
Abstract
Background
The type 1 interferon (IFN) response is part of the innate immune response and best known for its role in viral and bacterial infection. However, this pathway is also induced in sterile inflammation such as that which occurs in a number of neurodegenerative diseases, including neuronopathic Gaucher disease (nGD), a lysosomal storage disorder (LSD) caused by mutations in GBA.
Methods
Mice were injected with conduritol B-epoxide, an irreversible inhibitor of acid beta-glucosidase, the enzyme defective in nGD. MyTrMaSt null mice, where four adaptors of pathogen recognition receptors (PRRs) are deficient, were used to determine the role of the IFN pathway in nGD pathology. Activation of inflammatory and other pathways was analyzed by a variety of methods including RNAseq.
Results
Elevation in the expression of PRRs associated with the IFN response was observed in CBE-injected mice. Ablation of upstream pathways leading to IFN production had no therapeutic benefit on the lifespan of nGD mice but attenuated neuroinflammation. Primary and secondary pathological pathways (i.e., those associated or not with mouse survival) were distinguished, and a set of ~210 genes including those related to sphingolipid, cholesterol, and lipoprotein metabolism, along with a number of inflammatory pathways related to chemokines, TNF, TGF, complement, IL6, and damage-associated microglia were classified as primary pathological pathways, along with some lysosomal and neuronal genes.
Conclusions
Although IFN signaling is the top elevated pathway in nGD, we demonstrate that this pathway is not related to mouse viability and is consequently defined as a secondary pathology pathway. By elimination, we defined a number of critical pathways that are directly related to brain pathology in nGD, which in addition to its usefulness in understanding pathophysiological mechanisms, may also pave the way for the development of novel therapeutic paradigms by targeting such pathways.
Similar content being viewed by others


Background
The lysosomal storage disease (LSD), Gaucher disease (GD), is caused by mutations in GBA, which encodes the lysosomal hydrolase, acid beta-glucosidase (GCase). GD is divided into neuronopathic (types 2 and 3) (nGD) and non-neuronopathic forms (type 1), depending on the involvement of symptoms associated with the central nervous system [1, 2]. Little is known about pathological mechanisms that lead to brain disease, but among these is neuroinflammation. Surprisingly, among the inflammatory pathways [3], activation of the type 1 interferon (IFN) response was demonstrated [4] in Gbaflox/flox (nestin-Cre mice) in which GCase deficiency in the brain is restricted to cells of neuronal lineage, with microglia displaying normal GCase levels [5]. In an unbiased gene profile analysis, genes associated with the type 1 IFN-related pathway were highly upregulated [4].
The IFN response is normally considered to occur as a result of pathogen recognition receptor (PRR) stimulation. The PRRs are part of the innate immune system and respond to either pathogen-associated molecular patterns (PAMPs) or to endogenous molecules secreted following tissue stress or injury, known as danger-associated molecular patterns (DAMPs) [6]. Several classes of PRRs are known, and a number can trigger the IFN response. The most well-defined PRRs are the Toll-like receptors (TLRs); after ligand binding, TLRs dimerize, the Toll/IL-1 receptor (TIR) domain associates with TIR domain-containing adaptor proteins, such as myeloid differentiation factor 88 (MyD88) and TIR domain-containing adaptor protein-inducing IFN-β (TRIF), which initiates downstream signaling [7]. The Nod-like receptor, NOD2, the RIG-I receptor retinoic acid-inducible gene I (RIG-I), and the melanoma differentiation-associated factor 5 (MDA5) use mitochondrial antiviral signaling protein (MAVS) as their adaptor [8]. The most important IFN-inducing cytosolic DNA sensing pathways is the cyclic guanosine monophosphate-adenosine monophosphate synthase (cGAS), stimulator of IFN gene (STING, TMEM173) axis. Upon binding of DNA, cGAS catalyzes the formation of the secondary messenger, 2′,3′-cyclic guanosine monophosphate-adenosine monophosphate (cGAMP), which binds STING and subsequently activates an antiviral cytokine response [9].
In the current study, we have examined the effect of inducing nGD, using a chemical inhibitor (conduritol B-epoxide, CBE [10, 11]), in a quadrat-deficient mouse with a combined deficiency of TLR, RIG-I like receptor (RLR) and STING signaling (Myd88-/-,Trif-/-,Mavs-/-,Tmem173-/-), referred to as the MyTrMaSt mouse [12]. These are the main PRRs that induce a type 1 IFN response, although it should be noted that IFN can also be induced by other PRRs such as cGAS [12]. We demonstrate that the IFN pathway is not the initial cause for pathology in nGD, but rather a secondary pathological pathway since the lifespan of MyTrMaSt null mice was not altered upon CBE injection compared with wild-type (WT) mice. We then go on to define a subset of genes and pathways as primary pathological pathways which may be responsible for the pathogenesis of nGD.
Methods
Mice
MyTrMaSt null mice were generated as described [12]. From postnatal day 8, C57BL/6JOlaHsd mice (Envigo Laboratories, Israel) or MyTrMaSt null mice were injected intraperitoneally (i.p.) daily with 25, 32, 37.5, or 100 mg CBE (Calbiochem Millipore, Darmstadt, Germany) per kilogram body weight, or with phosphate buffered saline (PBS). Genotyping was performed by PCR using genomic DNA extracted from mouse tails. Mice were maintained in the experimental animal center of the Weizmann Institute of Science in specific pathogen-free conditions. All animal experiments were approved by the Weizmann Institute Institutional Animal Care and Use Committee.
RNA extraction and quantitative PCR
Mice were euthanized using CO2 and brains were rapidly removed. Total RNA from half brain was isolated using the RNeasy mini kit (Qiagen GmbH, Hilden, Germany) according to manufacturer’s instructions. cDNA synthesis was performed using a qScript cDNA synthesis kit (Quanta Biosciences, Gaithersburg, MD, USA). Quantitative PCR (qPCR) was performed using the PerfeCT SYBR Green FastMix (Quanta BioSciences, Gaithersburg, MD, USA) and an ABI Prism 7300 Sequence Detection System (Applied Biosystems, Foster City, CA, USA). The primer concentration was 20 nM in a reaction volume of 10 μl. Primer sequences are listed in Additional file 1. IFNα2, TNFα, and IL1β primers were purchased from Quantitect (Qiagen). Each reaction was performed in duplicate. The ΔΔCt method was used to calculate relative changes in gene expression, with hypoxanthine phosphoribosyltransferase 1 (HPRT1) used as a housekeeping gene [13]. p values were calculated using a two-tailed, two-independent sample Student’s t test.
Western blotting
Brain tissue was lysed in lysis buffer (50 mM Tris HCl pH 7.6, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, and 0.1% sodium dodecyl sulfate (SDS)) supplemented with a protease inhibitor mixture (1:100, Sigma-Aldrich) using a GentleMACS dissociator (Miltenyi Biotec, Bergisch Gladbach, Germany). Following homogenization, samples were centrifuged at 14,000 × gav for 10 min at 4 °C, and the supernatants collected. Protein was quantified using the BCA protein assay reagent. Seventy micrograms of protein in the sample buffer was electrophoresed on a 10% SDS-polyacrylamide gel and transferred to a nitrocellulose membrane. Blots were incubated with the following primary antibodies: rat anti-MAC2 (1:1000, Cedarlane, Ontario, Canada), mouse anti-tubulin (1:5000, Santa Cruz, Dallas, TX, USA), followed by a horseradish peroxidase-conjugated secondary antibody. Bound antibodies were detected using the Westar Chemiluminescent substrate (Cyanagen, Bologna, Italy). Western blots were analyzed by densitometry and values quantified using a ratio of Mac2 to Tubulin.
RNAseq
Brain tissue was homogenized using a GentleMACS dissociator and mRNA isolated using the RNeasy mini kit. RNA concentrations (260/230- and 260/280-nm ratios) were measured using a NanoDrop ND-1000 (Thermo Scientific, Waltham, MA, USA). RNA integrity was evaluated using an RNA screen tape on a Tapestation 2200 (Agilent, Santa Clara, CA, USA). A bulk variation of MARSseq [14] was used to construct RNAseq libraries. Sequencing was performed using an Illumina Nextseq-500 75 cycle high output kit (Illumina, San Diego, CA, USA; paired end sequencing). Raw reads were mapped to the Mus musculus genome (mm10) using hisat (version 0.1.6). Only reads with unique mapping were considered for further analysis. Differentially expressed genes (DEGs) were selected using a 2-fold change cutoff between two populations and adjusted p value for multiple gene testing of < 0.05 [15]. Principle component analysis (PCA) of all DEGs and heatmaps were generated using Partek Genomics Suite® software, version 7.0 (St. Louis, MO, USA), or RStudio (Integrated Development for R. RStudio, Inc., Boston, USA). The BioVenn tool [16] was used to identify common and exclusively expressed genes between groups. Pathway analysis was done using Metascape [17] or Gene Analytics [18].
Results
Elevation of PRR expression in nGD mice
Genes encoding various PRRs were elevated in the brain of a genetic model of nGD [4], and we now show a similar elevation in PRR mRNA levels in a chemically induced nGD mouse. Upon injection of C57BL/6JOlaHsd mice with a high dose of CBE (100 mg/kg body weight [10]) for 10 days, a significant elevation in the expression of PRR family members related to IFN signaling was observed, with the highest elevation in a cell surface TLR, Tlr2 (Table 1). Tlr1 and Tlr4, which are also located on the cell surface, were also significantly elevated. Levels of expression of endosomal TLRs [19], Tlr3, Tlr7, Tlr8, Tlr9, and Tlr13 were likewise elevated. The elevation of Tlr expression implicates signaling via their downstream adaptors, namely MyD88 (used for all TLRs except TLR3) and TRIF (required for TLR3 and TLR4). NOD-like receptor (NLR) family members, such as NOD2, which utilizes the adaptor protein MAVS, can also induce IFN production [20], as can members of the RLR family, RIG-I and MDA5, which also use MAVS. STING, which functions both as a PRR and as a signaling adaptor, induces expression of type 1 IFN via the NFκB and IRF3 pathways [21]. All of these PRRs were elevated (Table 1), suggesting that GlcCer accumulation, or a downstream effector, acts as a DAMP.
The IFN response does not play a role in nGD mouse lifespan
We previously demonstrated that ablation of the type 1 IFN receptor (IFNAR1) attenuated neuroinflammation but had no effect on nGD mouse viability [4]. IFN is nevertheless secreted in IFNAR null mice. To determine the effect of inhibiting pathways upstream to IFNAR, we injected MyTrMaSt null mice with a low dose of CBE (25 mg/kg, since higher doses (i.e., 100 mg/kg CBE) cause extremely severe disease, with mice not surviving beyond 20 days of age [10]. The mean lifespan of MyTrMaSt null mice was similar to that of WT mice injected with CBE (Fig. 1a) although there was a significant variability between individual mice. Likewise, no effect was seen on the lifespan using higher doses [10] of CBE (32 mg/kg and 37.5 mg/kg). Levels of expression of Irf7, Usp18, and Ifnα, which play critical roles in IFN signaling, were elevated upon CBE injection in WT mice, as previously observed [4], but not elevated in MyTrMaSt null mice upon CBE injection (Fig. 1b). This demonstrates that the IFN pathway is not required for nGD pathology.

Inhibition of the type 1 IFN response has no effect on lifespan. a Kaplan-Meier survival curve of WT mice injected i.p. with PBS (n = 4) or 25 mg/kg CBE (n = 8) or MyTrMaSt null mice injected with PBS (n = 6) or 25 mg/kg CBE (n = 13). b RT-PCR from brain homogenates (day 96) of WT and MyTrMaSt null mice injected with PBS (n = 2) or CBE (25 mg/kg) (n = 4). Cycle threshold values were normalized to levels of hypoxanthine phosphoribosyltransferase 1 (HPRT1). * p < 0.05, ** p < 0.01
Attenuated neuroinflammation in MyTrMaSt null mice
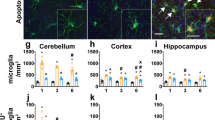
We next determined the relationship between the PRR pathway and components of inflammatory mediators that are altered in nGD such as chemokines, Il1β, and TNFα [22]. Levels of F4/80, which is expressed on microglia and macrophages, as well as the inflammatory chemokine Ccl5 (Rantes), were significantly reduced in MyTrMaSt null mice injected with 25 mg/kg CBE compared with control mice injected with CBE, but in contrast, no differences were observed in levels of Gfap, Ccl3 (Mip1α), IL1β, and TNFα (Fig. 2a) even by using higher doses of CBE (32 mg/kg and 37.5 mg/kg; data not shown). Likewise, the main inducers of the IFN pathway did not affect levels of an activated astrocyte marker (Gfap), which remained elevated when MyTrMaSt null mice were injected with doses of CBE as high as 100 mg/kg body weight (Fig. 2a, b). Levels of Mac2, which is expressed by cells of the macrophage/microglia lineage (Fig. 2b), were reduced in CBE-injected MyTrMaSt null mice similar to the reduction in F4/80. Since levels of macrophage/microglia markers (F4/80, Mac2) and the inflammatory marker Ccl5 were reduced in response to the inhibition of the IFN response, while levels of other inflammatory markers (Ccl3, IL1β, TNFα) and an astrocyte marker (Gfap) remain unchanged, this suggests that some pathways of neuroinflammation are affected upon loss of the IFN response, while others are not.

Attenuated neuroinflammation in MyTrMaSt null mice. a mRNA levels in brain homogenates from 96–130-day-old CBE-injected mice (25 mg/kg; n = 6–11) and PBS injected mice (n = 4–6). RNA expression is shown as arbitrary units (AU). * p < 0.05. b A representative western blot from brain homogenates is shown (70 μg of protein) of 19-day-old WT or MyTrMaSt null mice injected with 100 mg/kg CBE or PBS from 8 days of age. Results are representative of 3 experiments for control mice and 5 for CBE-treated mice, which all gave similar results. Tubulin was used as loading control and was similar in each lane. Quantification is normalized to tubulin. *p < 0.05
RNAseq distinguishes between primary and secondary pathological pathways
To distinguish between genes involved in pathophysiology that are associated with mouse death (which we define as “primary pathological pathways”) and those that are unrelated to mouse death (“secondary pathological pathways”), we performed RNAseq [MARSseq, [14]] on brain homogenates from four groups of mice, namely WT mice injected with PBS (referred to as WT + PBS), WT mice injected with 100 mg/kg body weight CBE (WT + CBE), MyTrMaSt null mice injected with PBS (MyTrMaSt + PBS) and MyTrMaSt null mice injected with 100 mg/kg body weight CBE (MyTrMaSt + CBE). Comparison of DEGs in WT mice injected with CBE versus PBS, compared with MyTrMaSt null mice injected with CBE versus PBS should distinguish between genes associated with putative primary or secondary pathological pathways.
A total of 13,866 genes were detected by RNAseq. Principle component analysis (PCA) revealed that PBS-injected mice clearly separate from CBE-injected mice, with the exception of one outlier WT mouse (Fig. 3a). A total of 767 genes were differentially expressed (DE) (fold change ≥ 2, p adjusted < 0.05) in WT + CBE versus WT + PBS (599 upregulated and 168 downregulated), and 467 genes were DE in MyTrMaSt + CBE versus MyTrMaSt + PBS (392 upregulated, 75 downregulated) (Fig. 3b). K-means clustering of the DEGs (Fig. 3b) indicated 5 gene clusters. The 215 genes in cluster 1 were elevated only in the WT + CBE group. The genes in clusters 2 (93 genes) and 5 (138 genes) display similar decreased expression in the two CBE-treated groups. The genes in clusters 3 and 4 (143 and 345 genes) displayed reduced expression in MyTrMaSt + CBE compared with the WT + CBE group. Comparison of the DEGs in the WT + CBE group versus WT + PBS with our previous high-throughput studies [4, 23] indicated similar changes in gene expression, even though previous studies were performed on different days of CBE injection and in different brain areas.

RNAseq discriminates three groups of genes. a PCA of DEGs in WT + PBS (n = 3) (red squares) versus WT + CBE (n = 3) (blue squares) and MyTrMaSt + PBS (n = 3) (red triangles) versus MyTrMaSt + CBE (n = 5) (blue triangles); fold-change > 2, p adjusted < 0.05. b K-means clustering of all DEGs (fold-change > 2, p adjusted < 0.05). Red (high, > 1) and blue (low, less than − 1) in the heat map represent relative gene expression. Five clusters are indicated
Analysis of DEGs in WT + CBE versus WT + PBS and in MyTrMaSt + CBE versus MyTrMaSt + PBS indicated 466 DEGs (767 − 301, Fig. 3b) in the WT+CBE versus WT + PBS group (mainly in cluster 1), whereas there are 301 DEGs common to the two groups (mainly in clusters 3 and 4). A total of 166 genes (467 − 301, Fig. 3b) are DE only in MyTrMaSt + CBE versus MyTrMaSt + PBS (clusters 2 and 3) (Fig. 3b). We define the group of 466 DEGs as associated with secondary pathological pathways, since the expression of these genes is altered in WT mice, whereas the 301 DEGs common to the two groups of CBE-injected mice are defined as primary pathological pathways, since the lifespan of the MyTrMaSt null mice is similar to WT mice injected with CBE.
Analysis of the 466 DEGs associated with secondary pathological pathways demonstrated enrichment in genes related to the type 1 IFN response (Fig. 4a) [4], with 8 of the 10 most upregulated genes induced by IFN. As expected, these genes were not DE in MyTrMaSt null mice (Fig. 4b). Various cytokines, chemokines, and Tnfα-induced genes were also enriched in the 466 genes (Fig. 4a). Of the 466 DEGs, approximately one third (149) were downregulated. Based on single-cell RNAseq analyses [24, 25], 42% of the downregulated genes can be assigned to neurons, consistent with neuronal loss by a “dying-back” mechanism in nGD [26, 27]. Among the downregulated genes in WT + CBE versus WT + PBS are Mapre3, Map1lc3a, and Mast2 microtubule genes, a pathway which underlies neurodegeneration via a dying-back mechanism [28]. The relevance of the 166 DEGs (110 upregulated and 56 downregulated) in MyTrMaSt + CBE versus MyTrMaSt + PBS mice (Fig. 3b) is somewhat ambivalent, particularly as a number of the DEGs showed a similar fold-change as in WT mice injected with CBE, but did not reach statistical significance (see clusters 2 and 3, Fig. 3b). Only 8 genes were exclusively upregulated in MyTrMaSt + CBE versus WT + CBE (Fig. 4c), and no obvious connection between them could be ascertained. Of the 56 downregulated genes, 27% are expressed in neurons.

Loss of IFN signaling in MyTrMaSt null mice. a Pathway analysis of the 466 DEGs in WT + PBS versus WT + CBE. Numbers of identified genes out of the total genes associated with each pathway are shown. b Heatmap of representative IFN genes. Red (high, > 1) and blue (low, < 1) in the heat map represent relative gene expression. Some of the DEGs were validated by PCR; WT + CBE versus WT + PBS, fold-change 27.5 ± 2.9 (Irf7), 168.4 ± 27.2 (Irg1); MyTrMaSt + CBE versus MyTrMaSt + PBS, fold-change 1.0 ± 0.1 (Irf7), 2.3 ± 0.2 (Irg1). c Heatmap of the 8 genes upregulated in MyTrMaSt null + CBE versus WT + CBE mice. Some of the DEGs were validated by PCR: MyTrMaSt + CBE versus WT + CBE, fold-change 2.35 ± 0.3 (Serpinf2), 1.93 ± 0.3 (Glycam1), 1.86 ± 0.2 (Apln). d Pathway analysis of the 301 common DEGs. Numbers of identified genes out of the total genes associated with each pathway are shown
Since MyTrMaSt null and WT mice displayed a similar life span when injected with CBE, the common 301 DEGs in both CBE-injected groups are likely to be associated with primary pathological pathways. Pathway analysis of the 301 DEGs revealed changes in the lysosome, complement cascades and in a variety of immune responses (Fig. 4d). However, it was possible to further divide the 301 DEGs in two groups, ~90 genes that differed more than 2-fold between MyTrMaSt and WT upon CBE injection (Table 2) and 210 genes whose levels are essentially similar (< 2-fold change) (Table 3). The ~90 genes are unlikely to be primary pathological components and encompass a number of pathways (Table 2). Thus, a number of chemokines and microglia genes were significantly lower in MyTrMaSt null mice compared with WT mice, as were genes associated with TNFα and TGFβ pathways. Expression of a number of cathepsin genes were also lower, as were some, but not all genes associated with complement, DAMs, and cholesterol metabolism. A number of genes associated with lipoproteins were elevated in WT + CBE mice but reduced in MyTrMaSt + CBE.
The most interesting set of genes are the 210 genes that were unchanged (< 2-fold) in MyTrMaSt mice compared with WT mice injected with CBE (Table 3). Although some of these genes are associated with similar pathways as in Table 2, they are nevertheless quite distinct. For instance, levels of expression of three biosynthetic genes associated with sphingolipid (SL) metabolism remain downregulated in MyTrMaSt null mice, indicating that blocking the IFN response does not revert or change the defects in SL metabolism, which is perhaps not surprising since GD is a SL LSD; likewise, genes associated with the lysosome were also unaltered. More unexpectedly, genes associated with cholesterol metabolism and lipoprotein metabolism were also unaltered; importantly, Plin4, which is found on lipid droplets [29], was one of the few genes whose expression was significantly elevated in MyTrMaSt null mice injected with CBE compared with WT mice injected with CBE. Thus, we conclude that primary pathological pathways include those associated with SLs, lipids, lipoproteins, and lysosomes, which is consistent with the known etiology of nGD.
In addition, and in line with data shown in Fig. 2, inflammation unrelated to IFN pathways is a key component in nGD pathology. Thus, expression of a variety of genes encoding chemokines and pathways associated with TNFα, TGFβ, and IL6 remain elevated in MyTrMaSt null mice, which is consistent with enhanced expression levels of genes associated with both microglia and with astrocytes (such as Gfap), supporting the notion that abrogating the IFN response does not attenuate key pathways in neuroinflammation related to nGD symptoms. This is supported by the elevated level of some (but not all, Table 2) components of the complement pathway (Table 3) and DAMs, including Trem2 and Tyrobp. Finally, three neuronal genes may play a critical role in nGD pathology since their levels were also similar in WT and in MyTrMaSt null mice injected with CBE, consistent with the unaltered levels of some metalloproteinases, of which at least one (Adam8) is involved in neurodegeneration [30, 31].
Discussion
Further to our recent study suggesting that the IFN pathway does not play a critical role in nGD pathology [4], we have now taken advantage of the availability of the MyTrMaSt null mouse, which has a deficiency of TLR, RLR, and STING, to differentiate between primary and secondary pathological pathways in nGD. Since the lifespan of MyTrMaSt mice was similar to that of WT mice injected with CBE, even though no IFNs were detected, pathways associated with PRRs and the IFN response cannot be directly involved in pathophysiology of the nGD brain.
Even though we are able to exclude a role for the IFN pathway in primary pathology, components of this pathway are nevertheless the top elevated pathway in the nGD brain and are also elevated in a number of other LSDs, including mucolipidosis type IV and Krabbe disease [32, 33], along with a number of other unrelated neuroinflammatory disorders [34]. This raises the question of how this pathway is activated. One possibility, at least in the three LSDs mentioned above, is that changes in membrane lipid composition result in the activation of PRRs such as TLRs, which upon dimerization associate with adaptor proteins, such as MyD88 and TRIF, to initiate downstream signaling [7]. Receptor dimerization is affected by membrane lipid composition [35]. In GD, changes in lipid composition affect a number of biophysical properties of membranes [36], and we suggest that altered GlcCer levels may directly impinge upon, and perhaps stimulate PRR dimerization, thus activating the IFN response. Nevertheless, since this response can be eliminated with no effect on the lifespan, at least in nGD mice, activation of the IFN pathway may be unrelated to primary disease pathophysiology. Having said that, a recent study has shown that activation of the IFN response in the nGD brain confers resistance to infection by neurotropic viruses (Melamed et al., submitted for publication).
By a process of elimination, we were able to cut down the number of genes associated with primary pathological pathways to ~210, by considering only genes whose expression changed consistently in both WT and MyTrMaSt mice injected with CBE, each compared with its respective PBS-injected control (Table 3). In addition, we have recently completed a study (Blumenreich et al., submitted for publication) in which WT mice were injected with CBE along with a small molecule inhibitor of the critical enzyme in the SL biosynthetic pathway, namely GlcCer synthase (UDP-glucose ceramide glucosyltransferase), in an approach known as substrate reduction therapy (SRT) [37]. SRT led to a decrease in levels of GlcCer and of GlcSph, the two lipids that accumulate in nGD, along with a significant extension of mouse lifespan. Analysis of gene expression by RNAseq revealed that SRT largely reversed the changes in genes and pathways that were DE upon CBE injection, suggesting that these pathways play a vital role in the mouse lifespan and thus pathophysiology, including pathways of GSL metabolism, lipoproteins, and other lipid metabolic pathways, lipid droplets, astrocyte activation, neuronal function, and to some extent, neuroinflammation.
The availability of this data allows us to further interrogate the ~210 genes associated with primary pathology from the current study, a selection of which are listed in Table 3, along with an indication of whether levels of expression of these genes is altered in the SRT study. We propose that genes that were not DE in the current study using MyTrMaSt null mice, along with those that were DE in the SRT study, are likely to be those that are critically involved in pathophysiology. By way of example, three DEGs in the SL metabolic pathway remained elevated in MyTrMaSt null mice but were reduced by SRT (Table 3); likewise some genes associated with cholesterol metabolism, and the three genes associated with lipoprotein metabolism and lipid droplets, including Plin4, whose expression actually increased in MyTrMaSt null mice but reverted to control levels in mice treated with SRT. Lipid droplets have been implicated in neurodegeneration [29, 38], in Parkinson’s disease [39], and in the aging brain [40] implying that they may play a broad role in neurodegenerative diseases. Similarly, lysosomal genes were unaffected in MyTrMaSt null mice but most reverted to control levels after SRT. Since all of these pathways are related, either directly or indirectly to changes in lysosomal SL metabolism, we suggest that they may be coupled to some of the earliest events that occur in nGD pathology, although we cannot determine the precise temporal sequence of events that leads to, or causes changes in their expression (Fig. 5).

Primary and secondary pathological pathways in nGD. Upon GlcCer/GlcSph accumulation, changes occur in a number of pathways related to altered SL, lipid or lysosomal function, which results in concomitant changes in a number of downstream pathways (green), which are classified as primary pathological pathways. IFN-associated pathways (red) can be eliminated without effect on mice lifespan, suggesting that they are secondary pathways. For further details, see text
As a result of these changes, or concomitant with these changes, a number of inflammatory pathways are activated including those associated with TNF, TGF, IL6, and associated chemokines, along with microglia and astrocyte activation, and upregulation of some components of the complement cascade (Fig. 5). It should be emphasized that the demarcation of genes as associated with primary and secondary pathological pathways is somewhat arbitrary with respect to a number of genes and pathways, with some components of the same pathways appearing in both. This is probably because many genes are components of multiple pathways that impinge upon each other at a number of signaling hubs that are downstream to more than one activator.
Concerning inflammatory pathways that appear associated with primary pathology, two chemokines may play critical roles in nGD, namely Ccl3 and Ccl4, along with other components of TNF signaling, noticeably Slamf9 and Cd109, associated with TGF signaling. Cd68 and Mpeg1 may be important for microglia activation, and Gfap critical in astrocytosis (Table 3). It is difficult to ascribe precise functions to each of these genes in nGD.
In contrast, functional significance can be advocated for genes associated with a novel microglia type, namely disease-associated microglia (DAM), which have been ascribed roles in Alzheimer’s disease [41] and in several other neurodegenerative diseases [42, 43], including the LSD, mucolipidosis type IV [33]. Several DAM signature genes were upregulated in our study, including Trem2 and Tyrobp (Dap12), which remained elevated in MyTrMaSt null mice but were reduced to some extent after SRT. Importantly, Trem2 overexpression attenuates neuroinflammation in Parkinson’s disease [44]. Phospholipids and lipoproteins (including Apoe which was reduced to some extent in response to SRT, Table 3) have been identified as ligands for TREM2 in Alzheimer’s disease, which promotes microglia activation and survival. Thus, TREM2 genetic variants, which interfere with this binding, increase the risk of Alzheimer’s disease [45]. Microglia, the only myeloid population in the brain in nGD [46], and its cell surface receptor, TREM2, the principle regulator that transforms microglia from a homeostatic to neuronal disease-associated state, appear critical in nGD pathology. It remains to be elucidated whether the TREM2 or TYROBP signaling is beneficial or detrimental [47].
Another critical gene family appears to be the complement system. The complement system is considered the first line of defense against pathogens, mediates the clearance of immune complexes and regulates inflammatory responses. Our data supports the notion that complement activation in nGD is through activation of the classical pathway [48]. The complement system has previously been implicated in GD, with complement activation suggested to be due to GlcCer accumulation, which leads to the break of tolerance and induction of GlcCer-specific IgG autoantibodies [49, 50].
Finally, three metalloproteases are involved with primary pathology, which may be related to neuronal function. Matrix metalloproteinases (MMPs) and their tissue inhibitors (TIMP) have been implicated in the pathology of Parkinson’s and Alzheimer’s diseases, where MMP can cleave amyloid beta [51]. Similarly, the induction of MT1/2 in the Alzheimer’s disease brain was suggested to be a defense cellular response against inflammatory signals, thus serving a neuroprotective effect [52, 53], though other studies suggest that MT1/2 may have detrimental consequences in amyloid beta clearance [54, 55].
MT1/2 were also implicated in Parkinson’s disease where their elevation was proposed to have a protective role against neurotoxicity [56]. MT1/2 was also unaltered in in MyTrMaSt null mice, suggesting their involvement in GD neuropathology.
Conclusions
We have differentiated between two types of pathways that are activated in nGD, namely those directly related to disease pathology and those that appear only to be secondarily related (Fig. 5). While this distinction might be somewhat artificial, since individual components of each pathway are activated in the same brains and certainly impact upon each other, our approach should nevertheless help distinguish between pathways that are valid therapeutic targets and those that are not, as well as providing mechanistic insight into how the three major cell types in the brain are affected in this devastating neurological disease.
Availability of data and materials
The RNAseq dataset generated during the current study was deposited in the Gene Expression Omnibus (GEO) database, http://www.ncbi.nlm.nih.gov/geo (accession no. GSE150266).
Abbreviations
- CBE:
-
Conduritol B-epoxide
- cGAMP:
-
2′,3′-cyclic guanosine monophosphate-adenosine monophosphate
- cGAS:
-
Cyclic GMP-AMP synthase
- DAM:
-
Novel microglia type associated with neurodegenerative diseases
- DAMP:
-
Danger-associated molecular patterns
- GCase:
-
Acid beta-glucosidase
- GD:
-
Gaucher disease
- GlcCer:
-
Glucosylceramide
- IFN:
-
Interferon
- LSD:
-
Lysosomal storage disorder
- MAVS:
-
Mitochondrial antiviral signaling protein
- MDA5:
-
Melanoma differentiation-associated factor 5
- MyD88:
-
Myeloid differentiation factor 88
- NOD:
-
Nod-like receptor
- PAMP:
-
Pathogen-associated molecular patterns
- PRR:
-
Pathogen recognition receptor
- RIG:
-
Retinoic acid-inducible gene I
- RLR:
-
RIG-I-like receptor
- SL:
-
Sphingolipid
- STING:
-
Stimulator of IFN genes
- TLR:
-
Toll-like receptors
- TRIF:
-
TIR-domain-containing adaptor protein-inducing IFN-β
References
Futerman AH, Zimran A. Gaucher disease. Taylor and Francis Group, Boca Raton, FL: CRC Press; 2006.
Mistry PK, Lopez G, Schiffmann R, Barton NW, Weinreb NJ, Sidransky E. Gaucher disease: progress and ongoing challenges. Mol Genet Metab Academic Press. 2017;120:8–21.
Vitner EB, Salomon R, Farfel-Becker T, Meshcheriakova A, Ali M, Klein AD, et al. RIPK3 as a potential therapeutic target for Gaucher’s disease. Nat Med. 2014;20:204–8.
Vitner EB, Farfel-Becker T, Ferreira NS, Leshkowitz D, Sharma P, Lang KS, et al. Induction of the type I interferon response in neurological forms of Gaucher disease. J Neuroinflammation BioMed Central. 2016;13:1–15.
Enquist IB, Bianco Lo C, Ooka A, Nilsson E, Månsson J-E, Ehinger M, et al. Murine models of acute neuronopathic Gaucher disease. Proc Natl Acad Sci U S A National Acad Sciences. 2007;104:17483–8.
Schaefer L. Complexity of Danger: The diverse nature of damage-associated molecular patterns. J Biol Chem American Society for Biochemistry and Molecular Biology. 2014;289:35237–45.
O'Neill LAJ, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol. 2007;7:353–64.
McNab F, Mayer-Barber K, Sher A, Wack A, O'Garra A. Type I interferons in infectious disease. Nat Rev Immunol Nature Research. 2015;15:87–103.
Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Röhl I, et al. cGAS produces a 2′-5′-linked cyclic dinucleotide second messenger that activates STING. Nature Nature Publishing Group. 2013;498:380–4.
Vardi A, Zigdon H, Meshcheriakova A, Klein AD, Yaacobi C, Eilam R, et al. Delineating pathological pathways in a chemically induced mouse model of Gaucher disease. J Pathol John Wiley & Sons, Ltd. 2016;239:496–509.
Kuo CL, Kallemeijn WW, Lelieveld LT, Mirzaian M, Zoutendijk I, Vardi A, et al. In vivo inactivation of glycosidases by conduritol B epoxide and cyclophellitol as revealed by activity-based protein profiling. The FEBS Journal. John Wiley & Sons, Ltd (10.1111); 2019;286:584–600.
Tegtmeyer P-K, Spanier J, Borst K, Becker J, Riedl A, Hirche C, et al. STING induces early IFN-β in the liver and constrains myeloid cell-mediated dissemination of murine cytomegalovirus. Nat. Commun. Nat Publ Group. 2019;10:1–12.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real time quantitative PCR and the 2^(-ΔΔC(T)) method. Methods. 2001;25:402–8.
Jaitin DA, Kenigsberg E, Keren-Shaul H, Elefant N, Paul F, Zaretsky I, et al. Massively parallel single-cell RNA-Seq for marker-free decomposition of tissues into cell types. Science. American Association for the Advancement of Science. 2014;343:776–9.
Kohen R, Barlev J, Hornung G, Stelzer G, Feldmesser E, Kogan K, et al. UTAP: User-friendly transcriptome analysis pipeline. BMC Bioinformatics. BioMed Central. 2019;20:1–7.
Hulsen T, Vlieg J, Alkema W. BioVenn - a web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genomics BioMed Central. 2008;9:488.
Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun Nature Publishing Group. 2019;10:1–10.
Ben-Ari Fuchs S, Lieder I, Stelzer G, Mazor Y, Buzhor E, Kaplan S, et al. GeneAnalytics: an integrative gene set analysis tool for next generation sequencing, RNAseq and microarray data. OMICS. Mary Ann Liebert, Inc. 140 Huguenot Street, 3rd Floor New Rochelle, NY 10801 USA; 2016;20:139–51.
Blasius AL, Beutler B. Intracellular Toll-like receptors. Immunity Elsevier Inc. 2010;32:305–15.
Sabbah A, Te HC, Harnack R, Frohlich V, Tominaga K, Dube PH, et al. Activation of innate immune antiviral responses by Nod2. Nat Immunol Nature Publishing Group. 2009;10:1073–80.
Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature Nature Publishing Group. 2008;455:674–8.
Vitner EB, Farfel-Becker T, Eilam R, Biton I, Futerman AH, Futerman A. Contribution of brain inflammation to neuronal cell death in neuronopathic forms of Gaucher’s disease. Brain. 2012;135:1724–35.
Klein AD, Ferreira NS, Ben-Dor S, Duan J, Hardy J, Cox TM, et al. Identification of modifier genes in a mouse model of Gaucher disease. Cell Rep Cell Press. 2016;16:2546–53.
Franzén O, Gan LM, Björkegren J. PanglaoDB: a web server for exploration of mouse and human single-cell RNA sequencing data. Database. 2019.
La Manno G, Gyllborg D, Codeluppi S, Nishimura K, Salto C, Zeisel A, et al. Molecular diversity of midbrain development in mouse, human, and stem cells. Cell Cell Press. 2016;167:566–580.e19.
Zigdon H, Meshcheriakova A, Becker TF, Volpert G, Sabanay H, Futerman AH. Altered lysosome distribution is an early neuropathological event in neurological forms of Gaucher disease. FEBS Lett John Wiley & Sons, Ltd. 2017;591:774–83.
Farfel-Becker T, Vitner EB, Pressey SNR, Eilam R, Cooper JD, Futerman AH. Spatial and temporal correlation between neuron loss and neuroinflammation in a mouse model of neuronopathic Gaucher disease. Hum Mol Genet Oxford University Press. 2011;20:1375–86.
Yaron A, Schuldiner O. Common and divergent mechanisms in developmental neuronal remodeling and dying back neurodegeneration. Current Biology Cell Press. 2016;26:R628–39.
Han X, Zhu J, Zhang X, Song Q, Ding J, Lu M, et al. Plin4-dependent lipid droplets hamper neuronal mitophagy in the MPTP/p-induced mouse model of Parkinson’s disease. Front Neurosci Frontiers. 2018;12:1257.
Schlomann U, Rathke-Hartlieb S, Yamamoto S, Jockusch H, Bartsch JW. Tumor necrosis factor α induces a metalloprotease-disintegrin, ADAM8 (CD 156): implications for neuron–glia interactions during neurodegeneration. J Neurosci Society for Neuroscience. 2000;20:7964–71.
Yoon-Seong Kim THJ. Matrix metalloproteinases, new insights into the understanding of neurodegenerative disorders. Biomol Ther Korean Society of Applied Pharmacology. 2012;20:133–43.
Weinstock LD, Furness AM, Herron SS, Smith SS, Sankar SB, DeRosa SG, et al. Fingolimod phosphate inhibits astrocyte inflammatory activity in mucolipidosis IV. Hum Mol Genet. 2018;27:2725–38.
Cougnoux A, Drummond RA, Fellmeth M, Navid F, Collar AL, Iben J, et al. Unique molecular signature in mucolipidosis type IV microglia. J Neuroinflammation BioMed Central. 2019;16:1–12.
McGlasson S, Jury A, Jackson A, Hunt D. Type I interferon dysregulation and neurological disease. Nat Rev Neurol Nature Publishing Group. 2015;11:515–23.
Wong SW, Kwon M-J, Choi AMK, Kim H-P, Nakahira K, Hwang DH. Fatty acids modulate Toll-like receptor 4 activation through regulation of receptor dimerization and recruitment into lipid rafts in a reactive oxygen species-dependent manner. J Biol Chem American Society for Biochemistry and Molecular Biology. 2009;284:27384–92.
Varela ARP, Gonçalves da Silva AMPS, Fedorov A, Futerman AH, Prieto M, Silva LC. Effect of glucosylceramide on the biophysical properties of fluid membranes. Biochim Biophys Acta Elsevier. 2013;1828:1122–30.
Vitner EB, Vardi A, Cox TM, Futerman AH. Emerging therapeutic targets for Gaucher disease. Expert Opin Ther Targets Informa UK, Ltd. 2014;19:321–34.
Wang C-W. Lipid droplets, lipophagy, and beyond. Biochim Biophys Acta Elsevier. 1861;2016:793–805.
Fanning S, Haque A, Imberdis T, Baru V, Barrasa MI, Nuber S, et al. Lipidomic analysis of α-synuclein neurotoxicity identifies stearoyl CoA desaturase as a target for Parkinson treatment. Mol Cell Cell Press. 2019;73:1001–8.
Marschallinger J, Iram T, Zardeneta M, Lee SE, Lehallier B, Haney MS, et al. Lipid droplet accumulating microglia represent a dysfunctional and pro-inflammatory state in the aging brain. bioRxiv. Cold Spring Harbor Laboratory. 2019;722827:454.
Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell Cell Press. 2017;169:1276–1290.e17.
Li J-T, Zhang Y. TREM2 regulates innate immunity in Alzheimer’s disease. J Neuroinflammation BioMed Central. 2018;15:1–7.
Zhou S-L, Tan C-C, Hou X-H, Cao X-P, Tan L, Yu J-T. TREM2 variants and neurodegenerative diseases: a systematic review and meta-analysis. Zhu L-Q, editor J Alzheimers Dis IOS Press. 2019;68:1171–84.
Ren M, Guo Y, Wei X, Yan S, Qin Y, Zhang X, et al. TREM2 overexpression attenuates neuroinflammation and protects dopaminergic neurons in experimental models of Parkinson's disease. Exp Neurol Academic Press. 2018;302:205–13.
Yeh FL, Hansen DV, Sheng M. TREM2, microglia, and neurodegenerative diseases. Trends Mol Med Elsevier Current Trends. 2017;23:512–33.
Cho SM, Vardi A, Platt N, Futerman AH. Absence of infiltrating peripheral myeloid cells in the brains of mouse models of lysosomal storage disorders. Kielian T, editor. J. Neurochem. John Wiley & Sons, Ltd (10.1111); 2019;148:625–38.
Konishi H, Kiyama H. Microglial TREM2/DAP12 signaling: a double-edged sword in neural diseases. Front Cell Neurosci Frontiers. 2018;e1002466:12.
Holers VM. Complement and its receptors: new insights into human disease. Annu Rev Immunol Annual Reviews. 2014;32:433–59.
Pandey MK, Grabowski GA, Köhl J. An unexpected player in Gaucher disease: the multiple roles of complement in disease development. Semin Immunol Academic Press. 2018;37:30–42.
Pandey MK, Burrow TA, Rani R, Martin LJ, Witte D, Setchell KD, et al. Complement drives glucosylceramide accumulation and tissue inflammation in Gaucher disease. Nature Nature Publishing Group. 2017;543:108–12.
Wang X-X, Tan M-S, Yu J-T, Tan L. Matrix metalloproteinases and their multiple roles in Alzheimer’s disease. Biomed Res Int. Hindawi; 2014;2014:1–8.
Reddy PH. Amyloid precursor protein-mediated free radicals and oxidative damage: implications for the development and progression of Alzheimer’s disease. J. Neurochem. John Wiley & Sons. Ltd. 2006;96:1–13.
Juárez-Rebollar D, Rios C, Nava-Ruíz C, Méndez-Armenta M. Metallothionein in brain disorders. Oxid Med Cell Longev Hindawi. 2017;2017:1–12.
Costa R, Ferreira-da-Silva F, Saraiva MJ. (null) IC. Transthyretin protects against A-beta peptide toxicity by proteolytic cleavage of the peptide: a mechanism sensitive to the Kunitz protease inhibitor. PLoS One. 2008;3.
Santos CRA, Martinho A, Quintela T, Gonçalves I. Neuroprotective and neuroregenerative properties of metallothioneins. IUBMB Life. John Wiley & Sons. Ltd. 2012;64:126–35.
Miyazaki I, Asanuma M, Hozumi H, Miyoshi K, Sogawa N. Protective effects of metallothionein against dopamine quinone-induced dopaminergic neurotoxicity. FEBS Lett No longer published by Elsevier. 2007;581:5003–8.
Acknowledgements
We thank Dr. Yael Pewzner-Jung for help in experimental design, and Drs. Ester Felsmesser, Noa Wigoda, and Ron Rotkopf for help with RNAseq analysis. A.H. Futerman is the Joseph Meyerhoff Professor of Biochemistry at the Weizmann Institute of Science.
Funding
This work was supported by the Children’s Gaucher Research Fund.
Author information
Authors and Affiliations
Contributions
AV performed the experiments and wrote the manuscript. SBD helped with RNAseq data analysis. SMC prepared the library and ran the RNAseq. UL and JS provided the MyTrMaSt mice and helped with data interpretation. AHF obtained funding, led the research and wrote the manuscript. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
Mice were maintained under specific pathogen-free conditions and handled according to protocols approved by the Weizmann Institute Animal Care Committee according to international guidelines.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1.
Primers used for RT-PCR
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Vardi, A., Ben-Dor, S., Cho, S.M. et al. Mice defective in interferon signaling help distinguish between primary and secondary pathological pathways in a mouse model of neuronal forms of Gaucher disease. J Neuroinflammation 17, 265 (2020). https://doi.org/10.1186/s12974-020-01934-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12974-020-01934-x