
Abstract
Background
Neuropathic pain is caused by damage to the nervous system, resulting in aberrant pain, which is associated with gene expression changes in the sensory pathway. However, the molecular mechanisms are not fully understood.
Methods
Wistar rats were employed for the establishment of the chronic constriction injury (CCI) models. Using the Illumina HiSeq 4000 platform, we examined differentially expressed genes (DEGs) in the rat dorsal horn by RNA sequencing (RNA-seq) between CCI and control groups. Then, enrichment analyses were performed for these DEGs using Gene Ontology (GO) function, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway, Hierarchical Cluster, and protein-protein interaction (PPI) network.
Results
A total of 63 DEGs were found significantly changed with 56 upregulated (e.g., Cxcl13, C1qc, Fcgr3a) and 7 downregulated (e.g., Dusp1) at 14 days after CCI. Quantitative reverse-transcribed PCR (qRT-PCR) verified changes in 13 randomly selected DEGs. GO and KEGG biological pathway analyses showed that the upregulated DEGs were mostly enriched in immune response-related biological processes, as well as 14 immune- and inflammation-related pathways. The downregulated DEGs were enriched in inactivation of mitogen-activated protein kinase (MAPK) activity. PPI network analysis showed that Cd68, C1qc, C1qa, Laptm5, and Fcgr3a were crucial nodes with high connectivity degrees. Most of these genes which have previously been linked to immune and inflammation-related pathways have not been reported in neuropathic pain (e.g., Laptm5, Fcgr3a).
Conclusions
Our results revealed that immune and defense pathways may contribute to the generation of neuropathic pain after CCI. These mRNAs may represent new therapeutic targets for the treatment of neuropathic pain.
Similar content being viewed by others


Background
Neuropathic pain is a chronic pain and may result from primary damage, disease or dysfunction of the peripheral or central nervous system, which is characterized by an increased responsiveness of nociceptive neurons in the nervous system [1]. The molecular mechanisms of neuropathic pain remain poorly understood, but it is known to involve nerve injury, inflammatory cytokine release, anatomical remodeling, and nociceptive receptors. [2, 3]. Thus, an improved understanding of pathogenesis from gene interactions in neuropathic pain is crucial for the development of the genetic and various neurobiological base therapeutic strategies to prevent neuropathic pain and improve the treatment effect.
The chronic constriction injury (CCI) rat model which simulates the symptoms of chronic nerve compression has been used as a model of neuropathic pain because rats subjected to CCI behave analogously to humans with neuropathic pain [4, 5]. Reportedly, CCI is highly associated with inflammation [6, 7]. Activation of immune and immune-like glial cells in the injured nerve, dorsal root ganglia, and spinal cord could generate a variety of mediators such as cytokines, chemokines, and other inflammatory mediators [8]. Interestingly, some of these mediators, such as cytokines and chemokines, can directly activate or sensitize nociceptors, contributing to the development of neuropathic pain [9].
Gene expression profile studies can be used to provide understanding of the molecular mechanisms underlying the development and maintenance of neuropathic pain [10,11,12]. Some studies using microarray and RNA sequencing (RNA-seq) analysis have been conducted to investigate the mechanism underlying the generation of neuropathic pain in spared nerve injury (SNI) model [13, 14]. Although they identified several crucial differentially expressed genes (DEGs) and different immune actions in SNI models, the alteration of gene expression and mechanisms on neuropathic pain are still unclear. Therefore, the present study was carried out to compare the different gene expression profiles of the dorsal horn of CCI rats and controls using the Illumina Hiseq 4000 to reveal the underlying regulatory mechanism of CCI rat models. Moreover, the molecular and cellular functions of the predicted mRNAs as well as the signaling pathways involved based on the present experiment will be further investigated.
Methods
Animals
Adult male Wistar rats weighing 200–250 g were obtained from the Animal Center of Taishan Medical University. All experimental procedures followed the guidelines of the Taishan Medical University Institutional Animal Care and Use Committee. Efforts were made to minimize the number of animals used and their sufferings.
CCI models
CCI to the sciatic nerve of the right hind limb in rats was performed based on previous description [15]. Briefly, animals were anesthetized with 4% chloral hydrate (10 ml/kg; i.p.). The sciatic nerve of the right hind limb was exposed at the middle of the thigh by blunt dissection. To prevent the interruption of blood circulation through the epineural vasculature, four chromic gut ligatures were loosely tied (4.0 silk) around the nerve with spacing at ~ 1 mm. In the control group, the right sciatic nerve was exposed for 2–3 min, but was not ligated. Following surgery, the skin was closed with a single suture, and the animals were allowed to recover for various period of time before behavioral testing.
Mechanical withdrawal threshold (MWT)
All behavioral tests were performed in a blinded manner. Mechanical allodynia and thermal hyperalgesia are reproducible and sensitive behavioral readouts of neuropathic pain. Behavioral testing was conducted prior to surgery and on days 1, 3, 7, 10, and 14 following surgery. Animals were allowed to acclimate to elevated cages (20 × 14 × 16 cm) with a wire mesh bottom. MWT was measured by assessing hind paw sensitivity to innocuous mechanical stimulation. Von Frey filaments (0.41–15.1 g; North Coast, Gilroy, CA) were applied to the plantar aspect of the right hind paw. Lifting, licking the paw, and running away were all considered as positive responses. The maximum applied pressure was recorded. The MWT of each animal was the average of six measurements taken at 5 min intervals.
Thermal withdrawal latency (TWL)
In this assay, rats were placed in a transparent, square, bottomless acrylic box (17 × 11.5 × 14 cm) and allowed to adapt for 20 min. Responses to thermal stimulation were evaluated using a 37,370 plantar test apparatus as a source of radiant heat. A beam of focused light set at 60 °C was directed towards the plantar surface of the hind paw, and the maximum latency time was recorded. The time to purposeful withdrawal of the foot from the beam of light was measured. A cutoff time was set at 40 s to prevent tissue damage. Every hind paw was tested alternately at 5 min intervals. The results obtained for each rat were expressed in second as the mean of six withdrawal latencies. Finally, the average value was used for statistical analysis.
Tissue collection, RNA isolation, cDNA library preparation, and sequencing
Animals were deeply anesthetized with isoflurane (3%) at 14 days after surgery and perfused through the ascending aorta with normal saline (100 ml, 4 °C). The L4–5 spinal cord segments that correspond to L4–5 spinal nerve roots and match L1 vertebral level were dissected. The dorsal horns of L4–5 spinal cord were collected. Total RNA was extracted from the dorsal horn tissue using Trizol reagent (Invitrogen, Carlsbad) according to the manufacturer’s protocol. RNA quantity and quality were measured using a NanoDrop ND-1000. The cDNA library was constructed using KAPA Stranded RNA-Seq Library Preparation Kit (Illumina) following the manufacturer’s protocol. Briefly, poly-(A) mRNA was isolated from total RNA using the NEBNext Oligo d(T) magnetic beads. Under an elevated temperature, mRNA was fragmented into small pieces after the fragmentation buffer was added. Using the mRNA fragments as templates, the first-strand cDNA was synthesized with random primers. Then, the second-strand cDNA was obtained using DNA polymerase I and RNase H. The synthetic cDNAs were end-repaired by polymerase and ligated with “A-tailing” base adaptors. Suitable fragments were selected for PCR amplification to construct the final cDNA library. The final double-stranded cDNA samples were verified with an Agilent 2100 Bioanalyzer (Agilent Technologies). After cluster generation (TruSeq SR Cluster Kit v3-cBot-HS, Illumina), sequencing was performed on an Illumina HiSeq 4000 sequencing platform.
RNA-seq data processing
Image analysis, base calling, and error estimation were performed using Illumina/Solexa Pipeline version 1.8 (Off-Line Base Caller software, version 1.8). Quality control was checked on the raw sequence data using FastQC (https://en.wikipedia.org/wiki/FASTQ_format). Raw data were pre-processed using Solexa CHASTITY and Cutadapt to remove adaptor sequences, ribosomal RNA, and other contaminants that may interfere with clustering and assembly. The trimmed reads are mapped to the corresponding reference genome using HISAT2 (version 2.0.4) for RNA-seq, and StringTie (version 1.2.3) was used to reconstruct the transcriptome [16, 17]. Then, Ballgown software was applied to calculate the fragments per kilobase of exon per million fragments mapped (FPKM) in RNA-seq data and analyze DEGs [18, 19], with the FPKM ≥ 0.5 (Cuffquant) considered statistically significant.
Bioinformatics analysis
The Gene Ontology (GO) functional and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis were performed for DEGs using the Database for Annotation, Visualization and Integrated Discovery (DAVID) and KEGG Orthology-Based Annotation System (KOBAS) online tools (http://www.geneontology.org and http://www.genome.jp/kegg). Hierarchical cluster analysis was performed for enriched genes by Cluster 3.0 software. The protein-protein interaction (PPI) network of the proteins encoded by the DEGs was searched using STRING online software (http://string-db.org/).
Quantitative reverse transcription-PCR (qRT-PCR) analysis
Thirteen DEGs (11 regulated and 2 downregulated genes) were randomly selected and detected by qRT-PCR. The expression of β-actin mRNA was also determined as an internal control. Total RNA was extracted from the dorsal horn tissue using Trizol reagent (Invitrogen) according to the manufacturer’s protocol. RNA concentration was determined spectrophotometrically. cDNA was synthesized using a cDNA synthesis kit (Invitrogen) according to the manufacturer’s instructions. Primer sequences are listed in the Table 1. qRT-PCR was performed in triplicates by using a 7300 real-time PCR system (Applied Biosystems, Foster City, CA) according to the manufacturer’s instructions. A comparative cycle of threshold fluorescence (∆Ct) method was used, and the relative transcript amount of target gene was normalized to that of β-actin using the 2−∆∆Ct method. The final results of qRT-PCR were expressed as the ratio of test mRNA to control.
Statistical analysis
Data are presented as the means ± SEM. The results from the behavioral study were statistically analyzed using repeated measures analysis of variance. qRT-PCR results were analyzed using one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparison test. Significance was set at p < 0.05.
Results
Model identification of neuropathic pain
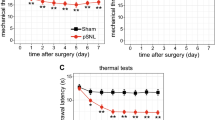
The neuropathic pain rat model was established by CCI to the sciatic nerve of the right hind limb in rats. Both mechanical allodynia and thermal sensitivity were determined in all CCI model rats at 0, 1, 3, 7, and 14 days after surgery, respectively. CCI rats exhibited higher sensitivity to mechanical and thermal stimuli from days 1 to 14. Both MWT and TWL reached a steady peak at day 14 after surgery (Fig. 1).

Nociceptive behavior developed in chronic constriction injury (CCI) model rats. Mechanical withdrawal threshold (MWT) in each time point (a) and thermal withdrawal latency (TWL) in each time point (b). n = 6, *p < 0.05 compared with controls
Differential gene expression in the spinal cord
To determine genes that are involved in the pathological process of neuropathic pain, the dorsal horn of L4–5 spinal cord of rats was analyzed using an Illumina HiSeq 4000 sequencing technique at 14 days after CCI surgery. Using the FPKM of ≥ 0.5, abundant expression levels were compared to those of CCI-induced neuropathic pain with control. We identified a total of 17,912 mRNA transcripts corresponding to 14,546 genes in CCI-induced neuropathic pain rat models after 14 days (Additional file 1: Table S1; Additional file 2: Table S2). Sixty-three genes were differentially expressed between CCI-induced neuropathic pain and control tissues (Table 2; Additional file 3: Table S3) using two criteria: a greater than 1.5 fold expression level change and p value ≤ 0.05 from ANOVA test. The related gene expression frequency and abundance in the dorsal horn of the CCI rat were showed in Fig. 2a. These 63 genes included 56 upregulated genes (e.g., Cxcl13, C1qc, Cgr3a) and 7 downregulated genes (e.g., Urgcp, Usp1) as shown in the volcano plot (Fig. 2b).

The differential expression of genes (DEGs) in the dorsal horn between control and chronic constriction injury (CCI) model was determined by RNA-seq technology. a Scatter plot showing the upregulated and downregulated genes (the red and green dots, respectively) in the dorsal horn of L4–5 spinal cord in the CCI rat with respect to the control. Black dots represent genes with no significant difference. b Volcano plot indicated upregulated and downregulated DEGs in the dorsal horn of CCI models. Red dots represent genes with significantly upregulated expression, green dots represent genes with significantly downregulated expression, while black dots represent genes with no significantly difference, respectively
GO functional analysis of DEGs
According to the functional annotation in GO database, the upregulated DEGs were mostly enriched in biological processes (BP) related to immune and defense responses (Additional file 4: Table S4), cellular component (CC) terms such as endocytic vesicle and phagocytic cup (Additional file 5: Table S5), and molecular function (MF) terms related to IgG binding and chemokine activity (Additional file 6: Table S6). The GO enrichment terms of BP, CC, and MF for upregulated DEGs are shown in Fig. 3a.

The Gene Ontology (GO) analysis of differentially expressed genes (DEGs) in the dorsal horn of chronic constriction injury (CCI) rats. GO annotation of upregulated DEGs (a) and downregulated DEGs (b) of CCI model versus control. Bar plots show the top ten enrichment score (−log10(p value)) values of the significant enrichment terms of DEGs involving biological process, cellular component, and molecular function
Meanwhile, the downregulated DEGs were enriched in BP terms such as regulation of spindle checkpoint and inactivation of mitogen-activated protein (MAP) kinase (MAPK) activity (Additional file 7: Table S7), CC terms such as Cul3-RING ubiquitin ligase complex (Additional file 8: Table S8), and MF terms such as MAP kinase phosphatase (MKP) kinase activity (Additional file 9: Table S9). The GO enrichment terms of BP, CC, and MF for downregulated DEGs are shown in Fig. 3b.
KEGG pathway enrichment analysis of DEGs
The DEGs between CCI model and control were subjected to KEGG pathway enrichment analysis using the software KOBAS. The p value < 0.05 was set as the threshold of significant enrichment. Based on the KEGG pathway enrichment analysis, the upregulated DEGs were significantly enriched in 14 signaling pathways, such as complement and coagulation cascades, B cell receptor signaling pathway, cytokine-cytokine receptor interaction, and Fc gamma R-mediated phagocytosis signaling pathway, which were mostly related to immune and inflammatory responses (Fig. 4a, Additional file 10: Table S10). However, none of the downregulated gene was significantly enriched in any KEGG pathway.

Kyoto Encyclopedia of Genes and Genomes (KEGG), hierarchical clustering, and protein-protein interaction (PPI) network analysis of differentially expressed genes (DEGs) in the dorsal horn of chronic constriction injury (CCI) rats. a Histogram of KEGG pathway enrichment distribution of DEGs. The bar plot shows the top ten enrichment score (−log10(p value)) value of the significant enrichment pathway. b Heat map of DEGs showing hierarchical clustering of changed DEGs of rats in CCI group compared with control group. In clustering analysis, upregulated and downregulated genes are colored in red and green, respectively. c STRING analysis for biological interactions within DEGs of RNA-seq datasets, involved in immune and inflammatory function. The line color indicates the type of interaction evidence, and line thickness indicates the strength of data support. d Quantitative reverse transcription-PCR (qRT-PCR) analysis for differences in expression levels of DEGs in the dorsal horn between CCI models and controls. Results were calculated by normalizing to β-actin in the same sample with the ΔCt method. Changes in relative levels of gene mRNAs expressed as folds of controls. All values were mean ± SEM. *p < 0.05 (n = 3)
Hierarchical cluster analysis of DEGs
To elucidate the role of DEGs in CCI model tissues, DEGs were hierarchically clustered dependent on the gene enrichment features of control against CCI model tissues (Fig. 4b). The most prominently upregulated genes consisted of families of chemokines (Cxcl13 and Ccl2), complement components (C1qc, Ccl2, C1qa, C3, C1qb, and C4a), Fc fragment receptors (Fcer1g, Fcgr3a, Fcgr1a, and Fcgr2b), cluster of differentiations (Cd22, Cd33, Cd53, and Cd68), and G protein-coupled receptors (Gpr31, Gpr34, and Gpr183). Strikingly, chemokine genes showed the greatest upregulation such as Cxcl13 (6.426 fold increase). Most of these genes which have previously been linked to immune and inflammation-related pathways have not been reported in neuropathic pain; and only 20 genes (e.g., Cxcl13, C1qc, Ccl2, C1qa, Fcer1g, Ngfr, Cd53, Cd68, Dusp1) have been demonstrated to be involved in this pathogenesis. This clustering analysis of RNA-seq data will indicate that the DEGs in CCI model are closely associated with the development of neuropathic pain.
PPI network analysis
To investigate the interaction and hub genes of DEGs involved in pathogenesis of neuropathic pain, the DEGs PPI network were constructed using STRING. The results demonstrated that the predicted PPI in CCI rats were driving the complex interaction network at 14 days after CCI (Fig. 4c). The established PPI network (PPI enrichment p value < 1.0e−16) contained 58 nodes (hub genes) and 77 edges (interactions). Five of the top genes with relatively high connectivity degrees (≥ 11) were highlighted: Cd68 (degree = 14), C1qc (degree = 12), C1qa (degree = 11), Laptm5 (degree = 11), and Fcgr3a (degree = 11). Many novel DEGs that we screened may play an important role through regulation of protein expression in neuropathic pain, but future in-depth studies are required.
Validation by qRT-PCR
To evaluate the reliability of the Illumina sequencing technology, 13 DEGs (11 upregulated and 2 downregulated genes) were randomly selected and detected by qRT-PCR. Figure 4d shows that the upregulation or downregulation trend of candidate genes between CCI-induced model and control group revealed by the qRT-PCR data is congruent with that revealed by RNA-seq method. The result of qRT-PCR analyses provides evidence that the RNA-seq method for the large-scale gene expression quantification was reliable.
Discussion
In this study, we profiled gene expression in the dorsal horn following CCI-induced neuropathic pain, using RNA-seq method. Sixty-three DEGs were identified in CCI rat model, including 56 upregulated and 7 downregulated genes. We also predicted potential functions of DEGs using GO, KEGG pathway, and PPI network analysis in the CCI model. These findings prompted the proposal that DEGs played a significant role in neuropathic pain processing, and sequencing analysis revealed a potential therapeutic target of neuropathic pain.
Accumulating evidence showed that Cxcl13 and Ccl2 are known to be involved in pathogenesis of neuropathic pain via different forms of neuron-glia interaction in the spinal cord [20, 21]. The chemokines by binding to the G protein-coupled receptors play an essential role in pathological pain conditions triggered by either peripheral inflammation or nerve injury [23, 24]. Our results demonstrated that chemokine genes showed the greatest upregulation, such as Cxcl13 (6.426 fold increase), suggesting that chemokines play a crucial role in the development of neuropathic pain. Furthermore, complements, a key component of the innate immune system and potentially important trigger of some types of neuropathic pain, have been associated with neuroinflammation [22, 25, 26]. Previous study showed that C1qc, C1qa, and Fcer1g might contribute to the generation of neuropathic pain after SNI via immune and defense pathways [26]. Activation of Cd68, the inflammatory microglia-dominant molecule, could result in neuropathic pain in mice with peripheral nerve injury [27]. Cd53, the inflammation-related gene, is chronically upregulated after spinal cord injury [28, 29]. The up-expression of nerve growth factor (NGF) and the NGF receptor is involved in regulating the function of sensory and the development of neuropathic pain [30]. In the present study, upregulated genes included chemokines, complement components, Fc fragment receptors, cluster of differentiations, G protein-coupled receptors, and NGF receptor. Most of the genes are well known in neuropathic pain (e.g., Cxcl13, C1qc, Ccl2) [20,21,22,23,24,25,26,27,28,29,30], suggesting DEGs with the differential functions in diverse cellular pathways, and many are involved in neuropathic pain development and progression.
Previous study demonstrated that Urgcp plays a critical role in glial cell cycle and cell proliferation [31]. Dusp1, a MKP-1, plays a pivotal role in controlling MAPK-dependent inflammatory responses [32]. In the present study, we found that Urgcp and Dusp1 displayed obvious down-expression in CCI rats, which would provide a better understanding of immune and glia cell proliferation, as well as MAPK-dependent inflammatory response abnormalities involved in the neuropathic pain of CCI rats.
In order to obtain insights into DEGs function, GO analysis annotation was applied to the DEG gene pool. GO terms for biological process categories included immune system process, phagocytosis, defense response, and response to external stimulus. Several studies using SNI rat model have reported that key higher expressed genes included those associated with immune and inflammatory pathways in neuropathic pain [20,21,22,23,24,25,26,27,28,29,30], similar to those in CCI model we observed. GO functional analysis showed that downregulated DEGs might associate with inactivation of MAPK activity in CCI models. Inhibition of MAPKs results in downregulation of downstream molecules (cytokines, chemokines, nitric oxide, etc.) in immunocompetent cells and depresses the excitability of neurons in the spinal cord [33, 34]. Therefore, spinal MAPK signaling pathway, such as p38-MAPK (p38) or extracellular receptor-activated kinases (ERKs), may play an important role in the development of chronic allodynia in CCI [35].
PPI network analysis showed that Cd68, C1qc, C1qa, Laptm5, and Fcgr3a were crucial nodes with high connectivity degrees. Laptm5 and Fcgr3a which have previously been linked to immune and inflammation-related pathways have not been reported in neuropathic pain [36, 37]; and other three genes (Cd68, C1qc, and C1qa) have been demonstrated to be involved in this pathogenesis [26, 27]. Previous study showed that Laptm5 is involved in the dynamics of lysosomal membranes associated with microglial activation after nerve injury [36]. Upregulation of Fcgr3a increased the microglial phagocytic capacity in neuroinflammation [37]. It might be inferred that Laptm5 and Fcgr3a are neuroinflammation-related genes that influence neuropathic pain behavior after CCI.
Conclusions
In conclusion, our results suggest that genes involved in immune and defense responses are affected most significantly after CCI. Genes like Cxcl13, Cd68, C1qc, Laptm5, and Fcgr3a are crucial for neuropathic pain after CCI in rat models. These genes could be used as novel diagnostic and therapeutic targets against CCI-induced neuropathic pain. However, the predicted expressions and interactions need to be further validated by extensive experiments.
Abbreviations
- BP:
-
Biological processes
- CC:
-
Cellular component
- CCI:
-
Chronic constriction injury
- DAVID:
-
Database for Annotation, Visualization and Integrated Discovery
- DEGs:
-
Differentially expressed genes
- ERKs:
-
Extracellular receptor-activated kinases
- FPKM:
-
Fragments per kilobase of exon per million fragments mapped
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- KOBAS:
-
KEGG Orthology-Based Annotation System
- MAPK:
-
Mitogen-activated protein (MAP) kinases
- MF:
-
Molecular function
- MKP:
-
MAP kinase phosphatase
- MWT:
-
Mechanical withdrawal threshold
- NGF:
-
Nerve growth factor
- PPI:
-
Protein-protein interaction
- qRT-PCR:
-
Quantitative reverse transcription-PCR
- RNA-seq:
-
RNA sequencing
- SNI:
-
Spared nerve injury
- TWL:
-
Thermal withdrawal latency
References
Baron R, Binder A, Wasner G. Neuropathic pain: diagnosis, pathophysiological mechanisms, and treatment. Lancet Neurol. 2010;9:807–19. https://doi.org/10.1016/S1474-4422(10)70143-5.
Hayashi Y, Kawaji K, Sun L, Zhang X, Koyano K, Yokoyama T, Kohsaka S, Inoue K, Nakanishi H. Microglial Ca(2+)-activated K(+) channels are possible molecular targets for the analgesic effects of S-ketamine on neuropathic pain. J Neurosci. 2011;31:17370–82. https://doi.org/10.1523/JNEUROSCI.4152-11.2011.
Leng C, Chen L, Gong X, Ma B, Gan W, Si Y, Xiao H, Li C. Upregulation of P2X2 and P2X3 receptors in rats with hyperalgesia induced by heroin withdrawal. Neuroreport. 2018;29:678–84. https://doi.org/10.1097/WNR.0000000000001018.
Sumizono M, Sakakima H, Otsuka S, Terashi T, Nakanishi K, Ueda K, Takada S, Kikuchi K. The effect of exercise frequency on neuropathic pain and pain-related cellular reactions in the spinal cord and midbrain in a rat sciatic nerve injury model. J Pain Res. 2018;11:281–91. https://doi.org/10.2147/JPR.S156326.
Da Silva JT, Evangelista BG, Venega RAG, Oliveira ME, Chacur M. Early and late behavioral changes in sciatic nerve injury may be modulated by nerve growth factor and substance P in rats: a chronic constriction injury long-term evaluation. J Biol Regul Homeost Agents. 2017;31:309–19.
Zhang Y, Chi D. Overexpression of SIRT2 alleviates neuropathic pain and neuroinflammation through deacetylation of transcription factor nuclear factor-kappa B. Inflammation. 2018;41:569–78. https://doi.org/10.1007/s10753-017-0713-3.
Ghasemzadeh Rahbardar M, Amin B, Mehri S, Mirnajafi-Zadeh SJ, Hosseinzadeh H. Anti-inflammatory effects of ethanolic extract of Rosmarinus officinalis L. and rosmarinic acid in a rat model of neuropathic pain. Biomed Pharmacother. 2017;86:441–9. https://doi.org/10.1016/j.biopha.2016.12.049.
Austin PJ, Moalem-Taylor G. The neuro-immune balance in neuropathic pain: involvement of inflammatory immune cells, immune-like glial cells and cytokines. J Neuroimmunol. 2010;229:26–50. https://doi.org/10.1016/j.jneuroim.2010.08.013.
Cook AD, Christensen AD, Tewari D, McMahon SB, Hamilton JA. Immune cytokines and their receptors in inflammatory pain. Trends Immunol. 2018;39:240–55. https://doi.org/10.1016/j.it.2017.12.003.
Liu H, Xia T, Xu F, Ma Z, Gu X. Identification of the key genes associated with neuropathic pain. Mol Med Rep. 2018;17:6371–8. https://doi.org/10.3892/mmr.2018.8718.
Kummer KK, Kalpachidou T, Kress M, Langeslag M. Signatures of altered gene expression in dorsal root ganglia of a Fabry disease mouse model. Front Mol Neurosci. 2018;10:449. https://doi.org/10.3389/fnmol.2017.00449.
Chen CJ, Liu DZ, Yao WF, Gu Y, Huang F, Hei ZQ, Li X. Identification of key genes and pathways associated with neuropathic pain in uninjured dorsal root ganglion by using bioinformatic analysis. J Pain Res. 2017;10:2665–74. https://doi.org/10.2147/JPR.S143431.
Vallejo R, Tilley DM, Cedeño DL, Kelley CA, DeMaegd M, Benyamin R. Genomics of the effect of spinal cord stimulation on an animal model of neuropathic pain. Neuromodulation. 2016;19:576–86. https://doi.org/10.1111/ner.12465.
Hong H, Hong Q, Liu J, Tong W, Shi L. Estimating relative noise to signal in DNA microarray data. Int J Bioinforma Res Appl. 2013;9:433–48. https://doi.org/10.1504/IJBRA.2013.056085.
Fox A, Kesingland A, Gentry C, McNair K, Patel S, Urban L, James I. The role of central and peripheral cannabinoid1 receptors in the antihyperalgesic activity of cannabinoids in a model of neuropathic pain. Pain. 2001;92:91–100.
Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12:357–60. https://doi.org/10.1038/nmeth.3317.
Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33:290–5. https://doi.org/10.1038/nbt.3122.
Frazee AC, Pertea G, Jaffe AE, Langmead B, Salzberg SL, Leek JT. Ballgown bridges the gap between transcriptome assembly and expression analysis. Nat Biotechnol. 2015;33:243–6. https://doi.org/10.1038/nbt.3172.
Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–8. https://doi.org/10.1038/nmeth.1226.
Zhang ZJ, Jiang BC, Gao YJ. Chemokines in neuron-glial cell interaction and pathogenesis of neuropathic pain. Cell Mol Life Sci. 2017;74:3275–91. https://doi.org/10.1007/s00018-017-2513-1.
Al-Mazidi S, Alotaibi M, Nedjadi T, Chaudhary A, Alzoghaibi M, Djouhri L. Blocking of cytokines signaling attenuates evoked and spontaneous neuropathic pain behaviours in the paclitaxel rat model of chemotherapy-induced neuropathy. Eur J Pain. 2018;22:810–21. https://doi.org/10.1002/ejp.1169.
Xu J, Zhang L, Xie M, Li Y, Huang P, Saunders TL, Fox DA, Rosenquist R, Lin F. Role of complement in a rat model of paclitaxel-induced peripheral neuropathy. J Immunol. 2018;200:4094–101. https://doi.org/10.4049/jimmunol.1701716.
Martínez-Muñoz L, Villares R, Rodríguez-Fernández JL, Rodríguez-Frade JM, Mellado M. Remodeling our concept of chemokine receptor function: from monomers to oligomers. J Leukoc Biol. 2018;104:323–31. https://doi.org/10.1002/JLB.2MR1217-503R.
Silva RL, Lopes AH, Guimarães RM, Cunha TM. CXCL1/CXCR2 signaling in pathological pain: role in peripheral and central sensitization. Neurobiol Dis. 2017;105:109–16. https://doi.org/10.1016/j.nbd.2017.06.001.
Kallio-Laine K, Seppänen M, Lokki ML, Lappalainen M, Notkola IL, Seppälä I, Koskinen M, Valtonen V, Kalso E. Widespread unilateral pain associated with herpes simplex virus infections. J Pain. 2008;9:658–65. https://doi.org/10.1016/j.jpain.2008.02.003.
Wang J, Ma SH, Tao R, Xia LJ, Liu L, Jiang YH. Gene expression profile changes in rat dorsal horn after sciatic nerve injury. Neurol Res. 2017;39:176–82. https://doi.org/10.1080/01616412.2016.1273590.
Kiguchi N, Kobayashi D, Saika F, Matsuzaki S, Kishioka S. Inhibition of peripheral macrophages by nicotinic acetylcholine receptor agonists suppresses spinal microglial activation and neuropathic pain in mice with peripheral nerve injury. J Neuroinflammation. 2018;15:96. https://doi.org/10.1186/s12974-018-1133-5.
Giardini AC, Dos Santos FM, da Silva JT, de Oliveira ME, Martins DO, Chacur M. Neural mobilization treatment decreases glial cells and brain-derived neurotrophic factor expression in the central nervous system in rats with neuropathic pain induced by CCI in rats. Pain Res Manag. 2017;2017:7429761. https://doi.org/10.1155/2017/7429761.
Byrnes KR, Washington PM, Knoblach SM, Hoffman E, Faden AI. Delayed inflammatory mRNA and protein expression after spinal cord injury. J Neuroinflammation. 2011;8:130. https://doi.org/10.1186/1742-2094-8-130.
Patel MK, Kaye AD, Urman RD. Tanezumab: therapy targeting nerve growth factor in pain pathogenesis. J Anaesthesiol Clin Pharmacol. 2018;34:111–6. https://doi.org/10.4103/joacp.JOACP_389_15.
Dodurga Y, Seçme M, Eroğlu C, Gündoğdu G, Avcı ÇB, Bağcı G, Küçükatay V, Lale Şatıroğlu-Tufan N. Investigation of the effects of a sulfite molecule on human neuroblastoma cells via a novel oncogene URG4/URGCP. Life Sci. 2015;143:27–34. https://doi.org/10.1016/j.lfs.2015.10.005.
Kim H, Woo JH, Lee JH, Joe EH, Jou I. 22(R)-hydroxycholesterol induces HuR-dependent MAP kinase phosphatase-1 expression via mGluR5-mediated Ca(2+)/PKCα signaling. Biochim Biophys Acta. 2016;1859:1056–70. https://doi.org/10.1016/j.bbagrm.2016.05.008.
Monneau YR, Luo L, Sankaranarayanan NV, Nagarajan B, Vivès RR, Baleux F, Desai UR, Arenzana-Seidedos F, Lortat-Jacob H. Solution structure of CXCL13 and heparan sulfate binding show that GAG binding site and cellular signalling rely on distinct domains. Open Biol. 2017;7. pii: 170133. https://doi.org/10.1098/rsob.170133.
Landry RP, Martinez E, DeLeo JA, Romero-Sandoval EA. Spinal cannabinoid receptor type 2 agonist reduces mechanical allodynia and induces mitogen-activated protein kinase phosphatases in a rat model of neuropathic pain. J Pain. 2012;13:836–48. https://doi.org/10.1016/j.jpain.2012.05.013.
Kawasaki Y, Kohno T, Zhuang ZY, Brenner GJ, Wang H, Van Der Meer C, Befort K, Woolf CJ, Ji RR. Ionotropic and metabotropic receptors, protein kinase A, protein kinase C, and Src contribute to C-fiber-induced ERK activation and cAMP response element-binding protein phosphorylation in dorsal horn neurons, leading to central sensitization. J Neurosci. 2004;24:8310–21.
Origasa M, Tanaka S, Suzuki K, Tone S, Lim B, Koike T. Activation of a novel microglial gene encoding a lysosomal membrane protein in response to neuronal apoptosis. Brain Res Mol Brain Res. 2001;88:1–13.
Sivagnanam V, Zhu X, Schlichter LC. Dominance of E. coli phagocytosis over LPS in the inflammatory response of microglia. J Neuroimmunol. 2010;227:111–9. https://doi.org/10.1016/j.jneuroim.2010.06.021.
Acknowledgements
The authors would like to thank Dr. Zhen Ye for skillful assistance in protein-protein interaction (PPI) network analysis. RNA-seq experiments were performed by KangChen Bio-tech, Shanghai, China.
Funding
This work was supported by the National Natural Science Foundation of China (31871215) and Scientific Research Projects of Colleges and Universities in Shandong Province (J15LK07).
Availability of data and materials
The key data are included in the figures, tables, and additional files. The full datasets that were analyzed are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
ZW, HD, and JS designed the overall project. JS, MW, SA, and XG performed the experiments. ZW, HD, and JS were responsible for the analysis and interpretation of the data and drafted the manuscript. All authors revised and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. All expressed transcripts in chronic constriction injury (CCI) model rats. (XLSX 3674 kb)
Additional file 2:
Table S2. All expressed genes in chronic constriction injury (CCI) model rats. (XLSX 2944 kb)
Additional file 3:
Table S3. Differentially expressed genes (DEGs) in chronic constriction injury (CCI) model rats. (XLSX 19 kb)
Additional file 4:
Table S4. Biological processes (BP) result of the upregulated differentially expressed genes (DEGs) in chronic constriction injury (CCI) model rats. (XLS 162 kb)
Additional file 5:
Table S5. Cellular component (CC) result of the upregulated differentially expressed genes (DEGs) in chronic constriction injury (CCI) model rats. (XLS 34 kb)
Additional file 6:
Table S6. Molecular function (MF) result of the upregulated differentially expressed genes (DEGs) in chronic constriction injury (CCI) model rats. (XLS 33 kb)
Additional file 7:
Table S7. Biological processes (BP) result of the downregulated differentially expressed genes (DEGs) in chronic constriction injury (CCI) model rats. (XLS 52 kb)
Additional file 8:
Table S8. Cellular component (CC) result of the downregulated differentially expressed genes (DEGs) in chronic constriction injury (CCI) model rats. (XLS 28 kb)
Additional file 9:
Table S9. Molecular function (MF) result of the downregulated differentially expressed genes (DEGs) in chronic constriction injury (CCI) model rats. (XLS 29 kb)
Additional file 10:
Table S10. Kyoto Encyclopedia of Genes and Genomes (KEGG) result of the upregulated differentially expressed genes (DEGs) in chronic constriction injury (CCI) model rats. (XLS 35 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Du, H., Shi, J., Wang, M. et al. Analyses of gene expression profiles in the rat dorsal horn of the spinal cord using RNA sequencing in chronic constriction injury rats. J Neuroinflammation 15, 280 (2018). https://doi.org/10.1186/s12974-018-1316-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12974-018-1316-0