
Abstract
Background
The neuroinflammatory response following traumatic brain injury (TBI) is known to be a key secondary injury factor that can drive ongoing neuronal injury. Despite this, treatments that have targeted aspects of the inflammatory pathway have not shown significant efficacy in clinical trials.
Main body
We suggest that this may be because classical inflammation only represents part of the story, with activation of neurogenic inflammation potentially one of the key initiating inflammatory events following TBI. Indeed, evidence suggests that the transient receptor potential cation channels (TRP channels), TRPV1 and TRPA1, are polymodal receptors that are activated by a variety of stimuli associated with TBI, including mechanical shear stress, leading to the release of neuropeptides such as substance P (SP). SP augments many aspects of the classical inflammatory response via activation of microglia and astrocytes, degranulation of mast cells, and promoting leukocyte migration. Furthermore, SP may initiate the earliest changes seen in blood-brain barrier (BBB) permeability, namely the increased transcellular transport of plasma proteins via activation of caveolae. This is in line with reports that alterations in transcellular transport are seen first following TBI, prior to decreases in expression of tight-junction proteins such as claudin-5 and occludin. Indeed, the receptor for SP, the tachykinin NK1 receptor, is found in caveolae and its activation following TBI may allow influx of albumin and other plasma proteins which directly augment the inflammatory response by activating astrocytes and microglia.
Conclusions
As such, the neurogenic inflammatory response can exacerbate classical inflammation via a positive feedback loop, with classical inflammatory mediators such as bradykinin and prostaglandins then further stimulating TRP receptors. Accordingly, complete inhibition of neuroinflammation following TBI may require the inhibition of both classical and neurogenic inflammatory pathways.
Similar content being viewed by others

Background
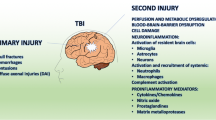
The role of inflammation in perpetuating the secondary injury response following traumatic brain injury (TBI) has received a significant amount of attention over the last two decades and is clearly an important factor in exacerbating neuronal injury. However, while many pre-clinical studies have shown that therapeutics targeting the immune response are effective in improving outcome when administered in the immediate aftermath of the injury [1–3], reports from clinical trials have been less promising. Agents with known anti-inflammatory properties such as corticosterone, progesterone, and erythropoietin (EPO) have all shown no benefit to date [4–6]. Specifically, the CRASH trial, a prospective, randomized, placebo-controlled multicenter trial of the corticosteroid, methylprednisolone, in TBI, reported an increased mortality following TBI [4]. The ProTECT III trial utilizing progesterone was halted due to failure to demonstrate improved outcome by the Glasgow Outcome Scale-Extended Score at 6 months post-injury [6], with similar findings in another clinical trial utilizing progesterone, the SyNAPse study [5]. In addition, promising anti-inflammatory agents identified in pre-clinical studies often have narrow therapeutic windows. For example, interleukin-1 antagonists appear most efficacious when first administered within hours following injury [1, 2], with minocycline also typically delivered within the first hour post-injury [7–9]. This may be, in part, due to the duality of the immune response following TBI, with some aspects of the inflammatory response necessary to promote repair [10]. In addition, this may also reflect that classical inflammation may be only be half the story, with neurogenic inflammation recently reported as playing a key initiating role [11–13], augmenting many aspects of the classical inflammatory response [14, 15]. This review will outline the interrelationship between classical and neurogenic inflammation, promoting a better understanding of the entire neuroinflammatory cascade and potentially facilitating the development of targeted anti-inflammatory regimes that can improve outcome following TBI.
Traumatic brain injury
TBI results from the head impacting with an object or from acceleration/deceleration forces that produce vigorous movement of the brain within the skull or varying combinations of these mechanical forces [16]. The resultant injury is caused by two mechanisms, either primary or secondary, although there is some degree of overlap [17, 18]. Primary injury is the result of mechanical forces (rotation, acceleration/deceleration, and direct force applied to the head) acting at the moment of the injury that damage the blood vessels, axons, nerve cells, and glia of the brain in a focal, multifocal, or diffuse pattern of involvement. The type and severity of the resulting injury depends upon the nature of the initiating force, as well as the site, direction, and magnitude of the force [19]. Contact forces generated when the head strikes or is struck by an object generally produce focal injuries, such as skull fractures, extradural hemorrhages, and contusions. In contrast, acceleration/deceleration forces that result from violent unrestrained head movement, such as in a motor vehicle accident, are associated with diffuse axonal injury (DAI) [20]. In contrast, secondary injury is a gradual process that occurs over minutes to days as the result of cellular, neurochemical, and metabolic alterations initiated by the primary insult [21]. Injury factors that contribute to this phenomenon include metabolic changes, edema formation, calcium influx, increased oxidative stress, excitotoxicity, inflammation, and ultimately, cell death via necrosis or apoptosis [22]. In particular, inflammation is thought to contribute to much of the secondary cell injury, directly injuring cells, and facilitating other injury factors such as oxidative stress [23] and edema formation [24, 25].
Classical inflammatory response following TBI
A robust inflammatory response develops acutely post-TBI and is characterized by the activation of resident cells, migration and recruitment of peripheral leukocytes, and the release of inflammatory mediators [26]. Cellular damage associated with the mechanical impact causes the release of a number of endogenous factors such as RNA, DNA, heat shock proteins, and HMGB1 (high mobility group box 1) which act as damage-associated molecular patterns (DAMPs) [27]. These bind to toll-like receptors (TLRs) activating the nuclear-factor-κB (NFκB) and MAPK pathways leading to the release of a variety of pro-inflammatory factors including cytokines (IL-1β, IL-6), chemokines, and immune receptors [28]. Members of the TLR family are expressed by a number of resident cells within the central nervous system (CNS), including astrocytes, microglia, and the cerebrovascular endothelium [29–31]. Apart from DAMPs, the classical inflammatory response is also initiated by the presence of extravasated blood products, complement fragments, and reactive oxygen and nitrogen species [27, 32, 33].
This inflammatory response is signaled by a rapid rise in the levels of cytokines and chemokines following TBI, with release from microglia, astrocytes, cerebrovascular endothelial cells, peripheral immune cells, and even neurons [32, 34, 35]. Following a moderate diffuse TBI in mice, for example, levels of IL-1β, tumor necrosis factor alpha (TNFα) and IL-6 within the cortex peak at 3–9 h post-injury, before gradually subsiding [36]. Similarly, within clinical studies, increased levels of IL-6, TNFα, IL-10, C-C motif chemokine ligand 2 (CCL2), and IL-8 peak within the first 2 days following moderate-severe TBI and then return to normal over a period of several weeks [37–39]. This spike in cytokine release has been correlated with astrogliosis, microglial activation, and axonal dysfunction, providing evidence of the association between the activated immune response and brain pathology [40].
Immune cells are recruited to the area of injury by the release of chemokines from the damaged neuronal tissue [41, 42]. This cellular response to injury appears to differ slightly depending on whether the initiating insult is primarily focal or diffuse in nature. A focal injury is characterized by the early infiltration of neutrophils (peaking within a few days), followed by the migration of microglia, astrocytes, macrophages, and lymphocytes to the injured site [43]. Flow cytometric analysis indicates that there is a 10- to 20-fold increase in numbers of microglia compared to peripheral macrophages, suggesting that this is predominantly a central rather than peripheral response [44]. In diffuse injury, little to no neutrophil infiltration is seen, with the early cellular response consisting of microglial accumulation and astrocytosis most prominent in the white matter tracts [45]. This response amplifies over-time with Hellewell et al. indicating that the highest numbers of microglia are present at 14 days post-injury, the latest time-point investigated in their study [45]. Even mild TBI is associated with the induction of an inflammatory response, with diffuse mTBI in pigs showing enhanced microglial activation associated with thalamic axonal injury at 6 h post-injury [46].
The function of this inflammatory response can be both detrimental and potentially beneficial. Both microglia and astrocytes can serve a neuroprotective role immediately following injury by clearing damaged cell debris by phagocytosis, releasing anti-inflammatory cytokines and neurotrophic factors, [33, 47, 48]. Indeed, numerous studies have shown that at least some inflammation is necessary following an insult to the CNS to assist in clearing damage and preparing for remodeling [10, 26]. For example, ablation of proliferating reactive astrocytes following a moderate controlled cortical impact injury significantly exacerbated cortical neuronal loss and inflammation [49]. A potential protective role for astrocytes following injury includes removal of glutamate to reduce the effects of excitotoxicity [50], with the glial scar also thought to act as a physical barrier to prevent spread of toxic molecules. [51] However, this glial scar can also have a later inhibitory effect on axonal regrowth and regeneration [52, 53]. Furthermore, even pro-inflammatory cytokines have an important role to play with Scherbel et al. showing that although knockout of TNFα was beneficial in the acute phase following injury, it had long-term deleterious consequences as demonstrated by TNFα−/− mice showing worsening of motor outcome at 1 month following a focal TBI, which was associated with enhanced cortical tissue loss [54]. This may relate to purported associated neuroprotective functions of TNFα, including the ability to reduce oxidative stress [55] and promote neurotrophic factor synthesis [56, 57], indicating that the presence of at least some TNFα is needed following injury.
In addition, activated microglia demonstrate phenotypic subpopulations, characterized by a specific molecular signature of gene expression: M1 microglia promote a classic pro-inflammatory state releasing pro-inflammatory cytokines and oxidative metabolites, while M2 microglia are important for tissue remodeling and suppress the inflammatory response [58–60]. Reports from TBI studies suggest that there is an early peak in M2-like activated microglia in the week following injury [61, 62], but this then shifts to a maladaptive M1-like activation at later time points. The importance of M2 microglia after TBI is demonstrated by a study by Kumar et al., where aged mice with an impaired M2 response had increased lesion size following a focal injury [63]. Indeed, M1 activation can exacerbate neuronal injury by triggering downstream pathways that culminate in oxidative damage, activation of apoptotic cell death, and increases in permeability of the blood-brain barrier (BBB) through modifications in its tight junctions (Tight junctions) and matrix metalloproteinase (MMP) activation [64, 65]. Furthermore, prolonged M1-like activation hampers repair and can allow tissue damage to persist for years after the initial injury; in a subset of TBI patients, there is incomplete resolution of the acute neuroinflammatory response [66]. A vicious cycle is initiated following the original insult, where the release of pro-inflammatory factors by resident glial cells promotes further glial activation, leading to a progressive chronic cycle of neuroinflammation [67], which can have neurotoxic effects on neurons through mechanisms such as oxidative stress, apoptosis, and excitotoxicity [68].
Neurogenic inflammation
Activation of sensory unmyelinated neurons by noxious stimuli causes the simultaneous release of neuropeptides such as SP, neurokinin A (NKA), neurokinin B (NKB), and calcitonin gene-related peptide (CGRP) [69]. Their release invokes neurogenic inflammation, a neurally elicited response with the typical features of an inflammatory response involving vasodilation and increased vascular permeability [70]. CGRP is chiefly responsible for promoting vasodilation, while SP primarily induces plasma extravasation, although it also produces a brief period of vasodilation [71]. Indeed, although NKA, NKB, and SP act synergistically [72], increases in capillary permeability are principally mediated by SP [73, 74].
SP is an 11-amino acid peptide that is a member of the tachykinin family which includes NKA and NKB [75]; both NKA and SP derived from the preprotachykinin-A (PPT-A) gene by alternative splicing [76]. SP is widely distributed throughout the CNS, peripheral nervous system (PNS), and enteric nervous systems. In the CNS, it is present in dorsal root ganglion (primary sensory) neurons [77] of the spinal cord and many regions of the brain including the hippocampus, cortex, basal ganglia, hypothalamus, amygdala, and caudate nucleus, being more abundant in the gray matter compared to the white matter [78]. The biological effects of SP are mediated by the tachykinin NK receptors, with SP preferentially binding to the NK1 receptor, although it has some affinity for the NK2 and NK3 receptors. NK1 receptors are expressed on endothelial cells, astrocytes, microglia, and various types of circulating and inflammation-activated immune cells [79]. Transduction of the SP signal through the NK1 receptor occurs via G protein signaling and the secondary messenger cAMP, ultimately leading to the regulation of ion channels, enzyme activity, and alterations in gene expression [80]. There are two versions of the NK1 receptor: the full-length version and a truncated form which lacks the 96 residues at the C terminus [81]. This truncated form has a diminished binding affinity for SP [79], and its activation produces a much diminished inflammatory response when compared to the full-length receptor [82]. Higher levels of expression of the shorter isoform are found in peripheral tissues, while in the brain, the longer isoform is expressed at much higher concentrations than the truncated version [83]. This suggests that for pathology within the CNS, the full-length version of the NK1 receptor is the most critical.
Role of SP following TBI
Extensive research has shown that levels of SP rise acutely following TBI in both pre-clinical animal models and in human tissue. Virtually, all blood vessels of the body are surrounded by sensory nerve fibers that contain SP [84]. Cerebral arteries, in particular, appear to receive a dense supply of these nerve fibers, and our studies in TBI have demonstrated that perivascular SP immunoreactivity increases in pre-clinical models, irrespective of injury model [13, 85, 86], and also in humans [87]. It appears that SP is released early following TBI, with increases noted in the plasma at 30 min following TBI in rodents [13]. Furthermore, this release of SP appears to depend on the magnitude of the insult, with a graded increase in SP immunoreactivity seen with increasing severity of injury [85, 88]. Indeed, SP appears to be a key injury marker, as levels in the plasma over the first 4 h following injury are significantly correlated with early mortality in clinical populations, with non-surviving TBI patients showing significantly higher levels than survivors [88].
Moreover, it has been shown that attenuating SP activity following TBI is beneficial to outcome [89]. The first demonstration of neurogenic inflammation in TBI showed that depletion of sensory neuropeptides by pre-treatment with capsaicin results in the attenuation of post-traumatic BBB permeability, edema formation, and improved functional outcome [11]. Later studies, specifically targeted SP by administering an NK1 antagonist showed beneficial effects in both male [13] and female rats [90], with a significant attenuation of post-traumatic BBB permeability and a resultant significant reduction in edema formation with improvement in motor and cognitive outcome.
What promotes the release of SP following TBI?
It appears likely that the initial release of SP from sensory neurons following TBI may be mediated by mechanical activation of members of the transient receptor potential (TRP) family, predominantly TRPV1 and TRPA1 [91]. Like all TRP receptors, TRPV1 and TRPA1 are comprised of six-transmembrane proteins that assemble as tetramers to form cation-permeable pores [92, 93]. Their activation allows the influx of cations, primarily sodium and calcium, triggering the release of neuropeptides [94, 95]. TRPV1 appears to be co-expressed in most if not all of TRPA1 expressing dorsal root ganglion neurons [96, 97], with co-expression of both these receptors with neuropeptides including SP [98]. Indeed, suppression of the TRPV1 receptor, with the antagonist capsazepine, has been shown to significantly reduce SP levels in a number of inflammatory models, including a model of sepsis [99], alcohol-induced gastric injury [100], and formalin-induced asthma [101], with the latter study showing a similar reduction in SP with administration of the TRPA1 antagonist, HC-030031 [101]. Furthermore, activation of both TRPV1 and TRPA1 [102–104] has been linked to increased vascular permeability, a key downstream effect of SP release. TRPV1 immunoreactivity is prominent in astrocytes and pericytes, which are closely associated with the vasculature, as well as neurons [105], with the administration of capsazepine able to reduce BBB disruption following an ischemia-reperfusion injury [106].
TRPV1 and TRPA1 channels are considered as polymodal receptors that are activated by a wide range of stimuli. For TRPV1, this includes capsaicin (the active ingredient in chilies) [107], heat (43–52 °C) [108], protons [107], bradykinin [109], prostaglandins [110], and arachidonic acid metabolites amongst others [111]. For TRPA1, agonists include exogenous noxious agents such as components of wasabi and cinnamon [98], oxidized lipids [112], protons [113], and potentially cold (<17 °C) [113, 114]. Notably, both TRPV1 and TRPA1 are also putative mechanoreceptors. Early reports identified that the long ankyrin repeat region within the N-terminal domain of TRPA1, which assists in anchoring the receptor to the plasma membrane, could form a spring-like structure to sense mechanical forces [115, 116]. Indeed, TRPA1 knockout mice were found to be less sensitive to low-intensity mechanical stimuli compared to wild-type mice, and responses to high-intensity mechanical stimulation were notably impaired [113]. Furthermore, inhibition of TRPA1 via either gene knockout or treatment with the antagonist HC-03001 reduced mechanically induced action firing in dermal C fibers [117, 118], wide dynamic range, and nociceptive-specific neurons within the spinal cord [119] and mechanosensitive visceral afferents in the colon [120, 121]. Studies have also suggested that TRPV1 detects mechanical stimuli in a variety of tissues including the urothelium cells of the bladder [122], colonic primary afferent neurons [123], renal pelvis [124], and retinal ganglion cells [125, 126]. The threshold for the stimulation of both TRPV1 and TRPA1 appears to be lowered when there is pre-existing inflammation. Indeed, blockade of TRPA1 was only effective in reducing low-intensity mechanical stimulation of spinal neurons in animals with inflammatory arthritis [119], and application of a TRPV1 antagonist reduced Aδ-fiber unit responses only in inflamed skin [127].
In regard to TBI, a report by Zacest et al. showed that TBI mechanical stimulation of sensory nerves, presumably via TRPV1 and TRPA1, facilitates the perivascular release of SP, with SP co-localizing with amyloid precursor protein positive (injured) neurons that supply the blood vessels [87]. Apart from the initial external mechanical insult to the brain, another acute mechanical stimulus could be related to a brief but highly significant spike in blood pressure (BP) seen following injury. In a sheep model of TBI, BP was reported to rise markedly immediately after impact, peaking at a MAP of 176 mmHg before gradually returning to baseline over a 10-min period [128]. Similar findings have been shown in other models of experimental TBI [129–131]. TRPV1 receptors have indeed been shown to respond to increased intraluminal pressure [132, 133], with Scotland et al. showing that the normal myogenic response to elevation of increased transmural pressure in mesenteric small arteries in vitro was suppressed with the application of the TRPV1 antagonist capsazepine. As such release of neuropeptides including SP may be one of the earliest responses to TBI, highlighting its integral role in facilitating the inflammatory response.
How does neurogenic inflammation potentiate classical inflammation?
Direct interaction between SP and the classical inflammatory response
SP induces and augments many aspects of the classical inflammatory response (Fig 1), including leukocyte activation, endothelial cell adhesion molecule expression, and the production of inflammatory mediators such as histamine, nitric oxide, cytokines (such as IL-6), and kinins [14, 15]. SP is a potent mast cell activator [134]. Mast cells are found within the brain on the adluminal side of the BBB and in the leptomeninges [135], with their degranulation found to potentiate excitotoxicity [136] and augment and prolong numerous vasoactive, neuroactive, and immunoactive cellular and molecular responses to injury [137, 138].

Interaction between the neurogenic and classical inflammatory response following TBI
SP also directly activates microglia and astrocytes. Injury induces the expression of NK1 receptors on astrocytes, and their activation is thought to contribute to the transformation to reactive astrocytes, with the resultant production of inflammatory mediators such as cytokines, prostaglandins, and thromboxane derivatives [139–141]. Similarly SP can promote microglial activation, initiating signaling via the NFκB pathway, which leads to the production of pro-inflammatory cytokines [142], with microglia producing IL-1 in response to SP [143]. Moreover, following TBI, NK1 antagonists have been shown to significantly reduce the production of the pro-inflammatory cytokine IL-6, as well as to decrease microglial proliferation [144].
Role of SP in altering BBB permeability
Apart from directly augmenting the local classical inflammatory response, SP-related BBB disruption may also play a role in perpetuating the inflammatory response. The BBB is a highly selective barrier formed by a layer of endothelial cells joined together by tight junctions including proteins such as claudins, occludin, junctional adhesion molecules (JAMs), and zonula occluden (ZO) proteins, which are supported by the end-feet of astrocytes that act to support and enhance the tight junctions [145]. Tight junctions are present at the apical end of the interendothelial junction and closely rely on the integrity of the corresponding cadherin junction located in the basolateral region. Ultrastructural studies of cerebral endothelial cells indicate the circumferential arrangement of the TJs which provide the barrier formation required to actively restrict the movement of small hydrophilic molecules such as sodium and particular permeability tracers between cells [146]. Given the restrictions on paracellular transport, cerebral endothelial cells have tightly controlled mechanisms that enable the bidirectional transcellular passage of essential molecules and the efflux of potentially neurotoxic substances and waste products [147]. Smaller molecules can be ferried across via carrier-mediated transport or ion pumps while macromolecules employ transcytosis, which can be either receptor-mediated transport (RMT) or adsorptive-mediated transport (AMT), where positively charged macromolecules are nonspecifically transported across the cerebral endothelium [148]. Although the cerebral endothelium has relatively few pinocytotic vesicles compared to peripheral endothelial cells [149], they can still transport macromolecules via one of three vesicles: clathrin-coated pits, the most numerous and through which most of the RMT occurs, the smaller and less numerous caveolae, capable of both AMT and RMT, and large macropinocytic vesciles with nonspecific cargo [150].
Following TBI, the BBB is disrupted and this encompasses not just potential disruption to tight junctions but also increased transcytosis and altered transport properties [151–153]. This increase in the transcellular permeability of the BBB permits the extravasation of proteins and solutes from the cerebral vasculature into the extracellular space within the brain, promoting edema formation, perpetuating the inflammatory response, and causing further neuronal injury. Importantly, it appears that this transcytotic activity is the initial alteration to the BBB following TBI, which is then followed by later loss of tight junctions and movement via the paracellular pathway [151, 152]. An early study by Povlishock (1978) found an increase in BBB permeability as early as 3 min following a mild central fluid percussion injury, despite the apparent integrity of the endothelial tight junctions [154]. Notably, endothelial vesicles assume a modified appearance and initiate the formation of extensive networks for intracellular transport after injury, particularly in the setting of inflammation within the CNS [155]. Indeed, ultrastructural studies have shown that within minutes of brain injury, endothelial caveolae are significantly increased in vessel segments showing loss of BBB integrity [152, 155], with evidence for increased vesicular formation in the BBB of TBI patients [156, 157]. In a rat cortical cold injury model that produces a pure vasogenic edema, an increase in expression of caveolin-1, the key protein found in caveolae, occurred prior to the loss of tight junctions, represented by a reduction in the expression of occludin and claudin-5 [152]. Notably, caveolin-1 expression was seen in blood vessels which exhibited increased permeability to proteins, indicating that this may be the initial mechanism by which the BBB is disrupted [152]. These findings were replicated in a controlled cortical impact model of TBI, where caveolin-1 was increased at 1 day following injury, prior to a decrease in claudin-5 which occurred at day 3 following injury [151].
SP is known to promote increased permeability of the BBB leading to increased extravasation of vascular protein into the brain extracellular space. Indeed, injured animals show a strong co-localization of SP immunofluorescence with a marker of vascular permeability, Evan’s blue, within the cortical vasculature at 5 h post-TBI [13]. Administration of an NK1 antagonist also leads to a dose-dependent decrease in permeability of the BBB following TBI, with an accompanying reduction in cerebral edema [13]. The exact mechanisms by which SP promotes an increase in BBB permeability are less clear. There are previous reports that application of SP can decrease the expression of ZO-1 and claudin-5 in cerebral capillary endothelial cells [158], but immediately following TBI tight junctions are intact. Alternatively, SP may initially act to increase transcytosis via activation of transport via caveolae.
Caveolae have been shown to mediate the transport of molecules such as albumin, transferrin, insulin, low-density lipoproteins, cytokines, and chemokines through the specific localization of corresponding receptors within the caveolae coats [159, 160]. Indeed, caveolin-1 knockout mice were unable to transcytose albumin as evaluated via gold-labeled immunostaining [161, 162], supporting a key role for caveolae in the early BBB changes seen following TBI, and consistent with the increases seen in expression of caveolin-1 in the immediate phase following injury [151–153]. Caveolae constitute an entire membrane system responsible for the formation of unique endo- and exocytotic compartments [163]. During transcytosis, caveolae “pinch off” from the plasma membrane to form vesicular carriers that rapidly and efficiently shuttle to the opposite membrane of the endothelial cells, fuse and release their contents via exocytosis [164]. In addition to the structural caveolin proteins, a number of signaling molecules, including specific receptors, are known to localize to the caveolae pit [160, 165]. Receptor tyrosine kinase, G-protein-coupled receptors, transforming growth factor-beta (TGF-β) type 1 and 2, certain steroid receptors and enzymes have a confirmed presence within endothelial caveolae and may play a role in facilitating macromolecular transcytosis [166–168]. Of particular note, the NK1 receptor, to which SP preferentially binds, has been reported to localize within the endothelial caveolae and can be manipulated to upregulate or relocate upon stimulation, indicating that its presence and function is dynamic and subject to the environment [169, 170]. Indeed, stimulation of the NK1 receptor within caveolae by SP causes PKCα to relocate to caveolae [170], with this process previously shown to regulate the internalization of caveolae [171], the first step in transcytosis. Although not directly studied for the NK1 receptor, in vitro studies have demonstrated increased receptor-mediated transendothelial transport of target molecules in the setting of inflammation [172]. This suggests that the activation of a caveolae-housed receptor may also internalize additional proteins in their cargo, such as albumin, facilitating their entry into the brain.
Within the CNS, albumin is able to activate microglia and astrocytes leading to the release of pro-inflammatory cytokines and chemokines (e.g., IL-1β, TNFα, MCP-1, CXC3L1), glutamate, and free radicals promoting neuronal injury and amplifying the classical inflammatory response [173–178]. Although the majority of these studies apply albumin to isolated glial cultures [174–177], Chacheaux et al. were able to demonstrate that direct application of a solution containing bovine serum albumin to the rat cortex produced similar changes in gene expression when compared to application of an agent (deoxcycholic acid) that directly opens the BBB [179]. Microarray analysis showed a comparable gene expression profile to either treatment, with an early and persistent upregulation of genes associated with immune response activation, including NF-κB pathway-related genes, cytokines, and chemokines (IL-6, CCL2, CCL7) [179]. Indeed, recent evidence suggests that following activation by albumin, microglia are neurotoxic, with media taken from microglial cultures exposed to albumin promoting the upregulation of caspase 3 and 7 in cerebellar granule neuronal cultures with an accompanying increase in cell death [174]. Furthermore, when exposure to albumin was combined with TLR4 activation via lipopolysaccharide (LPS), a synergistic increase in the release of pro-inflammatory cytokines was noted from cultured microglia [174]. As the neuronal injury associated with TBI leads to the production of DAMPs [180] that act as TLR4 agonists, influx of albumin through a disrupted BBB would lead to more potent activation of microglia. This provides another key intersection between the classical inflammatory response, in TLR4 activation by DAMPs, and the neurogenic inflammatory response, with SP release promoting albumin influx via caveolae promoting further activation of resident immune cells.
The effects of SP on the BBB relates not only to increases in permeability but also on promoting immune cell trafficking into the CNS. Activation of SP’s preferred receptor, NK1, has been shown to enhance leukocyte migration by exerting a chemotactic effect on monocytes and neutrophils [181–183], increasing the expression of adhesion molecules on endothelial cells [184–187] and augmenting local chemokine production [188]. Indeed, application of SP to cerebral endothelial cultures led to a dose-dependent increase in ICAM-1 expression [184], with this associated with an increase in T lymphocyte adherence, suggesting facilitation of immune cell movement across the BBB [187]. Thus, the release of SP following TBI may facilitate the influx of peripheral immune cells like T cells, macrophages, and neutrophils into the CNS, which then further augments the local neuroinflammatory response via production of pro-inflammatory cytokines, reactive oxygen species, and metalloproteinases.
Exacerbation of neurogenic inflammation by classical inflammation
Although this discussion has focused on how neurogenic inflammation can facilitate the classical inflammatory response, this is a two-way interaction with classical inflammation further enhancing the release of neuropeptides (Fig 1). As discussed previously, TPRV1 and TRPA1 are responsive to a number of stimuli including the classical inflammatory mediators bradykinin [109] and prostaglandins [110], which are produced as part of the inflammatory response following TBI [189, 190]; their production would potentiate further SP release. For example, the kallikrein-kinin system is believed to be one of the first inflammatory pathways activated after tissue damage [191]. Bradykinin, a nonpeptide found in blood and tissue, is cleaved from its pro-form kininogen by the protease kallikrein [192], with levels found to peak within the brain at 2 h following a focal brain injury [193]. TBI is also known to increase the activity of cyclooxygenase (COX) enzymes, principally Cox-2 found in microglia and endothelial cells, increasing the synthesis of prostaglandins such as PGE2 and PGF2α. Indeed, levels of PGE2 have been shown to be elevated within 5 min following TBI [194]. In addition to these direct activators or TRP channels, cytokines like IL-1, IL-6, and TNFα may sensitize sensory neurons, lowering the threshold for release of neuropeptides [195, 196].
Conclusion
Following TBI activation of a classical inflammatory response only represents part of the neuroinflammatory response. The mechanical and shear stress associated with TBI also activates TRP receptors leading to the release of neuropeptides, including SP, instigating a neurogenic inflammatory response. SP both directly and indirectly, through alterations in BBB permeability with influx of plasma proteins, augments the classical inflammatory response, with both classical and neurogenic inflammation providing positive feedback to the other to amplify and propagate inflammation with the release of pro-inflammatory mediators, oxidative metabolites, and metalloproteinases amongst others, which cause further neuronal damage. As such, modulation of neuroinflammation following TBI may require addressing both inflammatory pathways with the aim to prevent the deleterious effects of the response, while facilitating repair.
Abbreviations
- AMT:
-
Absorptive-mediated transcytosis
- BBB:
-
Blood-brain barrier
- CNS:
-
Central nervous system
- EPO:
-
Erythropoietin
- RMT:
-
Receptor-mediated transcytosis
- SP:
-
Substance P
- TLR:
-
Toll-like receptor
- TRP channels:
-
Transient receptor potential cation channels
- TBI:
-
Traumatic brain injury
References
Jones NC, Prior MJ, Burden-Teh E, Marsden CA, Morris PG, Murphy S. Antagonism of the interleukin-1 receptor following traumatic brain injury in the mouse reduces the number of nitric oxide synthase-2-positive cells and improves anatomical and functional outcomes. Eur J Neurosci. 2005;22:72–8.
Sanderson KL, Raghupathi R, Saatman KE, Martin D, Miller G, McIntosh TK. Interleukin-1 receptor antagonist attenuates regional neuronal cell death and cognitive dysfunction after experimental brain injury. J Cereb Blood Flow Metab. 1999;19:1118–25.
Shohami E, Gallily R, Mechoulam R, Bass R, Ben-Hur T. Cytokine production in the brain following closed head injury: dexanabinol (HU-211) is a novel TNF-alpha inhibitor and an effective neuroprotectant. J Neuroimmunol. 1997;72:169–77.
Roberts I, Yates D, Sandercock P, Farrell B, Wasserberg J, Lomas G, Cottingham R, Svoboda P, Brayley N, Mazairac G, et al. Effect of intravenous corticosteroids on death within 14 days in 10008 adults with clinically significant head injury (MRC CRASH trial): randomised placebo-controlled trial. Lancet. 2004;364:1321–8.
Skolnick BE, Maas AI, Narayan RK, van der Hoop RG, MacAllister T, Ward JD, Nelson NR, Stocchetti N, Investigators ST. A clinical trial of progesterone for severe traumatic brain injury. N Engl J Med. 2014;371:2467–76.
Wright DW, Yeatts SD, Silbergleit R, Palesch YY, Hertzberg VS, Frankel M, Goldstein FC, Caveney AF, Howlett-Smith H, Bengelink EM, et al. Very early administration of progesterone for acute traumatic brain injury. N Engl J Med. 2014;371:2457–66.
Bye N, Habgood MD, Callaway JK, Malakooti N, Potter A, Kossmann T, Morganti-Kossmann MC. Transient neuroprotection by minocycline following traumatic brain injury is associated with attenuated microglial activation but no changes in cell apoptosis or neutrophil infiltration. Exp Neurol. 2007;204:220–33.
Teng YD, Choi H, Onario RC, Zhu S, Desilets FC, Lan S, Woodard EJ, Snyder EY, Eichler ME, Friedlander RM. Minocycline inhibits contusion-triggered mitochondrial cytochrome c release and mitigates functional deficits after spinal cord injury. Proc Natl Acad Sci U S A. 2004;101:3071–6.
Wells JE, Hurlbert RJ, Fehlings MG, Yong VW. Neuroprotection by minocycline facilitates significant recovery from spinal cord injury in mice. Brain. 2003;126:1628–37.
Russo MV, McGavern DB. Inflammatory neuroprotection following traumatic brain injury. Science. 2016;353:783–5.
Nimmo AJ, Cernak I, Heath DL, Hu X, Bennett CJ, Vink R. Neurogenic inflammation is associated with development of edema and functional deficits following traumatic brain injury in rats. Neuropeptides. 2004;38:40–7.
Donkin JJ, Cernak I, Blumbergs PC, Vink R. A substance P antagonist reduces axonal injury and improves neurologic outcome when administered up to 12 hours after traumatic brain injury. J Neurotrauma. 2011;28:217–24.
Donkin JJ, Nimmo AJ, Cernak I, Blumbergs PC, Vink R. Substance P is associated with the development of brain edema and functional deficits after traumatic brain injury. J Cereb Blood Flow Metab. 2009;29:1388–98.
Marriott I, Bost KL. Substance P receptor mediated macrophage responses. Adv Exp Med Biol. 2001;493:247–54.
Quinlan KL, Song IS, Naik SM, Letran EL, Olerud JE, Bunnett NW, Armstrong CA, Caughman SW, Ansel JC. VCAM-1 expression on human dermal microvascular endothelial cells is directly and specifically up-regulated by substance P. J Immunol. 1999;162:1656–61.
Finnie JW, Blumbergs PC. Traumatic brain injury. Vet Pathol. 2002;39:679–89.
Maas AI, Stocchetti N, Bullock R. Moderate and severe traumatic brain injury in adults. Lancet Neurol. 2008;7:728–41.
Gaetz M. The neurophysiology of brain injury. Clin Neurophysiol. 2004;115:4–18.
Smith DH, Meaney DF, Shull WH. Diffuse axonal injury in head trauma. J Head Trauma Rehabil. 2003;18:307–16.
Blumbergs P. Pathology. In: Reilly P, Bullock R, editors. Head injury: pathophysiology and management of severe closed injury. London: Chapman & Hall Medical; 1997. p. 39–70.
Hall ED, Gibson TR, Pavel KM. Lack of a gender difference in post-traumatic neurodegeneration in the mouse controlled cortical impact injury model. J Neurotrauma. 2005;22:669–79.
Saatman KE, Bozyczko-Coyne D, Marcy V, Siman R, McIntosh TK. Prolonged calpain-mediated spectrin breakdown occurs regionally following experimental brain injury in the rat. J Neuropathol Exp Neurol. 1996;55:850–60.
Liao Y, Liu P, Guo F, Zhang ZY, Zhang Z. Oxidative burst of circulating neutrophils following traumatic brain injury in human. PLoS One. 2013;8:e68963.
Vink R, Young A, Bennett CJ, Hu X, Connor CO, Cernak I, Nimmo AJ. Neuropeptide release influences brain edema formation after diffuse traumatic brain injury. Acta Neurochir Suppl. 2003;86:257–60.
Chodobski A, Zink BJ, Szmydynger-Chodobska J. Blood-brain barrier pathophysiology in traumatic brain injury. Transl Stroke Res. 2011;2:492–516.
Ziebell JM, Morganti-Kossmann MC. Involvement of pro- and anti-inflammatory cytokines and chemokines in the pathophysiology of traumatic brain injury. Neurotherapeutics. 2010;7:22–30.
Manson J, Thiemermann C, Brohi K. Trauma alarmins as activators of damage-induced inflammation. Br J Surg. 2012;99 Suppl 1:12–20.
Buchanan MM, Hutchinson M, Watkins LR, Yin H. Toll-like receptor 4 in CNS pathologies. J Neurochem. 2010;114:13–27.
Farina C, Aloisi F, Meinl E. Astrocytes are active players in cerebral innate immunity. Trends Immunol. 2007;28:138–45.
Gurley C, Nichols J, Liu S, Phulwani NK, Esen N, Kielian T. Microglia and astrocyte activation by toll-like receptor ligands: modulation by PPAR-gamma agonists. PPAR Res. 2008;2008:453120.
Nagyoszi P, Wilhelm I, Farkas AE, Fazakas C, Dung NT, Hasko J, Krizbai IA. Expression and regulation of toll-like receptors in cerebral endothelial cells. Neurochem Int. 2010;57:556–64.
Woodcock T, Morganti-Kossmann MC. The role of markers of inflammation in traumatic brain injury. Front Neurol. 2013;4:18.
Corps KN, Roth TL, McGavern DB. Inflammation and neuroprotection in traumatic brain injury. JAMA Neurol. 2015;72:355–62.
Bergold PJ. Treatment of traumatic brain injury with anti-inflammatory drugs. Exp Neurol. 2016;275(Pt 3):367–80.
Hellewell S, Semple BD, Morganti-Kossmann MC. Therapies negating neuroinflammation after brain trauma. Brain Res. 2016;1640:36–56.
Bachstetter AD, Rowe RK, Kaneko M, Goulding D, Lifshitz J, Van Eldik LJ. The p38alpha MAPK regulates microglial responsiveness to diffuse traumatic brain injury. J Neurosci. 2013;33:6143–53.
Csuka E, Morganti-Kossmann MC, Lenzlinger PM, Joller H, Trentz O, Kossmann T. IL-10 levels in cerebrospinal fluid and serum of patients with severe traumatic brain injury: relationship to IL-6, TNF-alpha, TGF-beta1 and blood-brain barrier function. J Neuroimmunol. 1999;101:211–21.
Semple BD, Bye N, Rancan M, Ziebell JM, Morganti-Kossmann MC. Role of CCL2 (MCP-1) in traumatic brain injury (TBI): evidence from severe TBI patients and CCL2−/− mice. J Cereb Blood Flow Metab. 2010;30:769–82.
Morganti-Kossman MC, Lenzlinger PM, Hans V, Stahel P, Csuka E, Ammann E, Stocker R, Trentz O, Kossmann T. Production of cytokines following brain injury: beneficial and deleterious for the damaged tissue. Mol Psychiatry. 1997;2:133–6.
Frugier T, Morganti-Kossmann MC, O'Reilly D, McLean CA. In situ detection of inflammatory mediators in post mortem human brain tissue after traumatic injury. J Neurotrauma. 2010;27:497–507.
Cardona AE, Gonzalez PA, Teale JM. CC chemokines mediate leukocyte trafficking into the central nervous system during murine neurocysticercosis: role of gamma delta T cells in amplification of the host immune response. Infect Immun. 2003;71:2634–42.
Wilson EH, Weninger W, Hunter CA. Trafficking of immune cells in the central nervous system. J Clin Invest. 2010;120:1368–79.
Gyoneva S, Ransohoff RM. Inflammatory reaction after traumatic brain injury: therapeutic potential of targeting cell-cell communication by chemokines. Trends Pharmacol Sci. 2015;36:471–80.
Al Nimer F, Lindblom R, Strom M, Guerreiro-Cacais AO, Parsa R, Aeinehband S, Mathiesen T, Lidman O, Piehl F. Strain influences on inflammatory pathway activation, cell infiltration and complement cascade after traumatic brain injury in the rat. Brain Behav Immun. 2013;27:109–22.
Hellewell SC, Yan EB, Agyapomaa DA, Bye N, Morganti-Kossmann MC. Post-traumatic hypoxia exacerbates brain tissue damage: analysis of axonal injury and glial responses. J Neurotrauma. 2010;27:1997–2010.
Lafrenaye AD, Todani M, Walker SA, Povlishock JT. Microglia processes associate with diffusely injured axons following mild traumatic brain injury in the micro pig. J Neuroinflammation. 2015;12:186.
Lull ME, Block ML. Microglial activation and chronic neurodegeneration. Neurotherapeutics. 2010;7:354–65.
Luo XG, Chen SD. The changing phenotype of microglia from homeostasis to disease. Transl Neurodegener. 2012;1:9.
Myer DJ, Gurkoff GG, Lee SM, Hovda DA, Sofroniew MV. Essential protective roles of reactive astrocytes in traumatic brain injury. Brain. 2006;129:2761–72.
Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, Kanai Y, Hediger MA, Wang Y, Schielke JP, Welty DF. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16:675–86.
Fitch MT, Silver J. Glial cell extracellular matrix: boundaries for axon growth in development and regeneration. Cell Tissue Res. 1997;290:379–84.
Ribotta MG, Menet V, Privat A. Glial scar and axonal regeneration in the CNS: lessons from GFAP and vimentin transgenic mice. Acta Neurochir Suppl. 2004;89:87–92.
Silver J, Miller JH. Regeneration beyond the glial scar. Nat Rev Neurosci. 2004;5:146–56.
Scherbel U, Raghupathi R, Nakamura M, Saatman KE, Trojanowski JQ, Neugebauer E, Marino MW, McIntosh TK. Differential acute and chronic responses of tumor necrosis factor-deficient mice to experimental brain injury. Proc Natl Acad Sci U S A. 1999;96:8721–6.
Bruce-Keller AJ, Geddes JW, Knapp PE, McFall RW, Keller JN, Holtsberg FW, Parthasarathy S, Steiner SM, Mattson MP. Anti-death properties of TNF against metabolic poisoning: mitochondrial stabilization by MnSOD. J Neuroimmunol. 1999;93:53–71.
Saha RN, Liu X, Pahan K. Up-regulation of BDNF in astrocytes by TNF-alpha: a case for the neuroprotective role of cytokine. J Neuroimmune Pharmacol. 2006;1:212–22.
Hattori A, Tanaka E, Murase K, Ishida N, Chatani Y, Tsujimoto M, Hayashi K, Kohno M. Tumor necrosis factor stimulates the synthesis and secretion of biologically active nerve growth factor in non-neuronal cells. J Biol Chem. 1993;268:2577–82.
Loane DJ, Kumar A. Microglia in the TBI brain: the good, the bad, and the dysregulated. Exp Neurol. 2016;275(Pt 3):316–27.
Colton CA. Heterogeneity of microglial activation in the innate immune response in the brain. J Neuroimmune Pharmacol. 2009;4:399–418.
David S, Kroner A. Repertoire of microglial and macrophage responses after spinal cord injury. Nat Rev Neurosci. 2011;12:388–99.
Jin X, Ishii H, Bai Z, Itokazu T, Yamashita T. Temporal changes in cell marker expression and cellular infiltration in a controlled cortical impact model in adult male C57BL/6 mice. PLoS One. 2012;7:e41892.
Wang G, Zhang J, Hu X, Zhang L, Mao L, Jiang X, Liou AK, Leak RK, Gao Y, Chen J. Microglia/macrophage polarization dynamics in white matter after traumatic brain injury. J Cereb Blood Flow Metab. 2013;33:1864–74.
Kumar A, Stoica BA, Sabirzhanov B, Burns MP, Faden AI, Loane DJ. Traumatic brain injury in aged animals increases lesion size and chronically alters microglial/macrophage classical and alternative activation states. Neurobiol Aging. 2013;34:1397–411.
da Fonseca AC, Matias D, Garcia C, Amaral R, Geraldo LH, Freitas C, Lima FR. The impact of microglial activation on blood-brain barrier in brain diseases. Front Cell Neurosci. 2014;8:362.
Huang WC, Sala-Newby GB, Susana A, Johnson JL, Newby AC. Classical macrophage activation up-regulates several matrix metalloproteinases through mitogen activated protein kinases and nuclear factor-kappaB. PLoS One. 2012;7:e42507.
Bigler ED. Neuroinflammation and the dynamic lesion in traumatic brain injury. Brain. 2013;136:9–11.
Lozano D, Gonzales-Portillo GS, Acosta S, de la Pena I, Tajiri N, Kaneko Y, Borlongan CV. Neuroinflammatory responses to traumatic brain injury: etiology, clinical consequences, and therapeutic opportunities. Neuropsychiatr Dis Treat. 2015;11:97–106.
Faden AI, Wu J, Stoica BA, Loane DJ. Progressive inflammation-mediated neurodegeneration after traumatic brain or spinal cord injury. Br J Pharmacol. 2016;173:681–91.
Escott KJ, Connor HE, Brain SD, Beattie DT. The involvement of calcitonin gene-related peptide (CGRP) and substance P in feline pial artery diameter responses evoked by capsaicin. Neuropeptides. 1995;29:129–35.
Maggi CA, Giuliani S. Role of tachykinins as excitatory mediators of NANC contraction in the circular muscle of rat small intestine. J Auton Pharmacol. 1995;15:335–50.
Schlereth T, Schukraft J, Kramer-Best HH, Geber C, Ackermann T, Birklein F. Interaction of calcitonin gene related peptide (CGRP) and substance P (SP) in human skin. Neuropeptides. 2016.
Brain SD, Williams TJ. Interactions between the tachykinins and calcitonin gene-related peptide lead to the modulation of oedema formation and blood flow in rat skin. Br J Pharmacol. 1989;97:77–82.
Inoue H, Nagata N, Koshihara Y. Involvement of substance P as a mediator in capsaicin-induced mouse ear oedema. Inflamm Res. 1995;44:470–4.
Lembeck F, Donnerer J, Tsuchiya M, Nagahisa A. The non-peptide tachykinin antagonist, CP-96,345, is a potent inhibitor of neurogenic inflammation. Br J Pharmacol. 1992;105:527–30.
Schaffer M, Beiter T, Becker HD, Hunt TK. Neuropeptides: mediators of inflammation and tissue repair? Arch Surg. 1998;133:1107–16.
Severini C, Improta G, Falconieri-Erspamer G, Salvadori S, Erspamer V. The tachykinin peptide family. Pharmacol Rev. 2002;54:285–322.
Hokfelt T, Pernow B, Wahren J. Substance P: a pioneer amongst neuropeptides. J Intern Med. 2001;249:27–40.
Ebner K, Singewald N. The role of substance P in stress and anxiety responses. Amino Acids. 2006;31:251–72.
Douglas SD, Leeman SE. Neurokinin-1 receptor: functional significance in the immune system in reference to selected infections and inflammation. Ann N Y Acad Sci. 2011;1217:83–95.
Lundy FT, Linden GJ. Neuropeptides and neurogenic mechanisms in oral and periodontal inflammation. Crit Rev Oral Biol Med. 2004;15:82–98.
Fong TM, Anderson SA, Yu H, Huang RR, Strader CD. Differential activation of intracellular effector by two isoforms of human neurokinin-1 receptor. Mol Pharmacol. 1992;41:24–30.
Lai JP, Lai S, Tuluc F, Tansky MF, Kilpatrick LE, Leeman SE, Douglas SD. Differences in the length of the carboxyl terminus mediate functional properties of neurokinin-1 receptor. Proc Natl Acad Sci U S A. 2008;105:12605–10.
Caberlotto L, Hurd YL, Murdock P, Wahlin JP, Melotto S, Corsi M, Carletti R. Neurokinin 1 receptor and relative abundance of the short and long isoforms in the human brain. Eur J Neurosci. 2003;17:1736–46.
Donkin JJ, Turner RJ, Hassan I, Vink R. Substance P in traumatic brain injury. Prog Brain Res. 2007;161:97–109.
Corrigan F, Vink R, Turner RJ. Inflammation in acute CNS injury: a focus on the role of substance P. Br J Pharmacol. 2016;173:703–15.
Gabrielian L, Helps SC, Thornton E, Turner RJ, Leonard AV, Vink R. Substance P antagonists as a novel intervention for brain edema and raised intracranial pressure. Acta Neurochir Suppl. 2013;118:201–4.
Zacest AC, Vink R, Manavis J, Sarvestani GT, Blumbergs PC. Substance P immunoreactivity increases following human traumatic brain injury. Acta Neurochir Suppl. 2010;106:211–6.
Lorente L, Martin MM, Almeida T, Hernandez M, Ramos L, Argueso M, Caceres JJ, Sole-Violan J, Jimenez A. Serum substance P levels are associated with severity and mortality in patients with severe traumatic brain injury. Crit Care. 2015;19:192.
Donkin JJ, Vink R. Mechanisms of cerebral edema in traumatic brain injury: therapeutic developments. Curr Opin Neurol. 2010;23:293–9.
Corrigan F, Leonard A, Ghabriel M, Van Den Heuvel C, Vink R. A substance P antagonist improves outcome in female Sprague Dawley rats following diffuse traumatic brain injury. CNS Neurosci Ther. 2012;18:513–5.
Geppetti P, Bertrand C, Ricciardolo FL, Nadel JA. New aspects on the role of kinins in neurogenic inflammation. Can J Physiol Pharmacol. 1995;73:843–7.
Minke B. TRP channels and Ca2+ signaling. Cell Calcium. 2006;40:261–75.
Pan Z, Yang H, Reinach PS. Transient receptor potential (TRP) gene superfamily encoding cation channels. Hum Genomics. 2011;5:108–16.
Parenti A, De Logu F, Geppetti P, Benemei S. What is the evidence for the role of TRP channels in inflammatory and immune cells? Br J Pharmacol. 2016;173:953–69.
Vriens J, Appendino G, Nilius B. Pharmacology of vanilloid transient receptor potential cation channels. Mol Pharmacol. 2009;75:1262–79.
Anand U, Otto WR, Facer P, Zebda N, Selmer I, Gunthorpe MJ, Chessell IP, Sinisi M, Birch R, Anand P. TRPA1 receptor localisation in the human peripheral nervous system and functional studies in cultured human and rat sensory neurons. Neurosci Lett. 2008;438:221–7.
Bautista DM, Jordt SE, Nikai T, Tsuruda PR, Read AJ, Poblete J, Yamoah EN, Basbaum AI, Julius D. TRPA1 mediates the inflammatory actions of environmental irritants and proalgesic agents. Cell. 2006;124:1269–82.
Jordt SE, Bautista DM, Chuang HH, McKemy DD, Zygmunt PM, Hogestatt ED, Meng ID, Julius D. Mustard oils and cannabinoids excite sensory nerve fibres through the TRP channel ANKTM1. Nature. 2004;427:260–5.
Ang SF, Moochhala SM, MacAry PA, Bhatia M. Hydrogen sulfide and neurogenic inflammation in polymicrobial sepsis: involvement of substance P and ERK-NF-kappaB signaling. PLoS One. 2011;6:e24535.
Gazzieri D, Trevisani M, Springer J, Harrison S, Cottrell GS, Andre E, Nicoletti P, Massi D, Zecchi S, Nosi D, et al. Substance P released by TRPV1-expressing neurons produces reactive oxygen species that mediate ethanol-induced gastric injury. Free Radic Biol Med. 2007;43:581–9.
Wu Y, You H, Ma P, Li L, Yuan Y, Li J, Ye X, Liu X, Yao H, Chen R, et al. Role of transient receptor potential ion channels and evoked levels of neuropeptides in a formaldehyde-induced model of asthma in BALB/c mice. PLoS One. 2013;8:e62827.
Trevisani M, Siemens J, Materazzi S, Bautista DM, Nassini R, Campi B, Imamachi N, Andre E, Patacchini R, Cottrell GS, et al. 4-Hydroxynonenal, an endogenous aldehyde, causes pain and neurogenic inflammation through activation of the irritant receptor TRPA1. Proc Natl Acad Sci U S A. 2007;104:13519–24.
Andre E, Campi B, Materazzi S, Trevisani M, Amadesi S, Massi D, Creminon C, Vaksman N, Nassini R, Civelli M, et al. Cigarette smoke-induced neurogenic inflammation is mediated by alpha, beta-unsaturated aldehydes and the TRPA1 receptor in rodents. J Clin Invest. 2008;118:2574–82.
Nassini R, Materazzi S, Andre E, Sartiani L, Aldini G, Trevisani M, Carnini C, Massi D, Pedretti P, Carini M, et al. Acetaminophen, via its reactive metabolite N-acetyl-p-benzo-quinoneimine and transient receptor potential ankyrin-1 stimulation, causes neurogenic inflammation in the airways and other tissues in rodents. FASEB J. 2010;24:4904–16.
Toth A, Boczan J, Kedei N, Lizanecz E, Bagi Z, Papp Z, Edes I, Csiba L, Blumberg PM. Expression and distribution of vanilloid receptor 1 (TRPV1) in the adult rat brain. Brain Res Mol Brain Res. 2005;135:162–8.
Hu DE, Easton AS, Fraser PA. TRPV1 activation results in disruption of the blood-brain barrier in the rat. Br J Pharmacol. 2005;146:576–84.
Vellani V, Mapplebeck S, Moriondo A, Davis JB, McNaughton PA. Protein kinase C activation potentiates gating of the vanilloid receptor VR1 by capsaicin, protons, heat and anandamide. J Physiol. 2001;534:813–25.
Huang J, Zhang X, McNaughton PA. Inflammatory pain: the cellular basis of heat hyperalgesia. Curr Neuropharmacol. 2006;4:197–206.
Chuang HH, Prescott ED, Kong H, Shields S, Jordt SE, Basbaum AI, Chao MV, Julius D. Bradykinin and nerve growth factor release the capsaicin receptor from PtdIns(4,5)P2-mediated inhibition. Nature. 2001;411:957–62.
Shibata T, Takahashi K, Matsubara Y, Inuzuka E, Nakashima F, Takahashi N, Kozai D, Mori Y, Uchida K. Identification of a prostaglandin D2 metabolite as a neuritogenesis enhancer targeting the TRPV1 ion channel. Sci Rep. 2016;6:21261.
Szallasi A, Di Marzo V. New perspectives on enigmatic vanilloid receptors. Trends Neurosci. 2000;23:491–7.
Choi SI, Yoo S, Lim JY, Hwang SW. Are sensory TRP channels biological alarms for lipid peroxidation? Int J Mol Sci. 2014;15:16430–57.
Kwan KY, Allchorne AJ, Vollrath MA, Christensen AP, Zhang DS, Woolf CJ, Corey DP. TRPA1 contributes to cold, mechanical, and chemical nociception but is not essential for hair-cell transduction. Neuron. 2006;50:277–89.
Caspani O, Heppenstall PA. TRPA1 and cold transduction: an unresolved issue? J Gen Physiol. 2009;133:245–9.
Howard J, Bechstedt S. Hypothesis: a helix of ankyrin repeats of the NOMPC-TRP ion channel is the gating spring of mechanoreceptors. Curr Biol. 2004;14:R224–226.
Sotomayor M, Corey DP, Schulten K. In search of the hair-cell gating spring elastic properties of ankyrin and cadherin repeats. Structure. 2005;13:669–82.
Kerstein PC, del Camino D, Moran MM, Stucky CL. Pharmacological blockade of TRPA1 inhibits mechanical firing in nociceptors. Mol Pain. 2009;5:19.
Kwan KY, Glazer JM, Corey DP, Rice FL, Stucky CL. TRPA1 modulates mechanotransduction in cutaneous sensory neurons. J Neurosci. 2009;29:4808–19.
McGaraughty S, Chu KL, Perner RJ, Didomenico S, Kort ME, Kym PR. TRPA1 modulation of spontaneous and mechanically evoked firing of spinal neurons in uninjured, osteoarthritic, and inflamed rats. Mol Pain. 2010;6:14.
Brierley SM, Castro J, Harrington AM, Hughes PA, Page AJ, Rychkov GY, Blackshaw LA. TRPA1 contributes to specific mechanically activated currents and sensory neuron mechanical hypersensitivity. J Physiol. 2011;589:3575–93.
Brierley SM, Hughes PA, Page AJ, Kwan KY, Martin CM, O'Donnell TA, Cooper NJ, Harrington AM, Adam B, Liebregts T, et al. The ion channel TRPA1 is required for normal mechanosensation and is modulated by algesic stimuli. Gastroenterology. 2009;137:2084–95. e2083.
Birder LA, Nakamura Y, Kiss S, Nealen ML, Barrick S, Kanai AJ, Wang E, Ruiz G, De Groat WC, Apodaca G, et al. Altered urinary bladder function in mice lacking the vanilloid receptor TRPV1. Nat Neurosci. 2002;5:856–60.
Jones 3rd RC, Xu L, Gebhart GF. The mechanosensitivity of mouse colon afferent fibers and their sensitization by inflammatory mediators require transient receptor potential vanilloid 1 and acid-sensing ion channel 3. J Neurosci. 2005;25:10981–9.
Feng NH, Lee HH, Shiang JC, Ma MC. Transient receptor potential vanilloid type 1 channels act as mechanoreceptors and cause substance P release and sensory activation in rat kidneys. Am J Physiol Renal Physiol. 2008;294:F316–325.
Sappington RM, Sidorova T, Long DJ, Calkins DJ. TRPV1: contribution to retinal ganglion cell apoptosis and increased intracellular Ca2+ with exposure to hydrostatic pressure. Invest Ophthalmol Vis Sci. 2009;50:717–28.
Sappington RM, Sidorova T, Ward NJ, Chakravarthy R, Ho KW, Calkins DJ. Activation of transient receptor potential vanilloid-1 (TRPV1) influences how retinal ganglion cell neurons respond to pressure-related stress. Channels (Austin). 2015;9:102–13.
Brederson JD, Chu KL, Reilly RM, Brown BS, Kym PR, Jarvis MF, McGaraughty S. TRPV1 antagonist, A-889425, inhibits mechanotransmission in a subclass of rat primary afferent neurons following peripheral inflammation. Synapse. 2012;66:187–95.
Byard RW, Gabrielian L, Helps SC, Thornton E, Vink R. Further investigations into the speed of cerebral swelling following blunt cranial trauma. J Forensic Sci. 2012;57:973–5.
Lewis SB, Finnie JW, Blumbergs PC, Scott G, Manavis J, Brown C, Reilly PL, Jones NR, McLean AJ. A head impact model of early axonal injury in the sheep. J Neurotrauma. 1996;13:505–14.
Marmarou A, Foda MA, van den Brink W, Campbell J, Kita H, Demetriadou K. A new model of diffuse brain injury in rats. Part I: pathophysiology and biomechanics. J Neurosurg. 1994;80:291–300.
McIntosh TK, Vink R, Noble L, Yamakami I, Fernyak S, Soares H, Faden AL. Traumatic brain injury in the rat: characterization of a lateral fluid-percussion model. Neuroscience. 1989;28:233–44.
Sun H, Li DP, Chen SR, Hittelman WN, Pan HL. Sensing of blood pressure increase by transient receptor potential vanilloid 1 receptors on baroreceptors. J Pharmacol Exp Ther. 2009;331:851–9.
Scotland RS, Chauhan S, Davis C, De Felipe C, Hunt S, Kabir J, Kotsonis P, Oh U, Ahluwalia A. Vanilloid receptor TRPV1, sensory C-fibers, and vascular autoregulation: a novel mechanism involved in myogenic constriction. Circ Res. 2004;95:1027–34.
Okabe T, Hide M, Koro O, Nimi N, Yamamoto S. The release of leukotriene B4 from human skin in response to substance P: evidence for the functional heterogeneity of human skin mast cells among individuals. Clin Exp Immunol. 2001;124:150–6.
Florenzano F, Bentivoglio M. Degranulation, density, and distribution of mast cells in the rat thalamus: a light and electron microscopic study in basal conditions and after intracerebroventricular administration of nerve growth factor. J Comp Neurol. 2000;424:651–69.
Patkai J, Mesples B, Dommergues MA, Fromont G, Thornton EM, Renauld JC, Evrard P, Gressens P. Deleterious effects of IL-9-activated mast cells and neuroprotection by antihistamine drugs in the developing mouse brain. Pediatr Res. 2001;50:222–30.
Hendrix S, Warnke K, Siebenhaar F, Peters EM, Nitsch R, Maurer M. The majority of brain mast cells in B10.PL mice is present in the hippocampal formation. Neurosci Lett. 2006;392:174–7.
Taiwo OB, Kovacs KJ, Larson AA. Chronic daily intrathecal injections of a large volume of fluid increase mast cells in the thalamus of mice. Brain Res. 2005;1056:76–84.
Fiebich BL, Schleicher S, Butcher RD, Craig A, Lieb K. The neuropeptide substance P activates p38 mitogen-activated protein kinase resulting in IL-6 expression independently from NF-kappa B. J Immunol. 2000;165:5606–11.
Lieb K, Schaller H, Bauer J, Berger M, Schulze-Osthoff K, Fiebich BL. Substance P and histamine induce interleukin-6 expression in human astrocytoma cells by a mechanism involving protein kinase C and nuclear factor-IL-6. J Neurochem. 1998;70:1577–83.
Lin RC. Reactive astrocytes express substance-P immunoreactivity in the adult forebrain after injury. Neuroreport. 1995;7:310–2.
Rasley A, Bost KL, Olson JK, Miller SD, Marriott I. Expression of functional NK-1 receptors in murine microglia. Glia. 2002;37:258–67.
Martin FC, Anton PA, Gornbein JA, Shanahan F, Merrill JE. Production of interleukin-1 by microglia in response to substance P: role for a non-classical NK-1 receptor. J Neuroimmunol. 1993;42:53–60.
Carthew HL, Ziebell JM, Vink R. Substance P-induced changes in cell genesis following diffuse traumatic brain injury. Neuroscience. 2012;214:78–83.
Huber JD, Egleton RD, Davis TP. Molecular physiology and pathophysiology of tight junctions in the blood-brain barrier. Trends Neurosci. 2001;24:719–25.
Ballabh P, Braun A, Nedergaard M. The blood-brain barrier: an overview: structure, regulation, and clinical implications. Neurobiol Dis. 2004;16:1–13.
Cecchelli R, Berezowski V, Lundquist S, Culot M, Renftel M, Dehouck MP, Fenart L. Modelling of the blood-brain barrier in drug discovery and development. Nat Rev Drug Discov. 2007;6:650–61.
Luissint AC, Artus C, Glacial F, Ganeshamoorthy K, Couraud PO. Tight junctions at the blood brain barrier: physiological architecture and disease-associated dysregulation. Fluids Barriers CNS. 2012;9:23.
Villegas JC, Broadwell RD. Transcytosis of protein through the mammalian cerebral epithelium and endothelium. II. Adsorptive transcytosis of WGA-HRP and the blood-brain and brain-blood barriers. J Neurocytol. 1993;22:67–80.
Preston JE, Joan Abbott N, Begley DJ. Transcytosis of macromolecules at the blood-brain barrier. Adv Pharmacol. 2014;71:147–63.
Badaut J, Ajao DO, Sorensen DW, Fukuda AM, Pellerin L. Caveolin expression changes in the neurovascular unit after juvenile traumatic brain injury: signs of blood-brain barrier healing? Neuroscience. 2015;285:215–26.
Nag S, Manias JL, Stewart DJ. Expression of endothelial phosphorylated caveolin-1 is increased in brain injury. Neuropathol Appl Neurobiol. 2009;35:417–26.
Nag S, Venugopalan R, Stewart DJ. Increased caveolin-1 expression precedes decreased expression of occludin and claudin-5 during blood-brain barrier breakdown. Acta Neuropathol. 2007;114:459–69.
Povlishock JT, Becker DP, Sullivan HG, Miller JD. Vascular permeability alterations to horseradish peroxidase in experimental brain injury. Brain Res. 1978;153:223–39.
Lossinsky AS, Shivers RR. Structural pathways for macromolecular and cellular transport across the blood-brain barrier during inflammatory conditions. Review. Histol Histopathol. 2004;19:535–64.
Castejon OJ. Formation of transendothelial channels in traumatic human brain edema. Pathol Res Pract. 1984;179:7–12.
Vaz R, Sarmento A, Borges N, Cruz C, Azevedo I. Ultrastructural study of brain microvessels in patients with traumatic cerebral contusions. Acta Neurochir (Wien). 1997;139:215–20.
Lu TS, Avraham HK, Seng S, Tachado SD, Koziel H, Makriyannis A, Avraham S. Cannabinoids inhibit HIV-1 Gp120-mediated insults in brain microvascular endothelial cells. J Immunol. 2008;181:6406–16.
Ge S, Song L, Serwanski DR, Kuziel WA, Pachter JS. Transcellular transport of CCL2 across brain microvascular endothelial cells. J Neurochem. 2008;104:1219–32.
Sowa G. Caveolae, caveolins, cavins, and endothelial cell function: new insights. Front Physiol. 2012;2:120.
Razani B, Engelman JA, Wang XB, Schubert W, Zhang XL, Marks CB, Macaluso F, Russell RG, Li M, Pestell RG, et al. Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J Biol Chem. 2001;276:38121–38.
Schubert W, Frank PG, Razani B, Park DS, Chow CW, Lisanti MP. Caveolae-deficient endothelial cells show defects in the uptake and transport of albumin in vivo. J Biol Chem. 2001;276:48619–22.
Anderson RG. The caveolae membrane system. Annu Rev Biochem. 1998;67:199–225.
Predescu SA, Predescu DN, Malik AB. Molecular determinants of endothelial transcytosis and their role in endothelial permeability. Am J Physiol Lung Cell Mol Physiol. 2007;293:L823–842.
Cameron PL, Ruffin JW, Bollag R, Rasmussen H, Cameron RS. Identification of caveolin and caveolin-related proteins in the brain. J Neurosci. 1997;17:9520–35.
Chen YG. Endocytic regulation of TGF-beta signaling. Cell Res. 2009;19:58–70.
Mitchell H, Choudhury A, Pagano RE, Leof EB. Ligand-dependent and -independent transforming growth factor-beta receptor recycling regulated by clathrin-mediated endocytosis and Rab11. Mol Biol Cell. 2004;15:4166–78.
Zschocke J, Manthey D, Bayatti N, Behl C. Functional interaction of estrogen receptor alpha and caveolin isoforms in neuronal SK-N-MC cells. J Steroid Biochem Mol Biol. 2003;84:167–70.
Kubale V, Abramovic Z, Pogacnik A, Heding A, Sentjurc M, Vrecl M. Evidence for a role of caveolin-1 in neurokinin-1 receptor plasma-membrane localization, efficient signaling, and interaction with beta-arrestin 2. Cell Tissue Res. 2007;330:231–45.
Monastyrskaya K, Hostettler A, Buergi S, Draeger A. The NK1 receptor localizes to the plasma membrane microdomains, and its activation is dependent on lipid raft integrity. J Biol Chem. 2005;280:7135–46.
Mineo C, Ying YS, Chapline C, Jaken S, Anderson RG. Targeting of protein kinase Calpha to caveolae. J Cell Biol. 1998;141:601–10.
Fillebeen C, Dehouck B, Benaissa M, Dhennin-Duthille I, Cecchelli R, Pierce A. Tumor necrosis factor-alpha increases lactoferrin transcytosis through the blood-brain barrier. J Neurochem. 1999;73:2491–500.
Calvo CF, Amigou E, Tence M, Yoshimura T, Glowinski J. Albumin stimulates monocyte chemotactic protein-1 expression in rat embryonic mixed brain cells. J Neurosci Res. 2005;80:707–14.
Hooper C, Pinteaux-Jones F, Fry VA, Sevastou IG, Baker D, Heales SJ, Pocock JM. Differential effects of albumin on microglia and macrophages; implications for neurodegeneration following blood-brain barrier damage. J Neurochem. 2009;109:694–705.
Hooper C, Taylor DL, Pocock JM. Pure albumin is a potent trigger of calcium signalling and proliferation in microglia but not macrophages or astrocytes. J Neurochem. 2005;92:1363–76.
Ralay Ranaivo H, Hodge JN, Choi N, Wainwright MS. Albumin induces upregulation of matrix metalloproteinase-9 in astrocytes via MAPK and reactive oxygen species-dependent pathways. J Neuroinflammation. 2012;9:68.
Ralay Ranaivo H, Wainwright MS. Albumin activates astrocytes and microglia through mitogen-activated protein kinase pathways. Brain Res. 2010;1313:222–31.
Tabernero A, Velasco A, Granda B, Lavado EM, Medina JM. Transcytosis of albumin in astrocytes activates the sterol regulatory element-binding protein-1, which promotes the synthesis of the neurotrophic factor oleic acid. J Biol Chem. 2002;277:4240–6.
Cacheaux LP, Ivens S, David Y, Lakhter AJ, Bar-Klein G, Shapira M, Heinemann U, Friedman A, Kaufer D. Transcriptome profiling reveals TGF-beta signaling involvement in epileptogenesis. J Neurosci. 2009;29:8927–35.
Hinson HE, Rowell S, Schreiber M. Clinical evidence of inflammation driving secondary brain injury: a systematic review. J Trauma Acute Care Surg. 2015;78:184–91.
Cao T, Pinter E, Al-Rashed S, Gerard N, Hoult JR, Brain SD. Neurokinin-1 receptor agonists are involved in mediating neutrophil accumulation in the inflamed, but not normal, cutaneous microvasculature: an in vivo study using neurokinin-1 receptor knockout mice. J Immunol. 2000;164:5424–9.
Schratzberger P, Reinisch N, Prodinger WM, Kahler CM, Sitte BA, Bellmann R, Fischer-Colbrie R, Winkler H, Wiedermann CJ. Differential chemotactic activities of sensory neuropeptides for human peripheral blood mononuclear cells. J Immunol. 1997;158:3895–901.
Souza DG, Mendonca VA, De A Castro MS, Poole S, Teixeira MM. Role of tachykinin NK receptors on the local and remote injuries following ischaemia and reperfusion of the superior mesenteric artery in the rat. Br J Pharmacol. 2002;135:303–12.
Annunziata P, Cioni C, Santonini R, Paccagnini E. Substance P antagonist blocks leakage and reduces activation of cytokine-stimulated rat brain endothelium. J Neuroimmunol. 2002;131:41–9.
Li PC, Chen WC, Chang LC, Lin SC. Substance P acts via the neurokinin receptor 1 to elicit bronchoconstriction, oxidative stress, and upregulated ICAM-1 expression after oil smoke exposure. Am J Physiol Lung Cell Mol Physiol. 2008;294:L912–920.
Toneatto S, Finco O, van der Putten H, Abrignani S, Annunziata P. Evidence of blood-brain barrier alteration and activation in HIV-1 gp120 transgenic mice. AIDS. 1999;13:2343–8.
Vishwanath R, Mukherjee R. Substance P promotes lymphocyte-endothelial cell adhesion preferentially via LFA-1/ICAM-1 interactions. J Neuroimmunol. 1996;71:163–71.
Ramnath RD, Bhatia M. Substance P treatment stimulates chemokine synthesis in pancreatic acinar cells via the activation of NF-kappaB. Am J Physiol Gastrointest Liver Physiol. 2006;291:G1113–1119.
Homayoun P, de Rodriguez Turco EB, Parkins NE, Lane DC, Soblosky J, Carey ME, Bazan NG. Delayed phospholipid degradation in rat brain after traumatic brain injury. J Neurochem. 1997;69:199–205.
Hellal F, Pruneau D, Palmier B, Faye P, Croci N, Plotkine M, Marchand-Verrecchia C. Detrimental role of bradykinin B2 receptor in a murine model of diffuse brain injury. J Neurotrauma. 2003;20:841–51.
Golias C, Charalabopoulos A, Stagikas D, Charalabopoulos K, Batistatou A. The kinin system—bradykinin: biological effects and clinical implications. Multiple role of the kinin system—bradykinin. Hippokratia. 2007;11:124–8.
Dobo J, Major B, Kekesi KA, Szabo I, Megyeri M, Hajela K, Juhasz G, Zavodszky P, Gal P. Cleavage of kininogen and subsequent bradykinin release by the complement component: mannose-binding lectin-associated serine protease (MASP)-1. PLoS One. 2011;6:e20036.
Trabold R, Eros C, Zweckberger K, Relton J, Beck H, Nussberger J, Muller-Esterl W, Bader M, Whalley E, Plesnila N. The role of bradykinin B(1) and B(2) receptors for secondary brain damage after traumatic brain injury in mice. J Cereb Blood Flow Metab. 2010;30:130–9.
Dewitt DS, Kong DL, Lyeth BG, Jenkins LW, Hayes RL, Wooten ED, Prough DS. Experimental traumatic brain injury elevates brain prostaglandin E2 and thromboxane B2 levels in rats. J Neurotrauma. 1988;5:303–13.
Liu B, Li H, Brull SJ, Zhang JM. Increased sensitivity of sensory neurons to tumor necrosis factor alpha in rats with chronic compression of the lumbar ganglia. J Neurophysiol. 2002;88:1393–9.
Richardson JD, Vasko MR. Cellular mechanisms of neurogenic inflammation. J Pharmacol Exp Ther. 2002;302:839–45.
Acknowledgements
None to declare.
Funding
The authors received funding from the Australian National Health and Medical Research Council (NHMRC) and the Neurosurgical Research Foundation (NRF).
Availability of data and materials
The results supporting the conclusions of this review are published and have been fully referenced in the reference list.
Authors’ contributions
FC, KM, and AL performed the literature review and drafted the paper under the supervision of RV. RV graphed the illustration. All authors discussed and edited the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
This review summarizes published studies involving human and animal material. Research studies included in this work were approved by appropriate ethics committees.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Corrigan, F., Mander, K.A., Leonard, A.V. et al. Neurogenic inflammation after traumatic brain injury and its potentiation of classical inflammation. J Neuroinflammation 13, 264 (2016). https://doi.org/10.1186/s12974-016-0738-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12974-016-0738-9