NORT of mice 12 weeks after immunization (n = 12). b 3 chamber test of mice 12 weeks after immunization (n = 15). c Mice were assessed in the open field for time spent in the center area (left) and for the total distance during a 30-min trial (n = 18). d Mice were assessed in the elevated plus-maze for time spent in the open arms (left) and close arms (right) during a 5 min trial (n = 14)
Discussion
Anti-NMDAR encephalitis is a common form of autoimmune encephalitis, predominantly affecting young adults [17]. Active immunization animal models mimic the process of AB production in autoimmune diseases and have played an important role in the study of neurological diseases [18, 19]. Previous investigations have revealed that peptide fragments can induce autoantibodies to glutamate receptors (AMPA GluN3B), decrease epileptic thresholds, and cause behavioral changes in mice [20]. In the present study, we demonstrated that active immunization with the GluN1356–385 peptide targeting the ATD of GluN1 is sufficient to induce high titers of pathogenic anti-GluN1 autoantibodies. This immunization also reproduced many typical anti-NMDAR encephalitis symptoms in mice.
We first determined whether the immunization with GluN1 peptides could induce the production of ABs against GluN1 in the CSF. The ABs from GluN1356–385 peptide-immunized mice specifically bound to GluN1-transfected HEK293 cells. In addition, a site mutation at N368Q in the ATD of GluN1 prevented the binding of GluN1356–385 ABs to GluN1, suggesting that GluN1356–385 ABs share similar epitopes with patient ABs.
Current data support the idea that autoantibodies targeting NMDARs are responsible for disease pathogenesis [21, 22] and CSF titers of NMDAR ABs correlate with clinical relapses of patients [23]. Twelve weeks after immunization, the mice in our study exhibited memory loss, which is consistent with one of the key symptoms of patients with anti-NMDAR encephalitis [1]. Further investigation revealed that treatment with GluN1356–385 ABs significantly reduced GluN1 density in the membrane of primary hippocampal neurons and impaired NMDAR function. NMDARs are essential for establishing synaptic plasticity and memory formation. When the hippocampus is targeted by GluN1 ABs, LTP formation at Schaffer collateral–CA1 synapses is severely disrupted [16]. Our electrophysiological results revealed that GluN1 ABs from immunized mice have similar effects. The LTP recordings from CA1 synapses of GluN1356–385 AB-treated brain slices showed severe impairment of hippocampal synaptic plasticity, which may explain the memory deficits observed in our animal model.
Studies have shown that autoantibodies targeting NMDARs can induce receptor internalization, thereby affecting NMDAR-mediated electrophysiological activity and transmitter metabolism [24, 25].
At present, the triggering factors for the autoimmune response to NMDARs are still unclear. Previous studies using holoprotein immunogens of tetrameric Xenopus laevis GluN1/GluN2B receptors or rat GluN1/GluN2A receptors to produce NMDAR ABs produced fulminant encephalitis [19]. Active immunization of ApoE−/− mice against peptide fragments of GluN1 led to high circulating levels of NMDAR ABs and was able to induce psychosis-like symptoms upon MK-801 challenge [21, 26]. Intranasal infection with herpes simplex virus has also been shown to induce circulating NMDAR ABs [27], which may explain the pathogenesis of secondary anti-NMDAR encephalitis in patients with herpes simplex virus encephalitis. In our study, a peptide from the GluN1 subunit was used as an immunogen. It was sufficient to induce high titers of pathogenic anti-GluN1 autoantibodies. Active immunization also reproduced memory deficits, a typical anti-NMDAR encephalitis symptom in mice. It may be a convenient alternate endogenous model of anti-NMDAR encephalitis that may be useful for further research into the pathogenesis of this disease and aid in the development of potential new therapies.
Our study has several limitations. Although immunized with GluN1356–385 peptide decreased NMDAR density in the hippocampus and impaired LTP, the mice did not exhibit mood disorders or spontaneous seizures. GluN1356–385 ABs showed binding of the molecular layer of the cerebellum, which is not seen in ABs from patients’ CSF, Additionally, the roles of B cells and T cells in disease induction is still unclear, which should be elucidated with further studies.
Conclusions
We established a novel anti-NMDAR encephalitis model using active immunization with the peptide GluN1356–385, which targets the ATD of GluN1. ABs from GluN1356–385 peptide-immunized mice had a similar pathogenic effect to ABs from patients. Compared with the passive AB transfer model, this active immune model better simulates the immunological characteristics of anti-NMDAR encephalitis. We will explore the pathological mechanisms of anti-NMDAR encephalitis using this active immune model in future studies.
Availability of data and materials
The datasets analyzed in this study are available from the corresponding authors on reasonable request.
Abbreviations
- NMDAR:
-
N-methyl-D-aspartate receptor
- CNS:
-
Central nervous system
- CSF:
-
Cerebrospinal fluid
- AB:
-
Antibody
- ATD:
-
Amino-terminal domain
- PBS:
-
Phosphate-buffered saline
- HEK293:
-
Human embryonic kidney 293
- SDS:
-
Sodium dodecyl sulfate
References
Titulaer MJ, McCracken L, Gabilondo I, Armangué T, Glaser C, Iizuka T, Honig LS, Benseler SM, Kawachi I, Martinez-Hernandez E, Aguilar E, Gresa-Arribas N, Ryan-Florance N, Torrents A, Saiz A, Rosenfeld MR, Balice-Gordon R, Graus F, Dalmau J. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol. 2013;12(2):157–65.
Wang R, Guan HZ, Ren HT, Wang W, Hong Z, Zhou D. CSF findings in patients with anti-N-methyl-D-aspartate receptor-encephalitis. Seizure. 2015;29:137–42.
Kayser MS, Dalmau J. Anti-NMDA receptor encephalitis, autoimmunity, and psychosis. Schizophr Res. 2016;176(1):36–40.
Dalmau J. NMDA receptor encephalitis and other antibody-mediated disorders of the synapse: The 2016 Cotzias Lecture. Neurology. 2016;87(23):2471–82.
Greene M, Lancaster E. Assessing the incidence of anti-NMDAR encephalitis. Arch Dis Child. 2015;100(6):512–3.
Manto M, Dalmau J, Didelot A, Rogemond V, Honnorat J. In vivo effects of antibodies from patients with anti-NMDA receptor encephalitis: further evidence of synaptic glutamatergic dysfunction. Orphanet J Rare Dis. 2010;5:31.
Moscato EH, Peng X, Jain A, Parsons TD, Dalmau J, Balice-Gordon RJ. Acute mechanisms underlying antibody effects in anti-N-methyl-D-aspartate receptor encephalitis. Ann Neurol. 2014;76(1):108–19.
Planaguma J, Leypoldt F, Mannara F, Gutierrez-Cuesta J, Martin-Garcia E, Aguilar E, Titulaer MJ, Petit-Pedrol M, Jain A, Balice-Gordon R, Lakadamyali M, Graus F, Maldonado R, Dalmau J. Human N-methyl D-aspartate receptor antibodies alter memory and behaviour in mice. Brain. 2015;138(Pt 1):94–109.
Bien CG, Vincent A, Barnett MH, Becker AJ, Blumcke I, Graus F, Jellinger KA, Reuss DE, Ribalta T, Schlegel J, Sutton I, Lassmann H, Bauer J. Immunopathology of autoantibody-associated encephalitides: clues for pathogenesis. Brain. 2012;135(5):1622–38.
Camdessanché JP, Streichenberger N, Cavillon G, Rogemond V, Jousserand G, Honnorat J, Convers P, Antoine JC. Brain immunohistopathological study in a patient with anti-NMDAR encephalitis. Eur J Neurol. 2011;18(6):929–31.
Gleichman AJ, Spruce LA, Dalmau J, Seeholzer SH, Lynch DR. Anti-NMDA receptor encephalitis antibody binding is dependent on amino acid identity of a small region within the GluN1 amino terminal domain. J Neurosci. 2012;32(32):11082–94.
Potier M, Georges F, Brayda-Bruno L, Ladépêche L, Lamothe V, Al Abed AS, Groc L, Marighetto A. Temporal Memory and Its Enhancement by Estradiol Requires Surface Dynamics of Hippocampal CA1 N-Methyl-D-Aspartate Receptors. Biol Psychiatry. 2016;79(9):735–45.
Graus F, Titulaer MJ, Balu R, Benseler S, Bien CG, Cellucci T, Cortese I, Dale RC, Gelfand JM, Geschwind M, Glaser CA, Honnorat J, Höftberger R, Iizuka T, Irani SR, Lancaster E, Leypoldt F, Prüss H, Rae-Grant A, Reindl M, Rosenfeld MR, Rostásy K, Saiz A, Venkatesan A, Vincent A, Wandinger K-P, Waters P, Dalmau J. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol. 2016;15(4):391–404.
Tachibana N, Shirakawa T, Ishii K, Takahashi Y, Tanaka K, Arima K, Yoshida T, Ikeda S. Expression of various glutamate receptors including N-methyl-D-aspartate receptor (NMDAR) in an ovarian teratoma removed from a young woman with anti-NMDAR encephalitis. Intern Med. 2010;49(19):2167–73.
Kaech S, Banker G. Culturing hippocampal neurons. Nat Protoc. 2006;1(5):2406–15.
Zhang Q, Tanaka K, Sun P, Nakata M, Yamamoto R, Sakimura K, Matsui M, Kato N. Suppression of synaptic plasticity by cerebrospinal fluid from anti-NMDA receptor encephalitis patients. Neurobiol Dis. 2012;45(1):610–5.
Dalmau J, Gleichman AJ, Hughes EG, Rossi JE, Peng X, Lai M, Dessain SK, Rosenfeld MR, Balice-Gordon R, Lynch DR. Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol. 2008;7(12):1091–8.
Hemmer B, Kerschensteiner M, Korn T. Role of the innate and adaptive immune responses in the course of multiple sclerosis. Lancet Neurol. 2015;14(4):406–19.
Jones BE, Tovar KR, Goehring A, Jalali-Yazdi F, Okada NJ, Gouaux E, Westbrook GL. Autoimmune receptor encephalitis in mice induced by active immunization with conformationally stabilized holoreceptors. Sci Transl Med. 2019;11(500):eaaw0044.
Goldberg-Stern H, Ganor Y, Cohen R, Pollak L, Teichberg V, Levite M. Glutamate receptor antibodies directed against AMPA receptors subunit 3 peptide B (GluR3B) associate with some cognitive/psychiatric/behavioral abnormalities in epilepsy patients. Psychoneuroendocrinology. 2014;40:221–31.
Castillo-Gomez E, Oliveira B, Tapken D, Bertrand S, Klein-Schmidt C, Pan H, Zafeiriou P, Steiner J, Jurek B, Trippe R, Pruss H, Zimmermann WH, Bertrand D, Ehrenreich H, Hollmann M. All naturally occurring autoantibodies against the NMDA receptor subunit NR1 have pathogenic potential irrespective of epitope and immunoglobulin class. Mol Psychiatry. 2017;22(12):1776–84.
Malviya M, Barman S, Golombeck KS, Planaguma J, Mannara F, Strutz-Seebohm N, Wrzos C, Demir F, Baksmeier C, Steckel J, Falk KK, Gross CC, Kovac S, Bonte K, Johnen A, Wandinger KP, Martin-Garcia E, Becker AJ, Elger CE, Klocker N, Wiendl H, Meuth SG, Hartung HP, Seebohm G, Leypoldt F, Maldonado R, Stadelmann C, Dalmau J, Melzer N, Goebels N. NMDAR encephalitis: passive transfer from man to mouse by a recombinant antibody. Ann Clin Transl Neurol. 2017;4(11):768–83.
Gresa-Arribas N, Titulaer MJ, Torrents A, Aguilar E, McCracken L, Leypoldt F, Gleichman AJ, Balice-Gordon R, Rosenfeld MR, Lynch D, Graus F, Dalmau J. Antibody titres at diagnosis and during follow-up of anti-NMDA receptor encephalitis: a retrospective study. Lancet Neurol. 2014;13(2):167–77.
Kreye J, Wenke NK, Chayka M, Leubner J, Murugan R, Maier N, Jurek B, Ly L-T, Brandl D, Rost BR, Stumpf A, Schulz P, Radbruch H, Hauser AE, Pache F, Meisel A, Harms L, Paul F, Dirnagl U, Garner C, Schmitz D, Wardemann H, Prüss H. Human cerebrospinal fluid monoclonalN-methyl-D-aspartate receptor autoantibodies are sufficient for encephalitis pathogenesis. Brain. 2016;139(10):2641–52.
Dalmau J, Lancaster E, Martinez-Hernandez E, Rosenfeld MR, Balice-Gordon R. Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. Lancet Neurol. 2011;10(1):63–74.
Pan H, Oliveira B, Saher G, Dere E, Tapken D, Mitjans M, Seidel J, Wesolowski J, Wakhloo D, Klein-Schmidt C, Ronnenberg A, Schwabe K, Trippe R, Mätz-Rensing K, Berghoff S, Al-Krinawe Y, Martens H, Begemann M, Stöcker W, Kaup FJ, Mischke R, Boretius S, Nave KA, Krauss JK, Hollmann M, Lühder F, Ehrenreich H. Uncoupling the widespread occurrence of anti-NMDAR1 autoantibodies from neuropsychiatric disease in a novel autoimmune model. Mol Psychiatry. 2019;24(10):1489–501.
Linnoila J, Pulli B, Armangué T, Planagumà J, Narsimhan R, Schob S, Zeller MWG, Dalmau J, Chen J. Mouse model of anti-NMDA receptor post-herpes simplex encephalitis. Neurol Neuroimmunol Neuroinflamm. 2019;6(2):e529.
Acknowledgements
Not applicable.
Funding
This work was funded by the National Natural Science Foundation of China (81673950, 81873158, 82004189).
Author information
Authors and Affiliations
Contributions
WHH, ZJ, and XW designed the study. DYW, ZZY, CJY, and PY performed the experiments and analyzed the data. DYW and ZZY prepared the manuscript. WHH, QW and WHT edited the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The experimental protocol was established according to the ethical guidelines of the Declaration of Helsinki and was approved by the Human Ethics Committee of Nanfang Hospital Southern Medical University. Written informed consent was obtained from individual participants or their guardians to use their remaining CSF samples and data.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Supplementary Methods.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ding, Y., Zhou, Z., Chen, J. et al. Anti-NMDAR encephalitis induced in mice by active immunization with a peptide from the amino-terminal domain of the GluN1 subunit. J Neuroinflammation 18, 53 (2021). https://doi.org/10.1186/s12974-021-02107-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12974-021-02107-0